
Abstract
Cancer, a major public health problem, is one of the world’s top leading causes of death. Common treatments for cancer include cytotoxic chemotherapy, surgery, targeted drugs, endocrine therapy, and immunotherapy. However, despite the outstanding achievements in cancer therapies during the last years, resistance to conventional chemotherapeutic agents and new targeted drugs is still the major challenge. In the present review, we explain the different mechanisms involved in cancer therapy and the detailed outlines of cancer drug resistance regarding multidrug resistance-associated proteins (MRPs) and their role in treatment failures by common chemotherapeutic agents. Further, different modulators of MRPs are presented. Finally, we outlined the models used to analyze MRP transporters and proposed a future impact that may set up a base or pave the way for many researchers to investigate the cancer MRP further.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
By advancing cancer topics, our knowledge of cancer biology is being updated every time. Cancer results in high proliferative growth of abnormal cells with marked changes in the genome properties. Besides, the cancer progression affects the normal biological process of neighboring healthy cells through the invasion of nearby tissues and metastasis to another (Macconaill and Garraway 2010).
There have been huge efforts to clarify the basis of cancer biology and develop anti-cancer drugs. Anti-cancer agents recorded a significant jump from conventional cytotoxic chemotherapeutics to targeted and immune-related therapies in the last few decades. The conclusive goal for anti-cancer drugs is to disrupt the tumor cells and control their proliferation to prolong the patient survival time and improve their lives. This can be achieved in many ways. Based on the mechanism of action of anti-cancer drugs, chemotherapeutics can be divided into the following: (1) alkylating agents, which induce intra-strand DNA crosslinks that subvert DNA during replication (e.g., cyclophosphamide); (2) antimetabolites, which inhibit the synthesis of DNA and RNA (e.g., 5-fluorouracil (5-FU)); (3) topoisomerase inhibitors (protein synthesis interferer), which hinder the correct transcription and translation (e.g., irinotecan); and (4) cytotoxic antibiotics, which exert an antineoplastic effect through DNA damage and generation of reactive oxygen species (e.g., bleomycin) (Werner et al. 2014). So far, drug industries have produced four main groups of anti-cancer drugs: (1) cytotoxic drugs (alkylating agents, antimetabolites, antibiotics, plant extracts, and various cytotoxic drugs), (2) targeted based agents, (3) hormones and hormones antagonists, and (4) immunomodulators (Arrondeau et al. 2010). In chemotherapy, following the administration of a particular drug, many patients become resistant to this drug. So, anti-cancer drug progress has developed from conventional nonspecific cytotoxic agents, which often kill proliferating normal cells to specific target-based cancer therapy designed to destroy tumor cells only, and on immune-related modulators that help the patient’s immune system to defeat tumor cells (Kummar et al. 2010; Mackall et al. 2014).
Targeted therapy works via interference with cancer’s specific genes, proteins, or the internal tissue environment (tissue microenvironment) contributing to cancer growth and proliferation. These genes and proteins are mainly settled in cancer or cancer growth-related cells. Various targeted anti-cancer agents have been approved for cancer treatment; these therapies include the following: (a) Hormone therapies are mainly for hormone-sensitive tumors that require specific hormones to proliferate. These therapies act by preventing hormone production or by interfering with its effect. They have been licensed for breast and prostate cancer. (b) Gene expression modulators inhibit protein translation that plays a role in gene expression. (c) Apoptosis inducers induce cancer cells to undergo controlled cell death (apoptosis). (d) Angiogenesis inhibitors prevent the proliferation of new blood vessels to the cancer cells by impairing the function of vascular endothelial growth factor (VEGF), a substance that stimulates new blood vessel formation. (e) Immunotherapies provoke the immune system to damage the cancer cells through monoclonal antibodies (MABs) that recognize particular receptors on the surface of the cancer cells (Aggarwal 2010; Benson et al. 2006; Cole 2014).
Despite the significant advances in understanding the etiology and progression of cancer and developing novel diagnostics and therapeutics, one of the leading causes of cancer treatment failure is multidrug resistance (MDR) (Chang 2011). In the present review, we explained the detailed outlines of cancer drug resistance regarding multidrug resistance-associated proteins (MRPs) and their role in treatment failure. Further, different modulators of MRPs are presented. Finally, we outlined the models used to analyze MRP transporters and proposed a future impact that may set up a base or pave the way for many researchers to investigate the cancer MRP further.
Multidrug Resistance-Associated Proteins (MRP)
Multidrug resistance is a drastic obstacle in cancer therapy and one of the primary reasons for none or little response to therapy. It is active against a single drug or chemically related ones and a whole spectrum of chemotherapeutic drugs, even to ones that have not yet been tabled to the patient. Numerous factors involved in mediating MDR may include host factors, tumor factors, as well as tumor-host interplays. The host factors include genetic variants and drug-drug interactions (Assaraf et al. 2019). The tumor factors are many and involve diminished drug uptake at most via reducing influx transporters, boosting drug efflux via overexpression of MDR efflux transporters of the ATP-binding cassette (ABC) superfamily, or triggering extracellular vesicle-induced drug efflux, deregulation of cell death mechanisms, increased DNA damage repair, deregulation of microRNAs, and epigenetic alterations (Assaraf et al. 2019). Other significant tumor factors are intratumor heterogeneity and cancer stem cell plasticity.
The discovery of these MDR proteins began in 1974 when Victor Ling and Larry Thompson described the stable colchicine-resistant cell clone and discovered that the resistant cells did not allow colchicine to get inside (Ling and Thompson 1974). Further studies reported that the main difference between sensitive and resistant cells was the MRP (Cole et al. 1992). It quickly became apparent that all these proteins have the same features and belong to the ABC superfamily of proteins.
The ABC proteins are energy-dependent efflux pumps with very conserved amino acid (AA) sequences at their nucleotide-binding domain (NBD). They comprise one of the most prominent protein families identified to date and are present in almost all cells of most tissue, mainly the lung, kidney, liver, heart, brain, and skin. The human ABC family includes 48 members. To fit human genetic nomenclature requirements, they were subdivided into seven families: ABCA to ABCG (each named ABC followed by a family letter and a number). Accordingly, ABCB1, known as P-gp, ABC-subfamily C (ABCC1-6,10,11, and 12 known as MDR-associated proteins 1–9 (MRP1–9 organic anion transporters), and ABCG2, identified as BCRP, are the primary efflux transporters mediating MDR (Baguley 2010) (Dean and Allikmets 2001; Li et al. 2016c).
Several studies declared that different mechanisms could be engaged in the evolution of MDR. They include increased drug detoxification or nucleotide damage, redistribution of intracellular drug accumulation, a shift in drug target molecules, and suppression of drug-induced apoptosis (Lage et al. 2001; Nezami 2019). Nevertheless, overexpression of P-gp and MRP1 is the leading cause of MDR in several tissue cancers (Komdeur et al. 2003). Both proteins are part of ATP-binding cassette protein superfamily (ABC transporters). They cause the outflow of anti-cancer molecules out of tumor cells, thereby lowering their intracellular concentrations (Dean and Allikmets 2001; He et al. 2011) (Fig. 1).

