
Abstract
Alzheimer’s disease (AD) is a paramount chronic neurodegenerative condition that has been affecting elderly people since the 1900s. It causes memory loss, disorientation, and poor mental function. AD is considered to be one of the most serious problems that dementia sufferers face. Despite extensive investigation, the pathological origin of Alzheimer’s disease remains a mystery. The amyloid cascade theory and the vascular hypothesis, which stresses the buildup of Aβ plaques, have dominated research into dementia and aging throughout history. However, research into this task failed to yield the long-awaited therapeutic miracle lead for Alzheimer’s disease. Perhaps a hypothetical fragility in the context of Alzheimer’s disease was regarded as a state distinct from aging in general, as suggested by the angiogenesis hypothesis, which suggests that old age is one state associated with upregulation of angiogenic growth factors, resulting in decreased microcirculation throughout the body. There has also been evidence that by controlling or inhibiting the components involved in the sequence of events that cause angiogenesis, there is a visible progression in AD patients. In Alzheimer’s disease, one such antiangiogenic drug is being used.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Alzheimer’s disease (AD) is a prime cause of dysfunction and reduced standards of living in the aged population (Kawas and Brookmeyer 2001). The principal pathological attributes are the generation of senile plaques and vasculature with amyloid-β angiopathy. Both comprise agglomerates of α-, β-, Γ-, and δ-secretase-derived 40–42 residue amyloid precursor protein (APP) segments (Beyreuther et al. 1991; Bayer et al. 2001). Additionally, neurofibrillary tangles (NFTs) are located in neuronal cells which chiefly comprise hyperphosphorylated tau proteins (Lowe et al. 1993). Accordingly, uncertainties in amyloid-β42 and tau protein concentration in the cerebral regions are typical hallmarks of AD (Kanai et al. 1998). Despite several factors that have been recognized to impact the pathological framework of AD, adjacent interactivity between the independent pathological mechanisms and even proteins is believed to mobilize the disease (Takashima et al. 1993). Lately, damaged cerebral blood flow has been recognized in both the etiology and pathophysiology of AD. Multiple pieces of research have manifested persistent cerebral hypoperfusion in patients with AD (De jong et al. 1997), and it was proposed that counting simultaneously with endothelial aberration, hypoperfusion is a central factor in the progression of AD. At least 1/3rd of AD cases possibly evince substantial cerebrovascular pathophysiology, which is condensed as small vessel disease (Vinters et al. 1996; Moody et al. 1997). Though Aβ itself is regarded to be supremely neurotoxic (Hardy et al. 1998), increased expression of cytokines and stimulation of microglial cells and astrocytes catalyze further neuronal degradation in the vicinage of Aβ plaques (Frautschy et al. 1998). Also, support for the Aβ-inflammation-angiogenesis relationship in AD arises from analytical research associated with expression degrees of certain genes under the aegis of DNA array and proteomic studies (Lukiw et al. 2000). Endostatin (ES) is a 20-kDa COOH-cleaved segment derived from structural protein, specifically type XVIII collagen, that functions as an endogenous angiogenic forbidder by modifying the pro-angiogenic action of numerous growth modulators. FGF (fibroblast growth factor) and vascular permeability factor (VEGF) (Ständker et al. 1997) prevent endothelial expansion and translocation in vitro and limit angiogenesis and tumor formation in vivo by accelerating endothelial cell apoptosis (Dhanabal et al. 1999a). Biochemical analysis disclosed that the capability of ES to impede angiogenesis is induced through zinc binding and elastase refining (Wen et al. 1999), as the general endostatin level was located in elastic fibers in the vessel walls (Miosge et al. 1999). Intriguingly, endostatin decreases intimal angiogenesis and plaque development in the apolipoprotein E (ApoE)–deficit model (Moulton et al. 1999). Such discoveries are of particular interest, as ApoE is a prime genetic peril for AD (Roses 1995). Dual-labeling studies disclosed the association of endostatin in Aβ plaques and tau deposits, enclosed by focal gliosis. Furthermore, the Western blotting technique discovered elevated endostatin expression in an AD patient in contrast to controls (Moulton et al. 1999). Preliminary data revealed that endostatin is liberated by neural cells to deposit in Aβ plaques in AD brains (Deininger et al. 2002).
This review targets the potential role of endostatin in pathological studies of Alzheimer’s disease in association with deposits of Aβ plaques, which are considered among the most potent angiogenic inhibitors. It also elaborates on the new hypothetical basis in the context of the pathophysiology of AD, namely, the angiogenesis hypothesis, which could be regarded as a novel approach in acknowledging the progression of AD, consequently disclosing the complex novel interactions among the angiogenic modulators and ES, unfolding new approaches for the expansion of novel treatment policies in context to AD. The writer aims to represent the significance of ES as a possible therapeutic target and the role of angiogenesis in the pathology of AD along with diverse modulators of angiogenesis interacting with endostatin in one or another way thus facilitating the advancement in a suitable therapeutic regime for AD.
Angiogenesis hypothesis
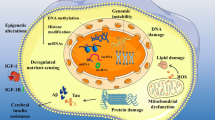
Among the diverse neurodegenerative diseases, Alzheimer’s disease, a persistent form of nerve cell atrophy, is well known to affect older humans and is the habitual origin of dementia (Weller and Budson n.d.). Alzheimer’s disease is regarded as a democratic process and as a persistent form of neuronal cell degeneration among several neurodegenerative diseases (Katzman 1993). The maximum number of AD cases has been outlined among individuals belonging to or above the 65-year age group (Evans et al. 1989; Lyketsos et al. 1997). AD outlines nearly around 2/3 of prevailing cases of senile dementia (Ol 1994). It is now the principal chore to recognize the preliminary risk factors for AD because several neurodegenerative processes underlying Alzheimer’s disease may start at midlife. Locating these risk factors may enlighten the pathophysiology of AD and also come up with novel possible avenues for its prevention and therapy (Kivipelto et al. 2001). Although the precise root of Alzheimer’s disease has been a concern so far, the amyloid cascade hypothesis is the most described and well-considered visional core of AD. After a while, this hypothesis has gone through several variations, firstly in the explanation of the pathogenic identity of Aβ that is proposed to start detrimental episodes giving rise to AD. The primary proposal that senile plaques are disease-causing is currently fired on a specific basis (Terry et al. 1991). Eventually, the recent idea that higher series soluble agglomerates of Aβ plaques are the chief disease-causing agents captivated considerable attention (Glabe 2005). The complete pathogenesis of AD is composite and multi-factorial. Despite that Aβ even now is distinctly connotated in AD, the field has been suggested to review another hypothesis besides Aβ as the central core of the disease (Kalaria 2002). There are arising manifestations that alterations in the vasculature of cerebral and peripheral regions lead the way to altered blood flow to the brain and may act as a condition for progressing AD (Dickstein et al. 2010). This reconsideration is demonstrated in the “angiogenesis hypothesis,” which suggests that old age is a part of a lacking state associated with a reduction in angiogenic growth factors, which successively generates decreased microcirculation all over the body (as shown in Fig. 1). This insufficiency is considered to be responsible for the development of early signs and symptoms of old age, namely decreased stamina, developing muscle weakness, cold intolerance, and insignificant memory lapses. The last may lead to the development of senile dementia. This possible theory of AD pathogenesis emerges via diverse investigational AD considerations, including blood-brain barrier dysfunction as well as compromised cerebral circulation (Bookheimer et al. 2000; Iadecola 2004), and has been assessed, proposing that impaired clearance processes at the BBB impact the aggregation of Aβ protein in the AD cerebrum (Bell and Zlokovic 2009). Novel studies have shown designate that pathological brain angiogenesis (abnormal evolution of new cells from the previous ones) potentially arises as a result of Aβ plaque deposition followed by BBB dysfunction in AD (Vagnucci Jr and Li 2003). A vital move in acknowledging the source of the onset of Alzheimer’s is distinguishing what the beginning ways are involved in the neurodegenerative event. The 2 chief vascular forerunners under the angiogenesis hypothesis for the neurodegenerative alterations and Aβ accumulation arising in AD are the failure of the BBB permeability (Ujiie et al. 2003) and impaired CBF (hypoperfusion) (Dickstein et al. 2006). Additionally, the inflammatory alterations noticed in AD patients provide a route for the advancement of angiogenic regulators such as VEGF, leading to abnormal angiogenesis resulting in Aβ plaques that might get advanced (Desai et al. 2009). Also, compromised BBB in AD may cause agglomeration of undesirable and active neurolysin entities in the brain (Zlokovic 2008). CBF residing in the brain acts as a chief factor affecting BBB permeability and Aβ concentration in the brain, thus giving rise to increased permeability when critically lowered. Aging and the existence of the ApoE ϵ4 allele also cause a reduction in CBF (Thambisetty et al. 2010). The case for angiogenic pathology in Alzheimer’s disease is reinforced by the direct relationship between dementia and cerebrovascular amyloid angiopathy (CAA), which occurs in around 90% of total AD cases (Vinters 1987). CAA was identified by utilizing the appearance of Aβ deposits in the neuronal and pial small capillaries or arteries in association with dementia (Holton et al. 2001; Jellinger 2007). Furthermore, the emission of angiogenic cytokines like TNF-α, VEGF, and thrombin through upregulation of inflammatory processes in AD implies an angiogenic process. The exact mechanism by which Aβ provokes angiogenesis is extremely preserved and may be intervened by the activity of notch signaling and Γ-secretase (Boscolo et al. 2007). The angiogenesis hypothesis supports that Alzheimer’s may occur due to an age-associated reduction in new blood vessel formation, which may affect blood flow in smaller vessels. As a consequence, it could be considered as a novel approach to understanding the progression of AD and dementia (Ambrose 2016) (as shown in Fig. 1).