Scheme of the general molecular structure of human ABC subfamily C (long MRP) transporters showing how drug efflux occurred. The transporters are energy-dependent, and upon substrate binding and ATP hydrolysis, they display conformational changes that lead to the substrate’s transport process. TMD, transmembrane domain; NBD, nucleotide-binding domain
The ATP-binding cassette transporters, subfamily C transporters (ABCC)
The molecular structure of ABCC transporters is one of the significant features of this family. The protein control unit consists of six hydrophobic membrane-crossing helical fragments founding a so-called transmembrane domain (TMD) and a hydrophilic cytoplasmic NBD that harbor specific AA sequences for ABC proteins. In most ABCC transporters, such structure is the same, forming a TMD1-NBD1-TMD2-NBD2 single polypeptide assembly (Fernandez et al. 2002) (Fig. 1).
MRP1/ABCC1
The MRP1 gene is in 16p13.1 chromosome and extended to around 200 kb. It holds 31 exons and 1531 AA with a 180–190 kDa molecular weight (Grant et al. 1997) (Table 1). MRP1 was first identified in MDR doxorubicin-selected lung cancer cell line called H69AR (Cole et al. 1992). MRP1 is composed of two NBDs and three membrane spanning domains (MSDs), namely MSD0, 1, and 2 consisting of 17 transmembrane helices (TM), making it different from the structure of specific ABC transporters that comprises two MSDs involving 6 TMs (Bakos et al. 2000). The two NBDs are distinguished by the existence of ABC accountable for turning ATP to ADP+P, which provides power for transmembrane delivery of solutes by MRP1. The MRP1 efflux characteristics and the substrates’ topological polar surface area are correlated (Fernandes and Gattass 2009). The protein molecules playing essential functions in identifying the specificity and selectivity of substrates are found in MSD1 and MSD2 as reported in mutagenesis, photo-labeling, and structure-activity relationship studies (He et al. 2011; Iram and Cole 2011).
AA amino acids
MRP1 encodes plasma membrane proteins which have a distinct and vital physiological transport function that mediates the active cellular efflux of different xenobiotics and their metabolites (hydrophobic and hydrophilic antineoplastic agents) against their concentration gradients (drug threshold) (Keppler 2011). MRP1/ABCC1 can transport a broad spectrum of substrates. It has been shown to transport organic anion conjugates, for example, glutathione S-conjugates (Jedlitschky et al. 1994), mono- and di-glucuronides, taurates, and sulfates of bile acids (Jedlitschky et al. 1996). Thus it tends to be the primary conjugate transporter involved in relatively normal detoxification processes (Balcerczyk et al. 2003). Since it can also transport glutathione disulfide, it plays a key role in cellular oxidation processes (Jedlitschky et al. 1997).
MRP1/ABCC1 appears to be expressed in most of all human tissues, higher levels in the testes, small intestine, lung, colon, kidney, skin, placenta, and skeletal muscle (Chang 2007) (Slot et al. 2011), with particular expression in blood-brain, blood test, and blood-cerebrospinal fluid (CSF) barriers (Mercier et al. 2004). MRP1 has been suggested to be overexpressed in leukemias, esophageal cancer, and non-small cell lung cancer (NSCLC) (Nooter et al. 1995). MRP1 levels in the liver are ordinarily low, while the dominant MRP transporter is MRP2 (Keppler 2011) (Table 1).
One of the essential features of MRP1 is the noticeable difference in its substrate specificity among primates and other species, although the AA identity is >90% for all species (Deeley et al. 2006). For example, MRP1 from humans and monkeys can transport estradiol glucuronide and anthracycline agents, such as doxorubicin, whereas this function is missed in mice, canines, and bovine.
MRP1 is involved in the resistance to anthracycline (doxorubicin), vinca alkaloid, and podophyllotoxin, mitoxantrone, methotrexate, as well as alkylating agents (Chen et al. 2003) (Table 2). MRP1 recognizes and outflows some of the newer “targeted” anti-cancer agents that work through modifying signal pathways of tumor growth, proliferation, and metastatic potential. One of these targeted agents is vandetanib (small-molecule tyrosine kinase inhibitor, TKI) that works by blocking the ATP-binding domains of ABC drug transporters. However, the mechanism is still unclear and misunderstood (Brozik et al. 2011). Other targeted anti-cancer agents that interact with MRP1 include the geldanamycin, the farnesyl protein transferase inhibitor lonafarnib, and nutlin-3 (Brozik et al. 2011; Michaelis et al. 2009; Pham et al. 2009; Wang and Johnson 2003). Also, MRP1 can be integrated into multiple physiological and pathophysiological pathways, including inflammatory changes, oxidative defenses (Cole 2014), and the pathogenesis and prognosis of cancer (Yang et al. 2019). Leukotriene C4 (LTC4, a pro-inflammatory arachidonic acid product that is incorporated in asthmatic and allergic reactions as well as smooth muscle constriction (Yonetomi et al. 2015)) is an essential physiological MRP1 substrate (Leier et al. 1994).
MRP2/ABCC2
MRP2 was first cloned from rat liver cells and named canalicular multiple organic anion transporter (cMOAT). It contains 1545 (long MRP) AA residues, is located at the 10q24 chromosome, and shows about 49% AA identity to MRP1 (Table 1). Similarly, with MRP1, MRP2 has three MSD and two NBD features. TM helices were proven necessary for substrates discrimination (Kool et al. 1997; Ryu et al. 2000).
MRP2 is overexpressed in the hepatocytes and intestine, with moderate tissue expression in the kidney, especially proximal tubules, nerves, urinary bladder, placenta, and CD4+ lymphocytes (Huisman et al. 2005; Kranz et al. 2014) (Table 1).
The physiological importance of MRP2 was detected using Mrp2−/− mice (Chu et al. 2006). Researchers suggested MRP2 plays a role in removing endogenous metabolites, xenobiotics, and their metabolites. In the mouse liver, MRP2 transports bilirubin glucuronides to the bile (Chu et al. 2006). MRP2 was reported to confer resistance to many anti-cancer agents such as anthracyclines, as well as vinca alkaloids, camptothecin, tamoxifen, methotrexate, and oxaliplatin, with remarkable resistance against cisplatin (which not for MRP1) (Kawabe et al. 1999) (Hjorth et al. 2019; Luo et al. 2018; Myint et al. 2019; Valinezhad Sani et al. 2020; Zhang et al. 2019b). Also, MRP2 shows a high affinity to LTC4 like MRP1 (Kawabe et al. 1999) (Table 2).