The angiogenesis hypothesis is known to be catalyzed by various pro-angiogenic growth factors as listed in the figure, which ultimately causes cell death as observed in Alzheimer’s disease
Endostatin and its architectural background
Endostatin (ES) is a derivative of protein, namely collagen XVIII, attained after cleavage from the COOH terminal, which is a member of the multiplexen subfamily and is mostly expressed as heparan sulfate proteoglycan collagen (HSPGs) in the vascular epithelium as well as the basement membrane. The collagen XVIII is commonly found in 3 forms—short, middle, and long isoform. ES is known to exhibit inhibition activity against angiogenesis that could be due to its heparin-binding and elastase processing (Ding et al. 1998). The molecular mechanism supporting the angiogenesis inhibiting activity of endostatin has not been explained so far (van Horssen et al. 2002). Recently, endostatin has been examined as aggregates in neuronal and paracellular Aβ deposits associated with AD through isolation from well-defined media of hemangioendothelioma (EOMA) cells (Bächinger 2005). Modern studies also suggest that endostatin suppresses several biological cycles and various pathways related to angiogenic actions. Endostatin is the foremost indigenous antiangiogenic agent that came across as a segment of the extracellular matrix and is the most researched among multiple angiogenic inhibitors (Abdollahi 2004). The development of endostatin involves 2 prime steps—first, a metal-reliant step producing endostatin-comprising large segments, which on further cleavage by a proteolytic enzyme, elastase, and the addition of an N-terminal section to the fragment, makes it identical to the functional endostatin (O'Reilly et al. 1997). The three distinct forms of cathepsins (proteases) that can produce ES are type B, type L, and type K, of which type L has maximal efficacy whereas type K has the least efficiency (Walia et al. 2015). Cathepsins L and B are also known to possess activity for the degradation of endostatin (Ferreras et al. 2000). The complex structure of ES is edged by an NH2-terminal domain and a COOH-terminal domain. It has 10 proteinaceous collagen domains distributed within 11 non-collagenous domains (as shown in Fig. 2). In addition, it also has 3 alpha-1 chains which on oligomerization form a homotrimeric collagen type XVIII, a hinge domain for protease target, and an antiangiogenic domain having 20 kDa of molecular weight (Digtyar et al. 2007). The human endostatin monomer form contains a 7-strand beta-sheet (E, F, A, P, M, O) which has a fibronectin-like plasma structure (as shown in Fig. 3) (Gordon and Hahn 2010). Recombinant endostatin (rES) isolated from transfected human nephrons exhibits marked affinity for heparin (Hohenester et al. 1998). The 3D examination of the ES fragment by X-ray chromatography at a resolution of 1.5 Å disclosed a closely packed molecule possessing a huge basic set of 11 arginine (Arg) remnants comprising the heparin attaching set assembled into 2 distinct zones: a large site consisting of Arg 24, 27, 53, and 139 and a small site comprising Arg 62 and 63. This particular set of residues was recently shown to exhibit the inhibiting activity of induced angiogenesis (Sasaki et al. 1999). At first, it was proposed that Zn-binding activity is necessary for angiogenic inhibiting activity. Subsequently, reports have failed to validate the relationship between Zn-binding and antiangiogenic activity or inhibition of endothelial cell proliferation (Yamaguchi et al. 1999). Lately, it was reported that zinc most probably plays a structural instead of a functional role in endostatin (Wickström et al. 2004; Faye et al. 2009). Though, it was supported that heparan coordinates with the arginine clumps of endostatin through sulfate anions, regulating the antiangiogenic activity (Blackhall et al. 2003). It was also suggested that sulfate is the most essential anion required for stabilizing the folding median at acidic pH so that heparan sulfate can adhere to endostatin (Ricard-Blum et al. 2004; Goto et al. 1990) (Fig. 2).

Schematic representation of generation and structure of endostatin. The endostatin is produced by the activity of several enzymes such as matrix metalloproteinases (MMPs), cathepsins, and elastases on NC1 domain of collagen XVIII

Pleiotropic effects of endostatin. The protective role of endostatin in Alzheimer’s disease is based upon its ability to downregulate and upregulate the pro-angiogenic factors and antiangiogenic factors, respectively, thus maintaining the range of normal activity of angiogenesis in the body
Emerging roles of endostatin as a dynamic antiangiogenic agent in AD
Endostatin has strong anti-endothelial cell activities which include cell multiplication, migration, binding, and survival, which are all essential for angiogenesis (Dhanabal et al. 1999b; Dixelius et al. 2000). The accurate molecular expression of endostatin, however, remains unclear, despite multiple ES attaching proteins have been implicated in its activity (Karumanchi et al. 2001). However, even if these receptors are required, the antiangiogenic action of endostatin remains an enigma. Endostatin binding proteins on the endothelial cell membrane. Nucleolin (NL) isolated from HMECs was cited as a vital part of regulating the antiangiogenic action of endostatin (Shi et al. 2007). Furthermore, endostatin binds to heparan sulfate, tropomyosin, caveolin-1, VEGFR-1, and VEGFR-2 and was the latest molecule to rely on E-selectin expression to be efficiently active (Ricard-Blum et al. 2004; Yu et al. 2004). Endostatin also affects basic fibroblast growth factor (bFGF) producing signal transmission, which inhibits endotheliocyte migration, thus provoking apoptosis via cyclin D1 inhibition and arresting the cell division of endotheliocytes in the G1 phase (Dixelius et al. 2002). Under the influence of ES, the TNF-α intervened actuation of the JNK pathway is inhibited, consequencing the repression of the pro-angiogenic gene expression controlled by JNK (Yin et al. 2002). The potential of endostatin to inhibit angiogenic stimulators by stimulating their antagonists appears to be an intermittent matter in the above considerations. A considerable realization emerging from such investigations is that distinct pathways are believed to work simultaneously to accomplish an organized antiangiogenic activity after endostatin therapy (Abdollahi et al. 2004). A possible role of endostatin in respective facets of the pathological studies of AD has been previously elucidated. HSPGs have the potential to upregulate fibril formation as indicated, both in vivo and in vitro in a rat model, along with the inhibition of proteolytic disintegration of amyloid plaques by stabilizing these deposits (Gupta-Bansal et al. 1995; van Horssen et al. 2002). Besides, immuno-chemical aspects discovered that heparan sulfate proteoglycans are associated with Aβ plaques, tau deposition, and cerebrovascular amyloid protein aggregates (Verbeek et al. 1999; Salza et al. 2015). Moreover, sufferers of Down’s syndrome have a higher probability of acquiring AD, having a considerably large extent of endogenous endostatin (Zorick et al. 2001). Multiple considerations were implicated in describing the specific role of endostatin in AD. A couple of novel bio-marker aspirants have been proposed for distinctive detection of Alzheimer’s in isolated studies under which the proportion of endostatin/Aβ42 in CSF was manifested to upgrade the discovery approaches of AD (Oeckl et al. 2016; Sato 2017). It has also been investigated in preliminary studies that endo-all-Ala (a contrasting transfected form of wild-type endostatin lacking disulfide pairs) instantly forms amyloid-like fibrils at a moderately low pH. This low pH constantly resides in older brains accompanied by cerebral acidosis (Yates et al. 1990). Contrastingly, endo-all-Ala was shown to possess larger hydrophobic stains at this pH along with inferior stability, which may contribute to its amyloid plaque deposition tendency. This discovery suggests that the disulfide linkages are of marked significance in preserving corroboration and hindering the fibrilization of ES (He et al. 2006). However, endostatin is usually more prone to partial denaturation and experiences subsequent aggregation steps directing to the formation of compactly arranged aggregates. Primarily, these partly unfolded proteins or their fragments combine and form small soluble aggregates, leading to the development of protofibrils. Eventually, mature fibrils assemble and are accumulated as plaques. Such fibrillar agglomerates are categorized as amyloid plaques based on the presence of cross-β form (piled β sheets consisting of flat and non-twirled β strands), not viewed in globular proteins. In Alzheimer’s disease, endostatin is related to Aβ plaques adjoining focal gliosis (Gebbink et al. 2004). Recently, research showed that endostatin is discharged by neural cells to deposit it in Aβ plaques in AD. Moreover, the current uncoverings suggest that the disturbances in endothelial functioning in such communities of AD victims resulted in distinct modifications in mediators of angiogenesis. In summation, peripheral endostatin concentrations allow intervention and association amid endothelial functioning and cognitive activity in a community at higher risk of acquiring AD. Ultimately, endostatin can certainly intervene in the connection between endothelial function and cognitive activity (Isaacs-Trepanier et al. 2020). Accordingly, endostatin is an example of an endogenous broad-spectrum antiangiogenic particle mediating several factors, as shown in Fig. 3.