MRP3/ABCC3
MRP3 is one of the MRPs in which complete coding sequences have been revealed, contains 1527 AA, and is in the 17q21.3 chromosome (Table 1). It has an AA identity of 58 % with MRP1. It is known that MRP3 was predominantly expressed in the pancreas, kidney, gut, liver, and adrenal gland (Kool et al. 1997). MRP3 plays a physiological role in the efflux of organic anions from the cholestatic liver and assists in the enterohepatic circulation of bile acids via the basolateral membranes of enterocytes (Belinsky et al. 2005; Kruh et al. 2001). MRP3 is an organic anion transporter that transports DNP-GS (dinitrophenyl-glutathione) such as MRP1 and MRP2 and may induce resistance to the anti-cancer drugs such as MTX (with a high concentration in a short time), etoposide, teniposide, and glycocholate (Kool et al. 1999). A low level of MRP3 was shown to mediate vincristine resistance in HEK293 cells (Belinsky et al. 2002; Zeng et al. 1999) and chemoresistance in cholangiocarcinoma (PDAC) (Lozano et al. 2020). Further, MRP3 was recently identified as a novel target for treating pancreatic cancer (Adamska et al. 2019b). It is overexpressed in PDAC cell lines and clinical samples. Knockdown of MRP3 in PDAC cell lines reduces cell proliferation by inhibiting STAT3 and HIF1α signaling pathways, critical regulators of PDAC progression (Adamska et al. 2019b). Moreover, the pharmacological targeting of MRP3 in pancreatic cancer was shown to slow down its progression in animal models and cell lines (Adamska et al. 2019a). These data confirm the role of MRP3 in drug resistance.
MRP4 and MRP5
MRP4 and MRP5 differ from each other in the AA identity. They are in chromosome 13q32 and 3q27.1 and have 1325 and 1437 AA, respectively (Table 1). They have only two MSDs and two NBDs. They are deficient in the extra N-terminal spanning domain present in MRP1 and MRP2 (Borst et al. 2000).
The overexpression of MRP4 and MRP5 mainly presents in the prostate, ovary, adrenal, pancreas, lung, and skeletal muscle with a low detectable level in the liver (Kruh and Belinsky 2003). A survey of the human expressed gene sequence database against the MRP4 showed that MRP4 mRNA exists in most tissues except for bone marrow, vascular endothelium cervix, thymus, and soft tissue. A unique characteristic of MRP4 is its dual localization in polarized cells: it can appear apically and basolaterally depending on the cell type (Lee et al. 2000). For example, MRP4 is found in the basolateral membrane of the human prostate cells. In contrast, it was in the apical membrane of cells of proximal renal tubules (Smeets et al. 2004).
MRP4 and MRP5 are incorporated in the physiological transport of cyclic adenosine monophosphate (cAMP), nucleoside diphosphate, or triphosphate (Reid et al. 2003) and cyclic guanosine monophosphate (cGMP) (Chen et al. 2001; Sampath et al. 2002). The substrates of MRP4 as MDR are effluxing anionic phosphate metabolites and glucuronides substances such as E217βG (Maeng et al. 2014), thiopurine analogs, MTX, and topotecan (Chen et al. 2002; Wielinga et al. 2002), while the substrates of MRP5 are dinitrophenyl-S-glutathione (DNP-SG), cisplatin, purine analogs, and GSH compounds (Wijnholds et al. 2000) (Table 2). Recently, MRP4 has been linked with cell proliferation in various types of cancer, including pancreas, ovary, lung, breast, colon, and osteosarcoma (Colavita et al. 2020; Low et al. 2020). In castration-resistant prostate cancer, MRP4 was overexpressed and mediated the acquired docetaxel resistance, reversed by antiandrogen treatment (Huang et al. 2020; Li et al. 2017). Further, in acute lymphoblastic leukemia, high MRP4 expression is associated with ex vivo MTX resistance (Jaramillo et al. 2019). In PDAC cells, MRP4 was found overexpressed and increased its proliferation by modulating the cAMP efflux, and its pharmacological targeting or silencing decreased cell proliferation through the stimulation of the cAMP/Epac/Rap1 signaling pathway (Carozzo et al. 2019). In ovarian cancer showing Myc overexpression, MRP4 contributes to its aggressiveness and is recommended as a potential therapeutic target (Jung et al. 2020). MRP4 was reported to be overexpressed in clear cell renal cell carcinoma and essential to regulate its cell proliferation (Colavita et al. 2020). MRP5 was recently shown to be overexpressed in GEM-resistant PDAC cells and successfully targeted via its nitration by nitric oxide–releasing gemcitabine pro-drugs that are encapsulated in liposomes (Masetto et al. 2020). These data suggest the possible roles of MRP4 and MRP5 in the resistance of different cancers to anti-cancer drugs.
MRP6/ABCC6
MRP6 is located in the 16p13 chromosome next to MRP1 with AA identity (40–45%) to other MRP and contains 1503 AA. Its expression occurs mainly in the liver, kidney, pancreas, intestine, and most exocrine tissues (Beck et al. 2005) (Table 1). Mutations in the MRP6 gene were related to pseudoxanthoma elasticum, a congenital disorder of the connective tissue involving impaired vision, skin lesions, and cardiovascular disorders (Bergen et al. 2000). MRP6 was described to transport and confer efflux of glutathione conjugates, anionic cyclopentapeptide BQ123 (the endothelin receptor antagonist), and epipodophyllotoxins (Belinsky et al. 1998; Madon et al. 2000). Recently, in intrahepatic cholangiocarcinoma, MRP6 was expressed and significantly upregulated upon gemcitabine exposure. Interestingly, MRP6 knockdown greatly enhanced gemcitabine-induced cytotoxicity (Yang et al. 2021).
MRP7, MRP8, and MRP9
ABCC10, ABCC11, and ABCC12 are the last identified three members of the MRP family. MRP7 is like MRP1, MRP2, MRP3, and MRP6 in possessing three MSDs and two NBDs (Kao et al. 2003), while the topology of MRP8 and MRP9 is similar to those of MRP4 and MRP5 (Madon et al. 2000). MRP7 is in the 6p12 chromosome, while the MRP8 and MRP9 are closely identical (47% identity), and their genes are at the same site of the 16q12 chromosome (Table 1). MRP7 is expressed in the testis, ovary, gut, kidney, and lung (Takayanagi et al. 2004), while MRP8 and MRP9 are expressed in both the brain and placenta and to a lesser extent in the liver and kidney (Bera et al. 2001).
MRP7 is a lipophilic negatively charged transporter (known as ATP-gated chloride channel). The most common physiological substrates selective for MRP7 are 17β-glucoronsyle estradiol (E217βG) and leukotriene C4 (Chen et al. 2001; Reid et al. 2003). It can confer resistance to some anti-cancer agents, specially taxanes (Huisman et al. 2005), as was determined using HEK293 cells for docetaxel, microtubule-stabilizing agent paclitaxel, vincristine, and vinblastine (Bortfeld et al. 2006). It is also associated with paclitaxel and vinorelbine resistance in NSCLC by increasing their efflux (Bessho et al. 2009; Oguri et al. 2008). The MRP7-mediated resistance to paclitaxel and vincristine could be reversed using TKIs, imatinib, and nilotinib (Shen et al. 2009) and tandutinib (Deng et al. 2013), as shown in HEK293 cell by blocking the efflux action of MRP7.
MRP8 has a broad spectrum of physiological substrate selectivity of lipophilic anions including glutathione conjugates, LTC4, DNP-SG, monoanionic bile acids glycocholate, E217βG, steroid sulfates dehydroepiandrosterone (DHEAS), estrone 3-sulfate (E13S), folic acid, as well as the substrate of MRP5 and MRP4 (cAMP-cGMP) (Bortfeld et al. 2006; Chen et al. 2001) (Table 2). The drug resistance-promoting ability of MRP8 was assured where it confers resistance to some anti-cancer agents such as 9′-2′- phosphonylmethoxyethyl adenine, the active metabolite of the prodrug adefovir (dipivoxil); zalcitabine; anti-HIV drugs; and 5-FU (Pratt et al. 2005; Uemura et al. 2019). In lung cancer, MRP8 was revealed to confer pemetrexed drug resistance in lung cancer (Uemura et al. 2010). Also, MRP8 expression was shown to significantly increase in alectinib-resistant anaplastic lymphoma kinase-rearranged NSCLC cell lines, and its inhibition increased the sensitivity to alectinib in vitro (Funazo et al. 2020). Overexpression of MRP8 in breast carcinoma subtypes was linked with its aggressiveness and poor prognosis (Yamada et al. 2013).
It has been suggested that MRP9 (ABCC12) may play a role as an anti-cancer resistance transporter protein after expression in human embryonic kidney cells. However, until now, the physiological part of MRP9 and its mechanism in cancer chemotherapy need more elucidation and further research. Recently, the MRP9 gene was identified as an anti-cancer drug-resistant protein (Shi et al. 2020). In lung cancer and colorectal cancer cells, upon exposure to histone deacetylase inhibitors, sodium butyrate, suberoylanilide hydroxamic acid, or trichostatin, the expression level of MRP9 was increased (Shi et al. 2020; Wang et al. 2019).
Role of epigenetic alterations in MDR development
Epigenetics alterations (EA) are changes in the DNA framework independent from sequence changes and are stably transmitted from cell to cell. It has been recognized as a substantial element in establishing non-genetic heterogeneity (Chang et al. 2008). Epigenetic alterations involve DNA methylation and modifications of histone, chromatin, and microRNA (miRNAs). It was reported to drive the drug resistance in cancer (Kagohara et al. 2018; Nowacka-Zawisza and Wisnik 2017). Following chemotherapy, epigenetic events frequently start and mutate the expression level of various MDR genes, such as drug transporters (ABCB1, MDR) (Spitzwieser et al. 2016), pro-apoptotic genes (Death-Associated Protein Kinase (DAPK) and Apoptotic Protease-Activating Factor 1 (APAF1)) (Wilting and Dannenberg 2012), DNA-repair proteins (MutL Homolog 1 (MLH1), Methylguanine-DNA Methyltransferase MGMT)) (Saghafinia et al. 2018), or histone modifiers (Ferraro 2016). DNA methylation, a primary EA in various malignancies (Giri and Aittokallio 2019), is triggered by DNA methyltransferases (DNMT) which contributes to the inhibition of gene transcription resulting in gene silencing (Bird 2002). Aberrant DNA methylation in cancer is linked with chemotherapy administration (Grasse et al. 2018; Yuan et al. 2019), affecting the genes responsible for cell differentiation and proliferation pathways and mitogen-activated protein kinases (MAPK), VEGF, Wingless/Integrated (Wnt), and Tumor Protein p53 (p53) signaling or expression of cell cycle inhibitors (Yuan et al. 2019). The inhibition of DNMT can reverse DNA methylation and restore the expression of essential silenced genes (Berdasco and Esteller 2010). Therefore, therapeutic targeting of EA in malignancies becomes an optimistic attitude to raise the efficiency of multi-treatment regimens and quash MDR (Ahuja et al. 2016).
Role of microRNAs in MDR development
MicroRNAs (miRNAs) are a group of small RNAs that do not encode for any protein but mainly downregulate gene expression at a post-transcriptional level and rarely activate mRNA translation. miRNAs have a role in cancer development, angiogenesis, metastasis, and drug resistance (Peng and Croce 2016).
It was shown that miRNAs regulate the expression of MDR efflux transporters of the ABC superfamily in different tumors. For example, the downregulation of ABCB1/MDR1 encoding for P-gp by expression of various miRNAs results in the reversal of drug resistance, such as miR-let-7 in ovarian cancer (Boyerinas et al. 2012), miR-200c (Chen et al. 2012) and miR-195 (Yang et al. 2013) in breast cancer, miR-200c in colorectal cancer (Sui et al. 2014), and miR-30a (Li et al. 2016a) and MiR-30 (Du et al. 2018) in advanced gastric cancer. Further, the upregulation of miRNAs in some cancers decreased MRP levels and increased their sensitivities to anti-cancer drugs. For example, in gallbladder cancer, overexpression of miR-145 reduced the level of ABCC1/MRP1 and sensitized it to cisplatin (Zhan et al. 2016); in the ovarian tumor, miR-490-3p decreased the level of ABCC2/MRP2 and elevated its response to cisplatin (Tian et al. 2017). Contrarily, in mammary tumors, the downregulation of miR-128 leads to ABCC5 overexpression and contributes to chemotherapeutic resistance (Zhu et al. 2011).
Interestingly, the miRNAs may also curb the response of malignancies to anti-cancer agents by managing DNA repair genes. In the ovarian tumor, it was revealed that miR-9 downregulated BRCA1 and restrained DNA damage repair, thus boosted its sensitivity to cisplatin and PARP inhibitors (Sun et al. 2013). The expression levels of miRNAs were found correlated with the response of cancer patients to chemotherapy. The expression values of miR-20b, miR-27a, and miR-181a regarding epirubicin/oxaliplatin/capecitabine chemotherapy regimen in a group of gastric cancer patients were analyzed (Danza et al. 2016). They reported that aberrant expression of these miRNAs associates with a chemotherapeutic response via modulation of ABCB1/MDR1, HIF1A, and HIPK2, suggesting a possible novel therapeutic strategy (Danza et al. 2016). These data reveal that targeting miRNAs might become charming therapeutic plans to target MDR. The roles of miRNAs in MDR, being a potential source of biomarkers and therapeutic targets to overcome MDR, are summarized in Table 3.
Where are we now?
Most researchers evoked many methods to overcome anti-cancer drug resistance. These strategies relied mainly on introducing new molecules that directly inhibit MRP (inhibitors/modulators). In this regard, we explain a short brief about typical modulators involved in MDR overcoming.
MDR modulators
Chemical drugs and analogs
Ibrutinib as MRP1 modulator
Ibrutinib is TKI that interferes with the cytoplasmic protein in B cell. Therefore, it is approved by FDA to treat chronic lymphocytic leukemia. Zhang et al. found that in human leukemia cell lines, doxorubicin-selected MRP1-overexpressing one (HL60/Adr) and MRP1-transfected cell line (HEK293/MRP1), ibrutinib could significantly boost the cytotoxicity of MRP1 substrates such as daunorubicin, vincristine, and vinblastine and raised their intracellular concentration via blocking the drug efflux function of MRP1 (Zhang et al. 2014). Further, they found that the coadministration of ibrutinib enhanced the vincristine efficacy to suppress the tumor growth of HEK293/MRP1 cells in vivo in nude mice.