Endostatin downregulates VEGF
Vascular endothelial growth factor (VEGF), or vascular permeability factor (VPF), was initially illustrated as an endothelial cell-distinct mitogenic agent that assists cell proliferation and is a powerful agent for mediating vascular porosity (Watson et al. 2000). The gene encoding for VEGF is located on chromosome 6p21.3 and is meant to be regulated by a collection of modulators, including cytokines, lipopolysaccharide (LPS), hormones (e.g., progesterone), and hypoxia. VEGF signaling is managed by crosstalk with multiple receptors largely. Receptor tyrosine kinases (RTKs), namely, VEGFR-1, VEGFR-2, and VEGFR-3, vary significantly in their signaling activities (Ferrara 1999). VEGF binds to the associated VEGF receptor (VEGFR) to cause receptor homodimerization/heterodimerization, directing to actuate the Tyr-kinases along with auto-phosphorylation of Tyr remnants in the intracellular domains of the receptor to mediate immediate responses. VEGFR-1 participates in an essential role in anatomical and pathological angiogenesis. VEGFR-2 is considered the initiator of endothelial cell proliferation that straightly controls tumor angiogenesis, whereas VEGF receptor-3 joins with VEGF-C and VEGF-D to increase endothelial cell signaling and reproduction (Yehya et al. 2018). VEGF signaling accelerates visual angiogenesis all over the brain infarcts. Possibly, the condition of brain hypoxia induces the expression of VEGF, which mediates angiogenesis. Contrastingly, only minute levels of VEGF can be traced in a healthy adult brain (Tarkowski et al. 2002). The polymorphism activity within the VEGF promoter lately was related to the possibility of acquiring AD. The degree of cognitive decline was affected by an evident interaction between the APOE 4 allele and the VEGF A allele in AD patients. A prominent degree of plasma VEGF has appeared in AD patients as compared to controls (Rogers and D'Amato 2006). VEGF appearance was remarked in overactive astrocytes and pericardiac aggregates in AD patients, indicating the existence of modulatory systems recompensing for inadequate vascularization and decreased psychical perfusion; elevated VEGF immune activity was located in the neocortex of AD patients, implicating major neurotrophic outcomes in the mental degeneration cascade. Though, the deposition of VEGF in the Aβ deposits in AD brains has been lately outlined (Del Bo et al. 2009). In the state of AD, there are merging manifestations concerning upregulation and downregulation of expression of the VEGF A gene in the blood, brain, and CSF. For instance, the degree of proteins of the VEGFA gene was described as being elated in the case of patients with AD as compared to healthy cases largely in plasma and the central parietal cortex (Miners et al. 2016). Furthermore, increased VEGF concentration in the case of AD has been identified with lack of pericytes, enhanced permeability of the BBB, and chronic basis of tangle pathophysiology. Besides, there is an indication of suppression of VEGF A in the case of Alzheimer’s pathology, provisional to the sampled area of the brain and the VEGF A isoform. For instance, they remarked superior VEGF189 levels and inferior VEGF121 levels in the hippocampus region of AD brains in contrast with the adult brain. Despite the complicated and varied indications of AD with regard to VEGF levels, there are multiple emerging indications that overexpression of VEGF may possess a neuroprotective function (Hohman et al. 2015). Consequently, its application could have major significance in the treatment of AD. There is advancing acknowledgment of the occurrence of factors that conquer neovascularization, consequently allowing a counterbalance to pro-angiogenic proliferating agents like VPF. For example, endostatin suppresses angiogenesis by acting indirectly downregulating MMP-9 and therefore hindering the discharge of ECM cohered VEGF. It can act directly on endothelial tissue via CD36 (membrane protein) as well (Im et al. 2005). This study provides major novel understandings into the mechanisms of action of this antiangiogenic agent, which may employ a direct effect on endothelial cells in the context of AD.
Endostatin mediated inhibition of MMPs
The matrix metallopeptidases/MMPs or matrixins belong to a family of membrane-coupled neutral endopeptidases constituting a broad range of substrates formed by numerous cell categories, namely, epithelial tissue, fibroblasts, and inflammatory cell lines. Matrixins are generated by multiple endothelial tissues, namely, MMP-1, MMP-2, MMP -9, and MMP -14 (Stetler-Stevenson 1999). The standard architecture of MMPs possesses an NH2-edged zymogenic propeptide region, a metal-based catalytic region, a linker domain, and a COOH-edged hemopexin-like region. The matrixin family can be sub-grouped into 6 sub-classes, namely, (1) gelatinases, (2) stromelysins, (3) collagenases, (4) MMP membrane-type, (5) matrilysins, and (6) remaining MMPs showing a vast range of functions and actions in different tissues (Quintero-Fabián et al. 2019). MMPs have also been manifested indirectly affecting the neovascularization process by adjusting EC adhesion, proliferation, and migration. Despite this, MMPs have been considered to be pro-angiogenic in nature, as some MMPs are also complex in the hindrance of angiogenesis by releasing matrix-based angiogenesis hindering factors, including endostatin from its precursor protein in the ECM (Öhlund et al. 2008). On the other hand, the result of various MMPs is to deteriorate APP, causing agglomeration of Aβ peptides along with the magnified expression of matrixins as regarded in association with postmortem brains of AD patients, proposing the action of MMPs in pathological studies of AD. Of these, the MMP-9 was observed to take part in the most essential step in the pathology of AD. Over-activity of MMP-9 was found to be associated with a matrix of neural cells, neurofibrillary tangles (NFTs), amyloid plaques, and vessels of the hippocampus and cerebral cortex of AD patients. Apart from the MMP-9 isoform, MMP-2 also possesses medicinal importance in the pathological study of AD. As designated in research, the anti-MMP agent, which is presumably largely specified for MMP-9 and MMP-2, inhibits Aβ-induced liberation of lactate dehydrogenase in primary cultured neurons, proposing the contribution of MMP-9 and MMP-2 in Aβ-induced neurotoxicity (Felbor et al. 2000; Brkic et al. 2015). Endostatin is intended for inhibiting the activation of proMMP-2 in endothelial cell culture. For the research on the association between anti-invasive actions of endostatin and blocking of activities of MMPs, the action of endostatin on the concentration and signaling of MMPs in human umbilical vein endothelial cells (HUVECs) was done. Gelatin zymography of the nutrient media was executed and the results were noted, which specifies that activation of proMMP-2 released from HUVECs can be blocked by endostatin. For further confirmation that the inhibition of proMMP-2 activation resulted from the existence of endostatin, endostatin was restrained from the purified endostatin system with the help of an anti-FLAG antibody bead accompanied by inspection of activation of MMPs. The endostatin drained sample entirely lost its inhibitory action on proMMP-2 activation as demonstrated by gelatin zymography and Western blotting technique. Additionally, it was unable to block endothelial cell invasion. Thus, it was confirmed that endostatin inhibits proMMP-2, emphasizing its role in the treatment of AD (Kim et al. 2000).
Endostatin mediated activity of angiopoietins
The earliest studies regarding tyrosine protein kinases (tyr-PrK) were demonstrated by endothelial cells (EC) and murine cardiogenesis guided to acknowledge the TEK gene for the Tie-1 and Tie-2 family of receptor Tyr-kinases (Fagiani and Christofori 2013). The angiopoietin (Ang)-Tie ligand receptor complex plays a central role in directing vascular adherence and latency. Despite its role in angiogenesis, it also acts as a principal modulator in states like inflammation. Activation of Ang-1-negotiated Tie2 is needed to support the latent phase of the endothelium, whereas Ang-2 sabotages the dormant endothelium, potentiating it to acknowledge any external stimulant, thus advancing the expression of inflammatory (TNF and interleukin-1) and proliferating (VEGF) cytokines (Fiedler and Augustin 2006). Angiopoietins largely Ang-1, Ang-2, Ang-3, and Ang-4 are regular substrates for the Tie-2 RTKs, which are highly expressed in EC and hematopoietic cells (Morisada et al. 2006). The considerable inspection of the present studies suggests that Angiopoeitin-1 plasma concentrations are atypically upregulated in Alzheimer’s cases in contrast to healthy brains. Secondly, considering the activity of abnormal Ang-1 plasma concentrations and MMSE concentrations in patients with AD, the extent of Ang-1 is inversely related to MMSE levels, assessed in the AD community, highlighting that a low grade of cognitive performance is linked to elevated Ang1 plasma concentrations (Schreitmüller et al. 2012; Gurnik et al. 2016). Angiopoietin-like 4 (ANGPTL-4), a hypoxia catalyzed regulator, is expressed largely in reactive astrocytes when signified in postmortem brains of AD patients, which consecutively increases migration and sprouting of endothelial cells. These discoveries suggest that the location of angiopoietin levels may assist the persisting diagnostic techniques for Alzheimer’s disease (Chakraborty et al. 2018). In addition, FOXA-2 has been traced in association with the existence and progression of Alzheimer’s disease. However, it remains enigmatic concerning the specific action of FOXA-2 in AD. The bioinformatic examination specified that FOXA-2 may serve as an upregulated goal in the Ang-1-prompted amyloid-β synthesis (Peng et al. 2020). Endostatin directly inhibits angiogenesis by binding to angiopoietin or indirectly by inhibiting angiopoietin activity with Flk-1 and Flt-1 to downregulate VEGF-mediated Tyr-phosphorylation of VEGF-receptors. Endostatin competitively inhibits angiopoietin’s attachment to its receptors and contrastingly behaves as an antagonizing angiogenic factor by blocking angiopoietin-activated EC propagation and relocation in the brain (Klagsbrun and Moses 1999; Oikonomou et al. 2011; Mohamed et al. 2011).