Nilotinib, vandetanib, imatinib, erlotinib, lapatinib, mastinib, and tandutinib as MRP7 modulators
They are TKI. They were reported as inhibitors of several ABC transporters such as P-gp, BCRP, and MRP7. They could reverse MRP7-mediated MDR in transfected HEK/MRP7 cells and interfere with the drug efflux function of MRP7 (Deng et al. 2013). The TKI, vandetanib, was shown to sensitize MRP1-overexpressing C-A120 cells toward vincristine and doxorubicin, increasing their accumulation inside the cell (Zheng et al. 2009).
Verapamil
Verapamil is an antiarrhythmic medication (calcium channel blocker). It sensitizes the lung cancer cell line (H69AR) overexpressing MRP1 transporter to many anthracyclines, such as daunorubicin (daunomycin), vinca alkaloids (vincristine and vinblastine), antifolates (methotrexate), and other cytostatic agents such as acivicin, colchicine, gramicidin-D, and mitoxantrone (Hooijberg et al. 1999).
Pyridine derivatives
Pyridine derivatives are known to target the MDR mediated by P-gp in vitro (Bourichi et al. 2018; Patel et al. 2019) and in vivo (Chen et al. 2004). Derivatives as nicardipine were shown to restore the sensitivity of MRP1-mediated doxorubicin resistance in MES-SA/DX-5 cell (Tatosian and Shuler 2009). It also blocked MRP1-mediated calcein-AM transport in Jurkat-SupT1/Vin cells, overexpressing MRP1 (Ivnitski-Steele et al. 2008). Dihydropyridine derivative, PAK-104P, was found to reverse MRP1-related MDR. It increased the response of doxorubicin-selected MRP1-overexpressing human leukemia cell line (HL60/ADR) to doxorubicin (Vanhoefer et al. 1996). Also, the compound reversed the paclitaxel resistance in this cell line and fibrosarcoma cell line, HT1080/DR4. Further, PAK-104P partially restored the sensitivity of the human epidermoid carcinoma cell line C-A120 overexpressing MRP1 against doxorubicin and completely against vincristine. However, Marbeuf-Gueye et al. reported that PAK-104P was a non-competitive inhibitor of the anthracyclines daunorubicin, pirarubicin, and calcein (Marbeuf-Gueye et al. 2000).
Cyclosporine A and immunosuppressant
It was reported that cyclosporin A enhanced chemosensitization and drug accumulation effects to vincristine and daunorubicin in human MDR large cell lung cancer cells expressing a 190k membrane protein distinct from P-gp (Barrand et al. 1993). Interestingly, Kim et al. used doxorubicin-selected bladder cancer cell line 5637/DR5.5 overexpressing MRP1, which showed to be cross-resistant toward several anti-cancer drugs, such as daunorubicin, vinblastine, or etoposide, and found that cyclosporine A not only enhanced the accumulation of daunorubicin but also reduced its efflux (Kim et al. 1995). Immunosuppressant agents and mTOR inhibitors (mammalian or mechanistic target of rapamycin) such as deforolimus, everolimus, sirolimus (rapamycin), and tacrolimus were reported to interfere with MRP1 and subsequently increased the intracellular anti-cancer drug concentration (Peterson et al. 2017). In T98G cells, with particular regard to tacrolimus, a marked reduction in the transcriptional levels of the MRP1 mRNA was shown, and the intracellular concentration of vincristine was increased 24 h after exposure (Garrido et al. 2011).
Indomethacin
Non-steroidal anti-inflammatory drug (NSAID), indomethacin, was reported to increase BCECF (carboxyfluorescein) accumulation in the murine MRP1-overexpressing PC-V4097 and human HL60/ADR cell lines and inhibit the efflux of the fluorescence dye (Draper et al. 1997). Indomethacin was shown to reverse the MDR by enhancing the toxicity of doxorubicin, daunorubicin, and epirubicin; vincristine; and etoposide compared to the administration of these anti-cancer agents alone (Benyahia et al. 2004).
Phytochemicals
The high biodiversity, bioavailability, and low intrinsic toxicity of natural products encourage the use of new chemical scaffolds for drug progress. Natural phytochemicals such as bioflavonoids and polyphenolic compounds found in curcumin, apigenin, quercetin, and naringenin are frequently favored for their health advantages and documented interaction with MRP1 and other drug-transporting ABC proteins (Li et al. 2010).
Curcumin
Curcumin is extracted from the Indian spice turmeric powder, comprised of curcumin (curcumin I), demethoxycurcumin (curcumin II), and bisdemethoxycurcumin (curcumin III). It has many biological activities, including anti-inflammatory (Bisht et al. 2010), anti-cancer (Aggarwal et al. 2003), and anti-viral properties (Shishodia et al. 2007). It plays an essential role in MDR-linked ABC transporters Pgp, MRP1, and ABCG2 (Limtrakul et al. 2007) (Chearwae et al. 2006). Tetrahydrocurcumin and curcumin I, the major in vivo metabolites of curcumin, showed sensitizing the MRP1-positive cancer cells toward most anti-cancer agents. However, the clinical prevalence of curcumin is limited due to its low oral bioavailability and rapid excretion from the body and less specificity for the target tissue.
Bioflavonoids
Flavonoids are among the most common natural product modulators and include flavonols, flavones, isoflavones, flavanones, and chalcones (Narayana et al. 2001). Many flavonoids are excellent modulators of major ABC drug transporters. They can alter the absorption, distribution, and excretion of drugs by altering the functions of ABC transporters (Cermak and Wolffram 2006; Morris and Zhang 2006).
The interaction of flavonoids with MRP1 was first reported by Versantvoort et al., who showed that the flavonoids genistein, apigenin, biochanin A, and quercetin might inhibit MRP1-mediated transport (Versantvoort et al. 1994). After that, several studies demonstrated that many other flavonoids aglycones such as baicalein, chalcone, chrysin, chrysoeriol, diosmetin, galangin, isorhamnetin, kaempferol, luteolin, morin, myricetin, naringenin, phloretin, robinetin, silymarin, and tamarixetin could all inhibit MRP1-mediated transport to a varying degree (Leslie et al. 2001; Nguyen et al. 2003; van Zanden et al. 2005).