Endostatin suppressed FGF expression
The first fibroblast growth factor (FGF), or commonly known as the basic fibroblast growth factor—bFGF, was recognized as the pro-angiogenic unit. Presently, the FGF family is familiar with possessing not less than 20 factors, out of which approx 30–70% possess similar orders of primary amino acids (Cross and Claesson-Welsh 2001). FGF-2 is well known to have a high capacity to prompt angiogenesis. The capability of FGF-2 to persuade cells to seize the basic matrix to form a capillary-like network was signified through a preclinical system of endothelial cells grown on a 3D gel of collagen. Exceptionally, FGF-2 was ascertained to be more potent than VEGF over distinct analysis (Lieu et al. 2011). FGFs execute distinct biological activities via inciting transmembrane Tyr-kinases and integrating their biological cycles into several transducing operations such as phosphoinositide-3-kinase (PI3K), phospholipase-C-Γ(PLC-Γ), and mitogen-actuated protein kinases (MAPKs) pathways (Chen et al. 2020). FGF-2 produces angiogenic action by upregulating VEGF signaling in stromal and endothelial cells. Moreover, FGF expression regulates the responsiveness of VEGFR-2 signaling (Murakami and Simons 2008). The indication of bFGF is upregulated in AD patients and is traced in association with the lesions characterizing this disease. In healthy cases, b-FGF was accounted to be broadly scattered largely in the 3 brain regions, namely, the prefrontal cortex, hippocampus, and hypothalamus, but is disrupted in respect of AD (Stopa et al. 1990). On the other hand, FGF-2 was also implicated in being advantageous in respect of Alzheimer’s disease by lowering the expression of the APP proteolyzing enzyme BACE-1, which is known to generate Aβ. This depletion was analogous to the reduction in Aβ agglomeration, tau-phosphorylation, and structural cognitive decline (Turner et al. 2016). Lately, it was also suggested that FGF-2 was not released from astrocytes/microglia but by oligomeric Aβ from hampered neurons (Noda et al. 2014). In the presence of endostatin, the disturbance in structural basis mediated by FGF-2 was specified as constituting docking of the vessel system, a remarkable decline in the plexus, thinning and stretching of the vessels, and a reduction in the number of EC to basal levels. Furthermore, alteration of FGF signaling by endostatin obstructs endothelial cell relocation by disturbing cell-matrix adhesiveness, cell-cell adhesions, and cytoskeletal organization (Eriksson et al. 2003; Sasaki et al. 1999).
Endostatin regulates expression of TNF-α
Modern analysis has shifted focus to cytokine-mediated neuroinflammation as a prime donor in the advancement of Alzheimer’s disease, and pieces of evidence show that inflammation advances pathological activities that catalyze AD (Tarkowski et al. 2003; Akiyama et al. 2000). Amid the cytokines concerned with neuroinflammation, tumor necrosis factor-α (TNF-α) is the most researched that essentially plays a role in the cytokine cascade in the course of inflammation. However, the expression of TNF-α in the peripheral and central nervous systems of healthy adults is kept at very low concentrations. The expression of this cytokine is remarkably raised in the blood and CNS of AD patients, demonstrated as a link by diverse clinical studies (Fillit et al. 1991). TNF-α initially acknowledged for its anti-neoplastic activity is one of the most researched inflammatory cytokines in association with the progression of inflammation. TNF-α (17 kDa) is a non-glycosylated, monomeric type 2 transmembrane protein belonging to a superfamily of receptor proteins known as the TNF-receptor (TNFR) superfamily proteins (Chang et al. 2017). TNF-α mediates its activity by joining with two different types of receptors, namely, type 1 (TNF-R1), high-affinity receptors ubiquitously expressed on cell surface except on erythrocytes, and type 2 (TNF-RII) majorly expressed in myeloid cells, endothelial cells, microglia, astrocytes, and universe population of neuronal cells (Wang and Al-Lamki 2013). Additionally, remarked angiogenesis located in brains with AD was found to be downregulated by endostatin. The AD brain is a hypoxic atmosphere that eventually upregulates the progression of neovascularization. The enhanced neuronal cell division urges a further stipulation on the vascular system, advancing asphyxia. This causes angiogenesis and consequently infiltration and dysplasia (Kasama et al. 2001). Analytical data suggest that endostatin has a potent protective action on the succession of AD that is associated with its suppressing activity on VEGF signaling along with downregulation of TNF-α synthesis. As a consequence, it was considered that ES is a potent agent for a novel therapeutic regime for AD due to its antiangiogenic property (Hu et al. 2012). This antiangiogenic activity of endostatin was derived from its distinct antagonizing effect on TNF-α synthesis by hepatic sinusoidal endothelium (HSE) concerned with tumor-released VEGF. It manifests that endostatin averts VEGF-prompted Tyr-phosphorylation of kinase insert domain receptor (KDR)/Fc receptor (Flk-1) primarily located in cultured HSE cells, obstructing VEGF-expressed signaling (Mendoza et al. 2004).
Endostatin downregulates HIF-1α
Hypoxia-inducible factor-1 (HIF-1) is a proteinaceous heterodimeric transcription component that controls the oxygen environment and biological response during the hypoxic state. In 2004, Moeller et al. disclosed that angiogenic signaling is controlled by a prominent angiogenic mediator, hypoxia-inducible factor-1. HIF-1 possesses 2 subunits, namely, HIF-1α (tightly mediated) and HIF-1β (loosely regulated), belonging to a family of basic helix-loop-helix (bHLH) transcription factors comprising a PAS (Per-ARNT-Sim) pattern. The 3 isomers of the α-unit, namely, HIF-1α, HIF-2α, and HIF-3α, have been established so far in the human genome (Rezvani et al. 2011). A progressive reduction in oxygen expression cellularly catalyzes the advancement of HIF-1 actions through fixation of the HIF-1α peptide. The binding of HIF-1α to DNA directs the transcriptional advance of genes that propitiate anaerobic metabolism, oxygen-carrying capability, and vasodilatation. HIF-1 can also couple with the gene encoding for VEGF and is necessary for transactivation of VEGF concerning hypoxia. Accordingly, HIF-1 is a fundamental transcriptional factor for metabolic adjustments and VEGF-intervened angiogenesis regarding hypoxia (Ravi et al. 2000). Also, tumor reoxygenation may induce nuclear aggregation of hypoxia-inducible factors in concerned reactive oxygen species and also increase transcription of HIF-1-mediated transcripts secondary to tense granule depolymerization (Moeller et al. 2004). The fusion of an angiogenesis inhibiting regime and antagonizing agent of HIF-1 may permit significant effectiveness as an angiogenic inhibitor that would discontinue the tumor’s vascular supply (Xu et al. 2013). It was also reported that endostatin can hinder angiogenesis indirectly by inhibiting HIF-1-activated involvement of VEGF signaling in Aβ deposited neural cells. However, the consequence of ES on HIF-1/VEGF signaling has not been acknowledged to date (Macpherson et al. 2003). Although detailed research in the last decade has evolved the hypoxia signaling pathway as a vital remedial target for several diseases, including AD. Even so, multiple questions still abide concerning the identification of the type of pro- or antiangiogenic factors elaborated in angiogenesis that can be described by acknowledging the signaling pathways catalyzing the downregulation of angiogenesis through a negative feedback mechanism. Contrastingly, the specific activities of these negative feedback regulators are an uplifting prospect for AD treatment (Messmer-Blust et al. 2009) (Table 1).
Future prospects of endostatin in AD
Angiogenesis is the process of forming new blood vessels from pre-existing ones, and it is now recognized as a critical process in a variety of developmental and pathological diseases. Alzheimer’s disease (AD), a progressive neurological illness, is one such condition. Several activities or angiogenesis hypotheses have been proposed to define emerging roles of angiogenesis in the pathogenesis of Alzheimer’s disease, implying the potential for future therapeutic interventions in the treatment of Alzheimer’s disease. Endostatin is deposited in neurons of all cortical layers and amyloid-beta plaques in AD patients, as evidenced by detecting the concentration of endostatin in CSF released by cerebellar Purkinje cells or the damaged blood-brain barrier, in contrast to normal instances. It may reveal a link between collagen type XVIII gene expression and the likelihood of developing Alzheimer’s disease, indicating a biological function for the extracellular matrix in the pathophysiology of Alzheimer’s disease. Despite this, endostatin has antiangiogenic properties and is currently undergoing stage 2 clinical trials to see if it can be used as a powerful angiogenesis inhibitor. Endostatin, also known as endostar, was acknowledged for its antiangiogenic activity as a therapeutic benefit. As a result, following the completion of its successful studies, it may be approved for use as an antiangiogenic agent in therapy.
Conclusion
The role of angiogenesis in cognitive decline has emerged as a critical factor to consider in understanding, preventing, treating, and reversing cognitive degeneration in Alzheimer’s disease. Angiogenesis is currently one of the most dangerous processes discovered in the pathophysiological underpinning of Alzheimer’s disease. Several current therapeutics are being developed to prevent or treat angiogenesis-related neurodegeneration and eventually death that occurs in Alzheimer’s disease. The role of angiogenesis in cognitive decline has emerged as a critical factor to consider in understanding, preventing, treating, and reversing cognitive degeneration in Alzheimer’s disease. Angiogenesis is currently one of the most dangerous processes discovered in the pathophysiological underpinning of Alzheimer’s disease. Several current therapeutics are being developed to prevent or treat angiogenesis-related neurodegeneration and eventually death that occurs in Alzheimer’s disease. Endostatin is one akin to the corresponding agent which is being researched for this preventive motivation because of its significant antiangiogenic activity as well as its link to the deposition of Aβ proteins, which is one of the causes of cognitive loss in Alzheimer’s disease. As a result, clinical trials are being conducted to determine its utility in the diagnosis and treatment of Alzheimer’s disease. As a result, the precise activities of endostatin should be fully known, or those that already exist should be further researched, to maximize endostatin’s potential as a treatment for Alzheimer’s disease.