Therapeutic targeting of epigenetic alterations to reverse MDR
Compared to genetic mutations, epigenetic alterations (EA) are reversible and often accompany diseases, particularly cancer. It influences gene expression without altering DNA sequences. During tumorigenesis, EA are complex and involve multiple steps such as DNA methylation, chromatin remodeling, histone modification, and noncoding RNA. Therefore, it is involved in different cellular processes such as proliferation, apoptosis, invasion, and senescence (Cheng et al. 2019). There have been considerable challenges to develop specific inhibitors to EA (epidrugs) to augment the efficacies of multiple therapy protocols and overcome MDR (Ahuja et al. 2016). Epidrugs are formulated to reprogram cancer cells to invert chemoresistance and classified based on their respective target enzyme. It includes inhibitors of DNA methyltransferase (DNMTi, e.g., 5-azacytidine, decitabine, SGI-110), isocitrate dehydrogenase inhibitors (e.g., ivosidenib and enasidenib), or histone deacetylase inhibitors (HDACi, e.g., vorinostat, romidepsin, or belinostat). DNMTi acts as anti-tumorigenic agents by inducing hypomethylating and breaking the double-strand DNA and, consequently, G2 phase arrest (Palii et al. 2008). Decitabine was used to restore cisplatin chemosensitivity in refractory patients with ovarian cancer (Zhang et al. 2017b). The targeting of EA by combining two or more agents resulted in optimum response and fewer adverse effects. The combination of DNMTi and HDACi epidrugs is recommended in several studies (García-Domínguez et al. 2018). For example, in sarcoma, using SAHA and HCI-2509 synergistically inhibited the cell viability of EWS-FLI1 and its tumor growth (García-Domínguez et al. 2018). In NSCLC, using DNMTis with HDACis revealed long-lasting and robust clinical responses in many patients (Schiffmann et al. 2016). In other malignancies like hematologic malignancies (Blagitko-Dorfs et al. 2019), colorectal cancer (Sharma et al. 2017), esophageal cancer (Schneider et al. 2017), and pancreatic cancer (Chan et al. 2018), the combined epidrugs were more efficacious. While several pre-clinical and clinical trials are running, FDA has already approved some EA inhibitors for clinical uses (Ball et al. 2017; Cheng et al. 2019) (Table 4).
Monoclonal antibodies
Monoclonal antibodies are often particular and can recognize only specific antigen or receptor sites. Therefore, coupling the active pharmaceutical ingredients with MABs holds a high capacity for site-specific drug delivery. MRP1-specific MABs such as QCRL-1, QCRL-2, QCRL-3, QCRL-4, and QCRL-6 have been chiefly used as valuable tools in experimental studies of different functions and sequences and essential domains of MRP1 (Cole 2014; Hipfner et al. 1994). Several MABs have been mapped to the extracellular epitopes of P-glycoprotein and are eligible to inhibit its drug efflux function (Ritchie et al. 2011). In addition to small-molecule compounds, MABs were developed to modulate or inhibit ABCB1 (Mechetner and Roninson 1992). Among the anti-P-glycoprotein MABs, MRK16 and MRK-17 were the first shown to stop the growth of human MDR tumors in immunodeficient mice (Yano et al. 1999). UIC2 is another MABs derived from BALB/c mice immunized with BALB/c 3T3-1000 cells that overexpress ABCB1 and efficiently block the transport of all investigated ABCB1 substrates (Alam et al. 2018). MRK16 blocks the transport of large substrates such as vincristine and actinomycin D, but not with small and flat molecules such as doxorubicin (Mechetner and Roninson 1992). Given the efficient delivery of MABs to intracellular epitopes, these MABs have little therapeutic potential, although they keep being special tools for experimental studies of MRP1 structure and function.
RNA interference therapy
RNA interference (RNAi), a novel established method in which RNA molecules interfere with target gene expression (Landesman-Milo et al. 2013), has latterly got proper research attention. RNAi demonstrates the potential for establishing new categories of molecular therapeutic drugs that interfere with genes-based diseases, especially those that encode so-called “undruggable” targets, which are not responsive to conventional therapeutics. Interestingly, Nieth et al. showed that transfecting pancreatic and gastric tumor cell lines with MDR1 small interfering RNA (siRNA) significantly inhibited mRNA and protein of MDR1 and lowered the resistance to daunorubicin (Nieth et al. 2003). This implies that RNAi can be used to drive back MDR and retrieve the response to chemotherapeutic agents. However, the delivery of siRNAs by the viral and non-viral vectors to diseased sites for gene therapy is a major challenge (Melamed et al. 2017). Recently, because of their merits, using nanoparticles to deliver RNAi molecules to the target sites has received attention (Xin et al. 2017).
Nanoparticles
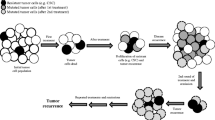
Nanomedicine describes the use of nano-objects to prevent, diagnose, and cure disease (Shi et al. 2017). Nanoparticles are fine dispersions or solid particles with particle sizes of 10–1000 nm. In the last two decades, nanomedicines emerged to inhibit the activity of efflux transporters. There are central three mechanisms by which nanoparticles achieve their roles: (1) targeting drugs to the specific tissue through increase local drug gradients, (2) engulfing the drug through endocytosis other than passive diffusion that exposes the drug to the ABC, and (3) blocking or inhibition of ABC expression (RNA interference) (Bar-Zeev et al. 2017; Corbo et al. 2016) (Fig. 2). Liu et al. developed a TAT-improved cell/tissue penetrating nanoparticle for MDR cancer therapy, by which silver nanoparticles showed outstanding antitumor activity in both MDR cells and non-resistant cells.

Scheme of MRP efflux transporter role and the developed novel approaches to overcome resistance
Moreover, in the in vivo study, these nanoparticles at a dose of 1 nmol/kg were able to strongly suppress the tumor growth in the mice with malignant melanoma, compared with the effective dose of doxorubicin (4.3 μmol/kg) and interestingly with low adverse effects (Liu et al. 2012). Advanced studies reported that Ag nanoparticles directly inhibit the efflux activity of MDR cancer cells (Kovacs et al. 2016). Au (gold) nanoparticles loaded with paclitaxel were endocytosed by both drug-sensitive human lung cancer H460 cells and MDR H460PTX cells overexpressing P-gp and MRP1 (Li et al. 2016b).
These data indicate that these nanoparticles could be a class of nano-drug for MDR cancer therapy.
Models to analyze MRP
Several models were used to study and analyze different MRPs (Fig. 3). These models include cell culture models, which involve cell lines and organoid models. Laboratory animals as immunocompromised mice were also used. Further, Drosophila melanogaster is also used.

Models to analyze the role of MRP in drug resistance
Cell culture models
Cell lines
Cell lines have a remarkable role in studying the physiological, pathophysiological, and biochemical processes of specific tissues or cells. It describes the changes in the structure, biology, and gene map of the cell under controlled conditions (Ulrich et al. 2002). From 1985 till 1990, the American National Cancer Institute (NCI) allowed the application of murine cell line P388 in studying the mechanisms, biology, and carcinogenic modifications in tumors of human origins. Later, this program has been canceled, and, as a substitute, in vitro primary tissue culture derived from human origin was developed to study these mechanisms (Shoemaker et al. 2002). The in vitro models are two types: either (1) human immortalized cancer cell lines, obtained from patients previously showing primary resistance, or (2) primary cell line cultures directly collected during diagnosis of human cancers, whose sensitivity or resistance to a specific targeted anti-cancer drug is not evaluated yet.
Cell lines are described as models of drug resistance through the administration of a clinical chemotherapeutical drug to study the mechanism of MDR to develop solutions to overcome it. For example, the first study in establishing an MDR cell line was established by Tsuruo and his colleagues. They used the K562 cell line, a human erythroleukemia cell line, to study the MDR that occurred against the clinical dose (60 nM) of vincristine to treat leukemia (Tsuruo et al. 1983). Amaral et al. reported that the total number of cancer cell lines used in experimental oncology for MDR and drug resistance analysis was 110 cell lines of solid and hematopoietic tumors. Most of them were for solid tumors with 97 cancer cell lines and only 13 cell lines for hematopoietic tumors (Amaral et al. 2019). Examples for cell lines used for the analysis of MDR-associated protein (MRP) are lung cell carcinoma cell line (H69AR), human ovarian cancer cell line A2780 for analysis of MRP1 (Cole et al. 1992; Mahdizadeh et al. 2016), human hepatic cell lines for analysis of MRP2 and MRP3 (Cizkova et al. 2005; Malinen et al. 2019) (Ceballos et al. 2018), colon cancer cell line for analysis of MRP3 (Kobayashi et al. 2016), and human embryonic kidney cell line (HEK293 cells) for MRP7 (Bortfeld et al. 2006).
Organoids models
Organoids, 3D-developed in vitro cellular structures, are derived mainly from stem cells with the capability of self-renewal and organization into a “mini-copy” of their original organ or tissue (Abugomaa and Elbadawy 2020; Abugomaa et al. 2020; Elbadawy et al. 2019a). Organoids can aptly and intimately recapitulate the in vivo architecture and the genetic and molecular signature of their parent tissues or organs (Elbadawy et al. 2018a, 2018b, 2019b; Usui et al. 2018b). In the last decade, organoids were essential tools to support basic medicine and translational research (Abugomaa and Elbadawy 2020) and analyzing the response to therapy (Abugomaa and Elbadawy 2020; Abugomaa et al. 2020; Elbadawy et al. 2021; Usui et al. 2018a).
Researchers began to use the 3D organoids for simulation and detection of the characterization of the ABC family transporter. For example, Zhang et al. were the first to use the 3D small intestinal organoids model (cultured from intestinal crypts) to investigate MRP2-mediated drug transport (Zhang et al. 2019a). They used their established organoids for the demonstration of modulators and inhibitors of MRP2 transporter. They select carboxy-dichlorofluorescein CDF (as MPR2 substrate) and probenecid (as MRP2 inhibitor) to validate the model, and the result assured the ability of MRP2 transport in the organoids (Zhang et al. 2019a). In another study, they continually described the role of intestinal organoids in estimating the function of BCRP transporter using the fluorescent dye as a substrate and Ko143 as an inhibitor. The mRNA expression and immunohistochemical analysis of BCRP revealed that organoid simulation was a valuable tool for detecting BCRP modulators (Zhang et al. 2017a).
Further, Lorenzi et al. employed the intestinal organoid to identify cellular response mediated by Fbxw7 (a member of the F-box protein family that is associated with drug resistance) to various concentrations of 5-FU and showed that the organoids from fbxw7∆G (specifically inactivated) are two to three times less sensitive to 5-FU therapy than fbxw7fl/fl (control floxed) ones (Lorenzi et al. 2016). Moreover, small hepatocyte organoids’ role in the expression of hepatic transporters and their capacity to convey organic anion substrates were investigated (Oshima et al. 2008). The result showed that transporters such as Oatp1, Oatp2, Ntcp, and MRP2 were increased with time in culture versus a decline in the expression of MRP3 and BCRP (Oshima et al. 2008).
Laboratory animals
Laboratory animal experimentation has been a part of biomedical and molecular research for several years. Animal research was introduced in Greece over 2000 years ago to understand how diseases, drugs, and organisms interact. The laboratory mice used as a model for MDR analysis must be immunocompromised to avoid rejection of the implanted cancer cells such as athymic nude mice (no thymus T cells) and SCID mice (no both B and T cells) (Richmond and Su 2008; Rosa et al. 2014). Rejinold et al. used mice to check the role of curcumin as a nanocarrier for doxorubicin delivery to MDR cancer cells and to compare the results with that obtained from using cell cultures such as NIH-3T3 (mouse embryonic fibroblast cells), HeLa (human cervical cancer cells), NCI-H460 (human lung carcinoma cells), and HFL1 (human normal lung cells) (Rejinold et al. 2018). Belinsky et al. previously used MRP3 null mice and transfected HEK293 cells to analyze the physiological role of MRP3 in enterohepatic circulation as an anionic transporter and its role to protect the normal tissue against etoposide and endobiotic (Belinsky et al. 2005). Karla et al. demonstrated the expression of MRP5 on the cornea and its role in drug efflux after using a male New Zealand white rabbit’s cornea (Karla et al. 2009). Finally, laboratory animals or generally in vivo experiments are the best methods (as they provide the native microenvironment in which tumors grow) to analyze the function and role of MRP transporter in cancer biology. However, due to animal welfare, livability, and ethical considerations, the need for tissue cells mimicking the in vivo experiment was the interest of scientists in the last decades, so organoids (3D tissue culture) were developed.
Drosophila melanogaster model
Drosophila is an alternative in vivo tool for high-throughput screening and validation of several anti-cancer drugs, including methotrexate, aminopterin, acivicin, gefitinib, and erlotinib due to high maintenance of its signaling pathways, brief life cycle, and genetic amenability (Yadav et al. 2016). D. melanogaster has a gene closely identical to human MRP1 that encodes a full ABC transporter containing three MSDs and two NBDs. Drosophila MRP (DMRP) is the only orthologue of the “long” human MRPs (MRP1, 3, 6, and 7). DMRP has been revealed to be a very active ABC transporter for different organic anions. It could transport MTX (Karasik et al. 2018), which can convey an MDR phenotype such as ABCG2, ABCB1, and ABCC transporters that often interfere with chemotherapy. Additionally, lowered expression of the endogenous DMRP has been revealed to decrease secretion of MRP1 substrate, daunorubicin, in the main insect excretory organ (Chahine et al. 2012). Such information points to the putative functional symmetry of DMRP and long ABCC transporters in vivo.
Summary and future perspectives
In the present review, nine MRPs with different structures, localization, tissue expression, substrate specificities, and functions have been discussed. Physiologically, most MRP family members protect different tissues and organs against several xenobiotic and endogenous substrates as well as their metabolites. In cancer therapeutics, MRPs have been demonstrated to confer resistance to many anti-cancer agents. Recently, there have been several attempts to reverse MRP-mediated MDR. Notably, small molecular inhibitors, especially TKIs, phytochemicals, miRNA-based therapy, nanomaterials, and MABs, are the main research spots in developing novel MRP modulators. The combination of more than one MRP modulator (hybrid drugs) results in a sophisticated understanding of the pharmacokinetics efflux of this combination by MRP. Progress in developing appropriate research models such as organoids and others will facilitate better understanding and developing effective MRP modulators.
Data availability
All relevant data are within the manuscript.