Data availability
Not applicable
References
Abdollahi A (2004) Antiangiogenic signaling network induced by endostatin
Abdollahi A, Hahnfeldt P, Maercker C, Gröne HJ, Debus J, Ansorge W, Folkman J, Hlatky L, Huber PE (2004) Endostatin’s antiangiogenic signaling network. Mol Cell 13(5):649–663. https://doi.org/10.1016/s1097-2765(04)00102-9
Akiyama H, Barger S, Barnum S, Bradt B, Bauer J, Cole GM, Cooper NR, Eikelenboom P, Emmerling M, Fiebich BL, Finch CE (2000) Inflammation and Alzheimer’s disease. Neurobiol Aging 21(3):383–421. https://doi.org/10.1016/s0197-4580(00)00124-x
Ambrose CT (2016) Angiogenesis, aging, and Alzheimer’s disease. Am Sci 104(2):82–85. https://doi.org/10.1511/2016.119.82
Bächinger HP (2005) Collagen: primer in structure, processing and assembly (Vol. 247). Springer Science & Business Media
Bayer TA, Wirths O, Majtényi K, Hartmann T, Multhaup G, Beyreuther K, Czech C (2001) Key factors in Alzheimer’s disease: β-amyloid precursor protein processing, metabolism and intraneuronal transport. Brain Pathol 11(1):1–11. https://doi.org/10.1111/j.1750-3639.2001.tb00376.x
Bell RD, Zlokovic BV (2009) Neurovascular mechanisms and blood–brain barrier disorder in Alzheimer’s disease. Acta Neuropathol 118(1):103–113. https://doi.org/10.1007/s00401-009-0522-3
Beyreuther K, Bush AI, Dyrks T, Hilbich C, KöNIG GE, MöNNING UR, Multhaup G, Prior R, Rumble B, Schubert W, Small DH (1991) Mechanisms of amyloid deposition in Alzheimer’s disease. Ann N Y Acad Sci 640(1):129–139. https://doi.org/10.1111/j.1749-6632.1991.tb00204.x
Blackhall FH, Merry CL, Lyon M, Jayson GC, Folkman J, Javaherian K, Gallagher JT (2003) Binding of endostatin to endothelial heparan sulphate shows a differential requirement for specific sulphates. Biochem J 375(1):131–139. https://doi.org/10.1042/bj20030730
Bookheimer SY, Strojwas MH, Cohen MS, Saunders AM, Pericak-Vance MA, Mazziotta JC, Small GW (2000) Patterns of brain activation in people at risk for Alzheimer’s disease. N Engl J Med 343(7):450–456. https://doi.org/10.1056/nejm200008173430701
Boscolo E, Folin M, Nico B, Grandi C, Mangieri D, Longo V, Scienza R, Zampieri P, Conconi MT, Parnigotto PP, Ribatti D (2007) β amyloid angiogenic activity in vitro and in vivo. Int J Mol Med 19(4):581–587. https://doi.org/10.3892/ijmm.19.4.581
Brkic M, Balusu S, Libert C, Vandenbroucke RE (2015) Friends or foes: matrix metalloproteinases and their multifaceted roles in neurodegenerative diseases. Mediat Inflamm 11:2015–2027. https://doi.org/10.1155/2015/620581
Chakraborty A, Kamermans A, van Het Hof B, Castricum K, Aanhane E, van Horssen J, Thijssen VL, Scheltens P, Teunissen CE, Fontijn RD, van der Flier WM (2018) Angiopoietin like-4 as a novel vascular mediator in capillary cerebral amyloid angiopathy. Brain. 141(12):3377–3388. https://doi.org/10.1093/brain/awy274
Chang R, Yee KL, Sumbria RK (2017) Tumor necrosis factor α inhibition for Alzheimer’s disease. J Cent Nerv Syst Dis 9:1179573517709278. https://doi.org/10.1177/1179573517709278
Chen M, Bao L, Zhao M, Cao J, Zheng H (2020) Progress in research on the role of FGF in the formation and treatment of corneal neovascularization. Front Pharmacol 11. https://doi.org/10.3389/fphar.2020.00111
Cross MJ, Claesson-Welsh L (2001) FGF and VEGF function in angiogenesis: signalling pathways, biological responses and therapeutic inhibition. Trends Pharmacol Sci 22(4):201–207. https://doi.org/10.1016/s0165-6147(00)01676-x
Damian AM, Jacobson SA, Hentz JG, Belden CM, Shill HA, Sabbagh MN, Caviness JN, Adler CH (2011) The Montreal Cognitive Assessment and the mini-mental state examination as screening instruments for cognitive impairment: item analyses and threshold scores. Dement Geriatr Cogn Disord 31(2):126–131. https://doi.org/10.1159/000323867
De Jong GI, De Vos RA, Steur EJ, Luiten PG (1997) Cerebrovascular hypoperfusion: a risk factor for Alzheimer’s disease? Animal model and postmortem human studies. Ann N Y Acad Sci 826(1):56–74. https://doi.org/10.1111/j.1749-6632.1997.tb48461.x
Deininger MH, Fimmen BA, Thal DR, Schluesener HJ, Meyermann R (2002) Aberrant neuronal and paracellular deposition of endostatin in brains of patients with Alzheimer’s disease. J Neurosci 22(24):10621–10626. https://doi.org/10.1523/jneurosci.22-24-10621.2002
Del Bo R, Ghezzi S, Scarpini E, Bresolin N, Comi GP (2009) VEGF genetic variability is associated with increased risk of developing Alzheimer’s disease. J Neurol Sci 283(1-2):66–68. https://doi.org/10.1016/j.jns.2009.02.318
Desai BS, Schneider JA, Li JL, Carvey PM, Hendey B (2009) Evidence of angiogenic vessels in Alzheimer’s disease. J Neural Transm 116(5):587–597. https://doi.org/10.1007/s00702-009-0226-9
Dhanabal M, Ramchandran R, Volk R, Stillman IE, Lombardo M, Iruela-Arispe ML, Simons M, Sukhatme VP (1999a) Endostatin: yeast production, mutants, and antitumor effect in renal cell carcinoma. Cancer Res 59(1):189–197. https://doi.org/10.1006/bbrc.1999.0595
Dhanabal M, Ramchandran R, Waterman MJ, Lu H, Knebelmann B, Segal M, Sukhatme VP (1999b) Endostatin induces endothelial cell apoptosis. J Biol Chem 274(17):11721–11726. https://doi.org/10.1074/jbc.274.17.11721
Dickstein DL, Biron KE, Ujiie M, Pfeifer CG, Jeffries AR, Jefferies WA (2006) Aβ peptide immunization restores blood-brain barrier integrity in Alzheimer disease. FASEB J 20(3):426–433. https://doi.org/10.1096/fj.05-3956com
Dickstein DL, Walsh J, Brautigam H, Stockton SD Jr, Gandy S, Hof PR (2010) Role of vascular risk factors and vascular dysfunction in Alzheimer’s disease. 77(1):82–102. https://doi.org/10.1002/msj.20155
Digtyar AV, Pozdnyakova NV, Feldman NB, Lutsenko SV, Severin SE (2007) Endostatin: current concepts about its biological role and mechanisms of action. Biochem Mosc 72(3):235–246. https://doi.org/10.1134/s0006297907030017
Ding YH, Javaherian K, Lo KM, Chopra R, Boehm T, Lanciotti J, Harris BA, Li Y, Shapiro R, Hohenester E, Timpl R (1998) Zinc-dependent dimers observed in crystals of human endostatin. Proc Natl Acad Sci 95(18):10443–10448. https://doi.org/10.1073/pnas.95.18.10443
Dixelius J, Larsson H, Sasaki T, Holmqvist K, Lu L, Engström A, Timpl R, Welsh M, Claesson-Welsh L (2000) Endostatin-induced tyrosine kinase signaling through the Shb adaptor protein regulates endothelial cell apoptosis. Blood: J Am Soc Hematol 95(11):3403–3411. https://doi.org/10.1182/blood.v95.11.3403.011k07_3403_3411
Dixelius J, Cross M, Matsumoto T, Sasaki T, Timpl R, Claesson-Welsh L (2002) Endostatin regulates endothelial cell adhesion and cytoskeletal organization. Cancer Res 62(7):1944–1947
Eriksson K, Magnusson P, Dixelius J, Claesson-Welsh L, Cross MJ (2003) Angiostatin and endostatin inhibit endothelial cell migration in response to FGF and VEGF without interfering with specific intracellular signal transduction pathways. FEBS Lett 536(1-3):19–24. https://doi.org/10.1016/s0014-5793(03)00003-6
Evans DA, Funkenstein HH, Albert MS, Scherr PA, Cook NR, Chown MJ, Hebert LE, Hennekens CH, Taylor JO (1989) Prevalence of Alzheimer’s disease in a community population of older persons: higher than previously reported. Jama 262(18):2551–2556. https://doi.org/10.1001/jama.1989.03430180093036
Fagiani E, Christofori G (2013) Angiopoietins in angiogenesis. Cancer Lett 328(1):18–26. https://doi.org/10.1016/j.canlet.2012.08.018
Faye C, Moreau C, Chautard E, Jetne R, Fukai N, Ruggiero F, Humphries MJ, Olsen BR, Ricard-Blum S (2009) Molecular interplay between endostatin, integrins, and heparan sulfate. J Biol Chem 284(33):22029–22040. https://doi.org/10.1074/jbc.m109.002840
Felbor U, Dreier L, Bryant RA, Ploegh HL, Olsen BR, Mothes W (2000) Secreted cathepsin L generates endostatin from collagen XVIII. EMBO J 19(6):1187–1194. https://doi.org/10.1093/emboj/19.6.1187
Ferrara N (1999) Molecular and biological properties of vascular endothelial growth factor. J Mol Med 77(7):527–543
Ferreras M, Felbor U, Lenhard T, Olsen BR, Delaissé JM (2000) Generation and degradation of human endostatin proteins by various proteinases. FEBS Lett 486(3):247–251. https://doi.org/10.1016/s0014-5793(00)02249-3
Fiedler U, Augustin HG (2006) Angiopoietins: a link between angiogenesis and inflammation. Trends Immunol 27(12):552–558. https://doi.org/10.1016/j.it.2006.10.004
Fillit H, Ding W, Buee L, Kalman J, Altstiel L, Lawlor B, Wolf-Klein G (1991) Elevated circulating tumor necrosis factor levels in Alzheimer’s disease. Neurosci Lett 129(2):318–320. https://doi.org/10.1016/0304-3940(91)90490-k
Frautschy SA, Yang F, Irrizarry M, Hyman B, Saido TC, Hsiao K, Cole GM (1998) Microglial response to amyloid plaques in APPsw transgenic mice. Am J Pathol 152(1):307–317
Gebbink MF, Voest EE, Reijerkerk A (2004) Do antiangiogenic protein fragments have amyloid properties? Blood 104(6):1601–1605. https://doi.org/10.1182/blood-2004-02-0433
Glabe CC (2005) Amyloid accumulation and pathogenesis of Alzheimer’s disease: significance of monomeric, oligomeric and fibrillar Aβ. In: Alzheimer’s Disease. Springer, Boston, pp 167–177. https://doi.org/10.1007/0-387-23226-5_8
Gordon MK, Hahn RA (2010) Collagens. Cell Tissue Res 339(1):247–257. https://doi.org/10.1007/s00441-009-0844-4
Goto Y, Takahashi N, Fink AL (1990) Mechanism of acid-induced folding of proteins. Biochemistry. 29(14):3480–3488. https://doi.org/10.1021/bi00466a009
Gupta-Bansal R, Frederickson RC, Brunden KR (1995) Proteoglycan-mediated inhibition of Aβ proteolysis. A potential cause of senile plaque accumulation. J Biol Chem 270(31):18666–18671. https://doi.org/10.1074/jbc.270.31.18666
Gurnik S, Devraj K, Macas J, Yamaji M, Starke J, Scholz A, Sommer K, Di Tacchio M, Vutukuri R, Beck H, Mittelbronn M (2016) Angiopoietin-2-induced blood–brain barrier compromise and increased stroke size are rescued by VE-PTP-dependent restoration of Tie2 signaling. Acta Neuropathol 131(5):753–773. https://doi.org/10.1007/s00401-016-1551-3
Hardy J, Duff K, Hardy KG, Perez-Tur J, Hutton M (1998) Genetic dissection of Alzheimer’s disease and related dementias: amyloid and its relationship to tau. Nat Neurosci 1(5):355–358. https://doi.org/10.1038/1565
He Y, Zhou H, Tang H, Luo Y (2006) Deficiency of disulfide bonds facilitating fibrillogenesis of endostatin. J Biol Chem 281(2):1048–1057. https://doi.org/10.1074/jbc.m507745200
Hohenester E, Sasaki T, Olsen BR, Timpl R (1998) Crystal structure of the angiogenesis inhibitor endostatin at 1.5 Å resolution. EMBO J 17(6):1656–1664. https://doi.org/10.2210/pdb1koe/pdb
Hohman TJ, Bell SP, Jefferson AL (2015) The role of vascular endothelial growth factor in neurodegeneration and cognitive decline: exploring interactions with biomarkers of Alzheimer disease. JAMA Neurol 72(5):520–529. https://doi.org/10.1001/jamaneurol.2014.4761
Holton JL, Ghiso J, Lashley T, Rostagno A, Guerin CJ, Gibb G, Houlden H, Ayling H, Martinian L, Anderton BH, Wood NW (2001) Regional distribution of amyloid-Bri deposition and its association with neurofibrillary degeneration in familial British dementia. Am J Pathol 158(2):515–526. https://doi.org/10.1016/s0002-9440(10)63993-4
Hu W, Xia LJ, Chen FH, Wu FR, Tang J, Chen CZ, Jiang S, Chen HH (2012) Recombinant human endostatin inhibits adjuvant arthritis by down-regulating VEGF expression and suppression of TNF-α, IL-1β production. Inflamm Res 61(8):827–835
Iadecola C (2004) Neurovascular regulation in the normal brain and in Alzheimer’s disease. Nat Rev Neurosci 5(5):347–360. https://doi.org/10.1038/nrn1387
Im E, Venkatakrishnan A, Kazlauskas A (2005) Cathepsin B regulates the intrinsic angiogenic threshold of endothelial cells. Mol Biol Cell 16(8):3488–3500. https://doi.org/10.1091/mbc.e04-11-1029
Isaacs-Trepanier C, Saleem M, Herrmann N, Swardfager W, Oh PI, Goldstein BI, Mitchell J, Sugamori KS, Lanctôt KL (2020) Endostatin as a mediator between endothelial function and cognitive performance in those at risk for vascular cognitive impairment. J Alzheimers Dis 76(2):601–611
Jellinger KA (2007) The enigma of vascular cognitive disorder and vascular dementia. Acta Neuropathol 113(4):349–388. https://doi.org/10.1007/s00401-006-0185-2
Kalaria RN (2002) Small vessel disease and Alzheimer’s dementia: pathological considerations. Cerebrovasc Dis 13(Suppl. 2):48–52. https://doi.org/10.1159/000049150
Kanai M, Matsubara E, Isoe K, Urakami K, Nakashima K, Arai H, Sasaki H, Abe K, Iwatsubo T, Kosaka T, Watanabe M (1998) Longitudinal study of cerebrospinal fluid levels of tau, Aβ1–40, and Aβ1–42 (43) in Alzheimer’s disease: a study in Japan. Ann Neurol: Off J Am Neurol Assoc Child Neurol Soc 44(1):17–26. https://doi.org/10.1002/ana.410440108
Karumanchi SA, Jha V, Ramchandran R, Karihaloo A, Tsiokas L, Chan B et al (2001) Cell surface glypicans are low-affinity endostatin receptors. Mol Cell 7(4):811–822. https://doi.org/10.1016/s1097-2765(01)00225-8
Kasama T, Shiozawa F, Kobayashi K, Yajima N, Hanyuda M, Takeuchi HT, Mori Y, Negishi M, Ide H, Adachi M (2001) Vascular endothelial growth factor expression by activated synovial leukocytes in rheumatoid arthritis: critical involvement of the interaction with synovial fibroblasts. Arthritis Rheum 44(11):2512–2524. https://doi.org/10.1002/1529-0131(200111)44:11%3C2512::aid-art431%3E3.0.co;2-o
Katzman R (1993 Jan.) Education and the prevalence of dementia and Alzheimer’s disease. Neurology 43:13. https://doi.org/10.1212/wnl.43.1_part_1.13
Kawas CH, Brookmeyer R (2001) Aging and the public health effects of dementia. N Engl J Med 344(15):1160–1161
Kim YM, Jang JW, Lee OH, Yeon J, Choi EY, Kim KW, Lee ST, Kwon YG (2000) Endostatin inhibits endothelial and tumor cellular invasion by blocking the activation and catalytic activity of matrix metalloproteinase 2. Cancer Res 60(19):5410–5413. https://doi.org/10.1016/j.atherosclerosis.2020.02.022
Kivipelto M, Helkala EL, Laakso MP, Hänninen T, Hallikainen M, Alhainen K, Soininen H, Tuomilehto J, Nissinen A (2001) Midlife vascular risk factors and Alzheimer’s disease in later life: longitudinal, population based study. Bmj 322(7300):1447–1451. https://doi.org/10.1212/wnl.56.12.1683
Klagsbrun M, Moses MA (1999) Molecular angiogenesis. Chem Biol 6(8):R217–R224. https://doi.org/10.1016/s1074-5521(99)80081-7
Kruskal WH, Wallis WA (1952) Use of ranks in one-criterion variance analysis. J Am Stat Assoc 47(260):583–621. https://doi.org/10.1080/01621459.1952.10483441
Lan HY, Mu W, Ng YY, Nikolic-Paterson DJ, Atkins RC (1996) A simple, reliable, and sensitive method for nonradioactive in situ hybridization: use of microwave heating to improve hybridization efficiency and preserve tissue morphology. J Histochem Cytochem 44(3):281–287. https://doi.org/10.1177/44.3.8648089
Lieu C, Heymach J, Overman M, Tran H, Kopetz S (2011) Beyond VEGF: inhibition of the fibroblast growth factor pathway and antiangiogenesis. Clin Cancer Res 17(19):6130–6139. https://doi.org/10.1158/1078-0432.ccr-11-0659
Lowe J, Mayer RJ, Landon M (1993) Ubiquitin in neurodegenerative diseases. Brain Pathol 3(1):55–65
Lukiw WJ, Carver LA, LeBlanc HJ, Bazan NG (2000) Analysis of 1184 gene transcript levels in Alzheimer CA1 hippocampus: synaptic signaling and transcription factor deficits and upregulation of pro-inflammatory pathways. Alzheimers Rep 3(3):161–167. https://doi.org/10.1016/s0197-4580(00)82895-x
Lyketsos CG, Steele C, Baker L, Galik E, Kopunek S, Steinberg M, Warren A (1997) Major and minor depression in Alzheimer’s disease: prevalence and impact. J Neuropsychiatry Clin Neurosci 9(4):556–561. https://doi.org/10.1176/jnp.9.4.556
Macpherson GR, Ng SS, Forbes SL, Melillo G, Karpova T, McNally J, Conrads TP, Veenstra TD, Martinez A, Cuttitta F, Price DK (2003) Anti-angiogenic activity of human endostatin is HIF-1-independent in vitro and sensitive to timing of treatment in a human saphenous vein assay. Mol Cancer Ther 2(9):845–854
Mann HB, Whitney DR (1947) On a test of whether one of two random variables is stochastically larger than the other. Ann Math Stat 1:50–60. https://doi.org/10.1214/aoms/1177730491
Mendoza L, Valcárcel M, Carrascal T, Egilegor E, Salado C, Sim BK, Vidal-Vanaclocha F (2004) Inhibition of cytokine-induced microvascular arrest of tumor cells by recombinant endostatin prevents experimental hepatic melanoma metastasis. Cancer Res 64(1):304–310. https://doi.org/10.1158/0008-5472.can-03-1829
Messmer-Blust A, An X, Li J (2009) Hypoxia-regulated angiogenic inhibitors. Trends Cardiovasc Med 19(8):252–256. https://doi.org/10.1016/j.tcm.2010.02.006
Miners JS, Palmer JC, Love S (2016) Pathophysiology of hypoperfusion of the precuneus in early Alzheimer’s disease. Brain Pathol 26(4):533–541. https://doi.org/10.1111/bpa.12331
Miosge N, Sasaki T, Timpl R (1999) Angiogenesis inhibitor endostatin is a distinct component of elastic fibers in vessel walls. FASEB J 13(13):1743–1750. https://doi.org/10.1096/fasebj.13.13.1743
Moeller BJ, Cao Y, Li CY, Dewhirst MW (2004) Radiation activates HIF-1 to regulate vascular radiosensitivity in tumors: role of reoxygenation, free radicals, and stress granules. Cancer Cell 5(5):429–441. https://doi.org/10.1016/s1535-6108(04)00115-1
Mohamed WA, Niyazy WH, Mahfouz AA (2011) Angiopoietin-1 and endostatin levels in cord plasma predict the development of bronchopulmonary dysplasia in preterm infants. J Trop Pediatr 57(5):385–388. https://doi.org/10.1093/tropej/fmq112
Moody DM, Brown WR, Challa VR, GHAZI-BIRRY HS, Reboussin DM (1997) Cerebral microvascular alterations in aging, leukoaraiosis, and Alzheimer’s disease. Ann N Y Acad Sci 826(1):103–116. https://doi.org/10.1111/j.1749-6632.1997.tb48464.x
Morisada T, Kubota Y, Urano T, Suda T, Oike Y (2006) Angiopoietins and angiopoietin-like proteins in angiogenesis. Endothelium. 13(2):71–79. https://doi.org/10.1080/10623320600697989
Moulton KS, Heller E, Konerding MA, Flynn E, Palinski W, Folkman J (1999) Angiogenesis inhibitors endostatin or TNP-470 reduce intimal neovascularization and plaque growth in apolipoprotein E–deficient mice. Circulation. 99(13):1726–1732. https://doi.org/10.1161/01.cir.99.13.1726
Murakami M, Simons M (2008) Fibroblast growth factor regulation of neovascularization. Curr Opin Hematol 15(3):215–220. https://doi.org/10.1097/moh.0b013e3282f97d98
Noda M, Takii K, Parajuli B, Kawanokuchi J, Sonobe Y, Takeuchi H, Mizuno T, Suzumura A (2014) FGF-2 released from degenerating neurons exerts microglial-induced neuroprotection via FGFR3-ERK signaling pathway. J Neuroinflammation 11(1):1–1. https://doi.org/10.1186/1742-2094-11-76
Oeckl P, Steinacker P, Feneberg E, Otto M (2016) Neurochemical biomarkers in the diagnosis of frontotemporal lobar degeneration: an update. J Neurochem 138:184–192. https://doi.org/10.1111/jnc.13669
Öhlund D, Ardnor B, Öman M, Naredi P, Sund M (2008) Expression pattern and circulating levels of endostatin in patients with pancreas cancer. Int J Cancer 122(12):2805–2810
Oikonomou KA, Kapsoritakis AN, Kapsoritaki AI, Manolakis AC, Tiaka EK, Tsiopoulos FD, Tsiompanidis IA, Potamianos SP (2011) Angiogenin, angiopoietin-1, angiopoietin-2, and endostatin serum levels in inflammatory bowel disease. Inflamm Bowel Dis 17(4):963–970. https://doi.org/10.1002/ibd.21410
Ol O (1994) Canadian study of health and aging: study methods and prevalence of dementia. Can Med Assoc J 150(6):899–913. https://doi.org/10.1017/s0714980800006334
O'Reilly MS, Boehm T, Shing Y, Fukai N, Vasios G, Lane WS, Flynn E, Birkhead JR, Olsen BR, Folkman J (1997) Endostatin: an endogenous inhibitor of angiogenesis and tumor growth. Cell 88(2):277–285. https://doi.org/10.1016/s0092-8674(00)81848-6
Peng Z, Luo Y, Xiao ZY (2020) Angiopoietin-1 accelerates Alzheimer’s disease via FOXA2/PEN2/APP pathway in APP/PS1 mice. Life Sci 246:117430. https://doi.org/10.1016/j.lfs.2020.117430
Qian S, Li R, Zhang C, Zhang R, Guo D, Bu X, Wang A, Peng H, Chen J, Zhang Y, He J (2020) Plasma endostatin levels at acute phase of ischemic stroke Are associated with post-stroke cognitive impairment. Neurotox Res 8:1–9. https://doi.org/10.1007/s12640-020-00173-5
Quintero-Fabián S, Arreola R, Becerril-Villanueva E, Torres-Romero JC, Arana-Argáez VE, Lara-Riegos J, Ramírez-Camacho MA, Alvarez Sanchez ME (2019) Role of matrix metalloproteinases in angiogenesis and cancer. Front Oncol 9:1370. https://doi.org/10.3389/fonc.2019.01370
Ravi R, Mookerjee B, Bhujwalla ZM, Sutter CH, Artemov D, Zeng Q, Dillehay LE, Madan A, Semenza GL, Bedi A (2000) Regulation of tumor angiogenesis by p53-induced degradation of hypoxia-inducible factor 1α. Genes Dev 14(1):34–44
Rezvani HR, Ali N, Nissen LJ, Harfouche G, De Verneuil H, Taïeb A, Mazurier F (2011) HIF-1α in epidermis: oxygen sensing, cutaneous angiogenesis, cancer, and non-cancer disorders. J Investig Dermatol 131(9):1793–1805. https://doi.org/10.1038/jid.2011.141
Ricard-Blum S, Féraud O, Lortat-Jacob H, Rencurosi A, Fukai N, Dkhissi F, Vittet D, Imberty A, Olsen BR, Van Der Rest M (2004) Characterization of endostatin binding to heparin and heparan sulfate by surface plasmon resonance and molecular modeling Role of divalent cations. J Biol Chem 279(4):2927–2936. https://doi.org/10.1074/jbc.m309868200
Rogers MS, D'Amato RJ (2006) The effect of genetic diversity on angiogenesis. Exp Cell Res 312(5):561–574. https://doi.org/10.1016/j.yexcr.2005.10.021
Roses AD (1995) On the metabolism of apolipoprotein E and the Alzheimer diseases. Exp Neurol 132(2):149–156. https://doi.org/10.1016/0014-4886(95)90019-5
Salza R, Oudart JB, Ramont L, Maquart FX, Bakchine S, Thoannès H, Ricard-Blum S (2015) Endostatin level in cerebrospinal fluid of patients with Alzheimer’s disease. J Alzheimers Dis 44(4):1253–1261. https://doi.org/10.3233/jad-142544
Sasaki T, Larsson H, Kreuger J, Salmivirta M, Claesson-Welsh L, Lindahl U, Hohenester E, Timpl R (1999) Structural basis and potential role of heparin/heparan sulfate binding to the angiogenesis inhibitor endostatin. EMBO J 18(22):6240–6248. https://doi.org/10.1093/emboj/18.22.6240
Sato Y (2017) Endostatin as a biomarker of basement membrane degradation. J Atheroscler Thromb 24(10):1014–1015. https://doi.org/10.5551/jat.ed077
Schreitmüller B, Leyhe T, Stransky E, Köhler N, Laske C (2012) Elevated angiopoietin-1 serum levels in patients with Alzheimer’s disease. Int J Alzheimers Dis 1:2012–2015. https://doi.org/10.1155/2012/324016
Shi H, Huang Y, Zhou H, Song X, Yuan S, Fu Y, Luo Y (2007) Nucleolin is a receptor that mediates antiangiogenic and antitumor activity of endostatin. Blood. 110(8):2899–2906. https://doi.org/10.1182/blood-2007-01-064428
Ständker L, Schrader M, Kanse SM, Jürgens M, Forssmann WG, Preissner KT (1997) Isolation and characterization of the circulating form of human endostatin. FEBS Lett 420(2-3):129–133. https://doi.org/10.1016/s0014-5793(97)01503-2
Stetler-Stevenson WG (1999) Matrix metalloproteinases in angiogenesis: a moving target for therapeutic intervention. J Clin Invest 103(9):1237–1241. https://doi.org/10.1172/jci6870
Stopa EG, Gonzalez AM, Chorsky R, Corona RJ, Alvarez J, Bird ED, Baird A (1990) Basic fibroblast growth factor in Alzheimer’s disease. Biochem Biophys Res Commun 171(2):690–696. https://doi.org/10.1016/0006-291x(90)91201-3
Takashima A, Noguchi K, Sato K, Hoshino T, Imahori K (1993) Tau protein kinase I is essential for amyloid beta-protein-induced neurotoxicity. Proc Natl Acad Sci 90(16):7789–7793. https://doi.org/10.1073/pnas.90.16.7789
Tarkowski E, Issa R, Sjögren M, Wallin A, Blennow K, Tarkowski A, Kumar P (2002) Increased intrathecal levels of the angiogenic factors VEGF and TGF-β in Alzheimer’s disease and vascular dementia. Neurobiol Aging 23(2):237–243. https://doi.org/10.1016/s0197-4580(01)00285-8
Tarkowski E, Andreasen N, Tarkowski A, Blennow K (2003) Intrathecal inflammation precedes development of Alzheimer’s disease. J Neurol Neurosurg Psychiatry 74(9):1200–1205. https://doi.org/10.1136/jnnp.74.9.1200
Terry RD, Masliah E, Salmon DP, Butters N, DeTeresa R, Hill R, Hansen LA, Katzman R (1991) Physical basis of cognitive alterations in Alzheimer’s disease: synapse loss is the major correlate of cognitive impairment. Ann Neurol: Off J Am Neurol Assoc Child Neurol Soc 30(4):572–580. https://doi.org/10.1002/ana.410300410
Thambisetty M, Beason-Held L, An Y, Kraut MA, Resnick SM (2010) APOE ε4 genotype and longitudinal changes in cerebral blood flow in normal aging. Arch Neurol 67(1):93–98. https://doi.org/10.1001/archneurol.2009.913
Turner CA, Eren-Kocak E, Inui EG, Watson SJ, Akil H (2016) Dysregulated fibroblast growth factor (FGF) signaling in neurological and psychiatric disorders. In: Seminars in cell & developmental biology, vol 53. Academic Press, pp 136–143. https://doi.org/10.1016/j.semcdb.2015.10.003
Ujiie M, Dickstein DL, Carlow DA, Jefferies WA (2003) Blood–brain barrier permeability precedes senile plaque formation in an Alzheimer disease model. Microcirculation. 10(6):463–470. https://doi.org/10.1080/mic.10.6.463.470
Vagnucci AH Jr, Li WW (2003) Alzheimer’s disease and angiogenesis. Lancet 361(9357):605–608. https://doi.org/10.1016/s0140-6736(03)12521-4
van Horssen J, Wilhelmus MM, Heljasvaara R, Pihlajaniemi T, Wesseling P, de Waal RM, Verbeek MM (2002) Collagen XVIII: a novel heparan sulfate proteoglycan associated with vascular amyloid depositions and senile plaques in Alzheimer’s disease brains. Brain Pathol 12(4):456–462. https://doi.org/10.1111/j.1750-3639.2002.tb00462.x
Verbeek MM, Otte-Höller I, van den Born J, van den Heuvel LP, David G, Wesseling P, de Waal RM (1999) Agrin is a major heparan sulfate proteoglycan accumulating in Alzheimer’s disease brain. Am J Pathol 155(6):2115–2125. https://doi.org/10.1016/s0002-9440(10)65529-0
Vinters HV (1987) Cerebral amyloid angiopathy. A critical review. Stroke. 18(2):311–324. https://doi.org/10.1161/01.str.18.2.311
Vinters HV, Wang ZZ, Secor DL (1996) Brain parenchymal and microvascular amyloid in Alzheimer’s disease. Brain Pathol 6(2):179–195. https://doi.org/10.1111/j.1750-3639.1996.tb00799.x
Walia A, Yang JF, Huang YH, Rosenblatt MI, Chang JH, Azar DT (2015) Endostatin’s emerging roles in angiogenesis, lymphangiogenesis, disease, and clinical applications. Biochim Biophys Acta (BBA)-Gen Subj 1850(12):2422–2438. https://doi.org/10.1016/j.bbagen.2015.09.007
Wang J, Al-Lamki RS (2013) Tumor necrosis factor receptor 2: its contribution to acute cellular rejection and clear cell renal carcinoma. Biomed Res Int 1:2013. https://doi.org/10.1155/2013/821310
Watson CJ, Webb NJ, Bottomley MJ, Brenchley PE (2000) Identification of polymorphisms within the vascular endothelial growth factor (VEGF) gene: correlation with variation in VEGF protein production. Cytokine. 12(8):1232–1235. https://doi.org/10.1006/cyto.2000.0692
Weller J, Budson A (2018) Current understanding of Alzheimer’s disease diagnosis and treatment. F1000Res 7:F1000 Faculty Rev–1161
Wen W, Moses MA, Wiederschain D, Arbiser JL, Folkman J (1999) The generation of endostatin is mediated by elastase. Cancer Res 59(24):6052–6056
Wickström SA, Alitalo K, Keski-Oja J (2004) An endostatin-derived peptide interacts with integrins and regulates actin cytoskeleton and migration of endothelial cells. J Biol Chem 279(19):20178–20185. https://doi.org/10.1074/jbc.m312921200
Xu H, Ge W, Jie F, Cao D, Ming P, Luo W, Song J, Li C (2013) Endostar enhances anti-tumor effects of radiation via inhibition of HIF-1α and bFGF in lung adenocarcinoma cell line A549. Chin-Ger J Clin Oncol 12(12):559–563. https://doi.org/10.1007/s10330-013-1255-2
Yamaguchi N, Anand-Apte B, Lee M, Sasaki T, Fukai N, Shapiro R, Que I, Lowik C, Timpl R, Olsen BR (1999) Endostatin inhibits VEGF-induced endothelial cell migration and tumor growth independently of zinc binding. EMBO J 18(16):4414–4423. https://doi.org/10.1093/emboj/18.16.4414
Yates CM, Butterworth J, Tennant MC, Gordon A (1990) Enzyme activities in relation to pH and lactate in postmortem brain in Alzheimer-type and other dementias. J Neurochem 55(5):1624–1630. https://doi.org/10.1111/j.1471-4159.1990.tb04948.x
Yehya AH, Asif M, Petersen SH, Subramaniam AV, Kono K, Majid AM, Oon CE (2018) Angiogenesis: managing the culprits behind tumorigenesis and metastasis. Medicina. 54(1):8. https://doi.org/10.3390/medicina54010008
Yin G, Liu W, An P, Li P, Ding I, Planelles V, Schwarz EM, Min W (2002) Endostatin gene transfer inhibits joint angiogenesis and pannus formation in inflammatory arthritis. Mol Ther 5(5):547–554. https://doi.org/10.1006/mthe.2002.0590
Yu Y, Moulton KS, Khan MK, Vineberg S, Boye E, Davis VM, O'Donnell PE, Bischoff J, Milstone DS (2004) E-selectin is required for the antiangiogenic activity of endostatin. Proc Natl Acad Sci 101(21):8005–8010. https://doi.org/10.1073/pnas.0402551101
Zlokovic BV (2008) The blood-brain barrier in health and chronic neurodegenerative disorders. Neuron. 57(2):178–201. https://doi.org/10.1016/j.neuron.2008.01.003
Zorick TS, Mustacchi Z, Bando SY, Zatz M, Moreira-Filho CA, Olsen B, Passos-Bueno MR (2001) High serum endostatin levels in Down syndrome: implications for improved treatment and prevention of solid tumours. Eur J Hum Genet 9(11):811–814. https://doi.org/10.1038/sj.ejhg.5200721
Author information
Authors and Affiliations
Contributions
DK and TB: Conceived the idea and wrote the first draft; SC, AS, SS, and NS: Figure work; VNB, CVDLC, SB, and AAH: Data compilation; AD, LA, and SBU: Proofread
Corresponding author
Ethics declarations
Ethics approval
Not applicable
Consent to participate
Not applicable
Consent for publication
All the authors have approved the manuscript for publication.
Competing interests
The authors declare no competing interests.
Additional information
Responsible Editor: Philippe Garrigues
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Kaur, ., Behl, T., Chigurupati, S. et al. Deciphering the focal role of endostatin in Alzheimer’s disease. Environ Sci Pollut Res 28, 61998–62011 (2021). https://doi.org/10.1007/s11356-021-16567-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11356-021-16567-7