References
Abugomaa A, Elbadawy M (2020) Patient-derived organoid analysis of drug resistance in precision medicine: is there a value? Expert Rev Precision Med Drug Dev 5:1–5
Abugomaa A, Elbadawy M, Yamawaki H, Usui T, Sasaki K (2020) Emerging roles of cancer stem cells in bladder cancer progression, tumorigenesis, and resistance to chemotherapy: a potential therapeutic target for bladder cancer. Cells 9:235
Adamska A, Domenichini A, Capone E, Damiani V, Akkaya BG, Linton KJ, Di Sebastiano P, Chen X, Keeton AB, Ramirez-Alcantara V, Maxuitenko Y, Piazza GA, De Laurenzi V, Sala G, Falasca M (2019a) Pharmacological inhibition of ABCC3 slows tumour progression in animal models of pancreatic cancer. J Exp Clin Cancer Res : CR 38:312
Adamska A, Ferro R, Lattanzio R, Capone E, Domenichini A, Damiani V, Chiorino G, Akkaya BG, Linton KJ, De Laurenzi V, Sala G, Falasca M (2019b) ABCC3 is a novel target for the treatment of pancreatic cancer. Adv Biol Regul 73:100634
Aggarwal S (2010) Targeted cancer therapies. Nat Rev Drug Discov 9:427–428
Aggarwal BB, Kumar A, Bharti AC (2003) Anticancer potential of curcumin: preclinical and clinical studies. Anticancer Res 23:363–398
Ahuja N, Sharma AR, Baylin SB (2016) Epigenetic therapeutics: a New weapon in the War against cancer. Annu Rev Med 67:73–89
Alam A, Kung R, Kowal J, McLeod RA, Tremp N, Broude EV, Roninson IB, Stahlberg H, Locher KP (2018) Structure of a zosuquidar and UIC2-bound human-mouse chimeric ABCB1. Proc Natl Acad Sci U S A 115:E1973–E1982
Amaral MVS, Desp AJ, Das EL, Deos L, Dasm JH, Dem MEA, Moreira-Nunes CA (2019) Establishment of drug-resistant cell lines as a model in experimental oncology: a review. Anticancer Res 39:6443–6455
Arrondeau J, Gan HK, Razak AR, Paoletti X, Le Tourneau C (2010) Development of anti-cancer drugs. Discov Med 10:355–362
Assaraf YG, Brozovic A, Goncalves AC, Jurkovicova D, Line A, Machuqueiro M, Saponara S, Sarmento-Ribeiro AB, Xavier CPR, Vasconcelos MH (2019) The multi-factorial nature of clinical multidrug resistance in cancer. Drug Resist Updat 46:100645
Baguley BC (2010) Multiple drug resistance mechanisms in cancer. Mol Biotechnol 46:308–316
Bakos E, Evers R, Calenda G, Tusnady GE, Szakacs G, Varadi A, Sarkadi B (2000) Characterization of the amino-terminal regions in the human multidrug resistance protein (MRP1). J Cell Sci 113(Pt 24):4451–4461
Balcerczyk A, Rychlik B, Kruszewski M, Burchell B, Bartosz G (2003) MRP1-transfected cells do not show increased resistance against oxidative stress. Free Radic Res 37:189–195
Ball B, Zeidan A, Gore SD, Prebet T (2017) Hypomethylating agent combination strategies in myelodysplastic syndromes: hopes and shortcomings. Leuk Lymphoma 58:1022–1036
Barrand MA, Rhodes T, Center MS, Twentyman PR (1993) Chemosensitisation and drug accumulation effects of cyclosporin A, PSC-833 and verapamil in human MDR large cell lung cancer cells expressing a 190k membrane protein distinct from P-glycoprotein. Eur J Cancer (Oxford, England : 1990) 29A:408–415
Bar-Zeev M, Livney YD, Assaraf YG (2017) Targeted nanomedicine for cancer therapeutics: towards precision medicine overcoming drug resistance. Drug Resist Updat 31:15–30
Beck K, Hayashi K, Dang K, Hayashi M, Boyd CD (2005) Analysis of ABCC6 (MRP6) in normal human tissues. Histochem Cell Biol 123:517–528
Belinsky MG, Bain LJ, Balsara BB, Testa JR, Kruh GD (1998) Characterization of MOAT-C and MOAT-D, new members of the MRP/cMOAT subfamily of transporter proteins. J Natl Cancer Inst 90:1735–1741
Belinsky MG, Chen ZS, Shchaveleva I, Zeng H, Kruh GD (2002) Characterization of the drug resistance and transport properties of multidrug resistance protein 6 (MRP6, ABCC6). Cancer Res 62:6172–6177
Belinsky MG, Dawson PA, Shchaveleva I, Bain LJ, Wang R, Ling V, Chen ZS, Grinberg A, Westphal H, Klein-Szanto A, Lerro A, Kruh GD (2005) Analysis of the in vivo functions of Mrp3. Mol Pharmacol 68:160–168
Benson JD, Chen YN, Cornell-Kennon SA, Dorsch M, Kim S, Leszczyniecka M, Sellers WR, Lengauer C (2006) Validating cancer drug targets. Nature 441:451–456
Benyahia B, Huguet S, Decleves X, Mokhtari K, Criniere E, Bernaudin JF, Scherrmann JM, Delattre JY (2004) Multidrug resistance-associated protein MRP1 expression in human gliomas: chemosensitization to vincristine and etoposide by indomethacin in human glioma cell lines overexpressing MRP1. J Neuro-Oncol 66:65–70
Bera TK, Lee S, Salvatore G, Lee B, Pastan I (2001) MRP8, a new member of ABC transporter superfamily, identified by EST database mining and gene prediction program, is highly expressed in breast cancer. Mol Med (Cambridge, Mass) 7:509–516
Berdasco M, Esteller M (2010) Aberrant epigenetic landscape in cancer: how cellular identity goes awry. Dev Cell 19:698–711
Bergen AA, Plomp AS, Schuurman EJ, Terry S, Breuning M, Dauwerse H, Swart J, Kool M, van Soest S, Baas F, ten Brink JB, de Jong PT (2000) Mutations in ABCC6 cause pseudoxanthoma elasticum. Nat Genet 25:228–231
Bessho Y, Oguri T, Ozasa H, Uemura T, Sakamoto H, Miyazaki M, Maeno K, Sato S, Ueda R (2009) ABCC10/MRP7 is associated with vinorelbine resistance in non-small cell lung cancer. Oncol Rep 21:263–268
Bird A (2002) DNA methylation patterns and epigenetic memory. Genes Dev 16:6–21
Bisht K, Wagner KH, Bulmer AC (2010) Curcumin, resveratrol and flavonoids as anti-inflammatory, cyto- and DNA-protective dietary compounds. Toxicology 278:88–100
Blagitko-Dorfs N, Schlosser P, Greve G, Pfeifer D, Meier R, Baude A, Brocks D, Plass C, Lubbert M (2019) Combination treatment of acute myeloid leukemia cells with DNMT and HDAC inhibitors: predominant synergistic gene downregulation associated with gene body demethylation. Leukemia 33:945–956
Borel F, Han R, Visser A, Petry H, van Deventer SJ, Jansen PL, Konstantinova P, Reseau Centre de Ressources Biologiques Foie F (2012) Adenosine triphosphate-binding cassette transporter genes up-regulation in untreated hepatocellular carcinoma is mediated by cellular microRNAs. Hepatology (Baltimore, Md) 55:821–832
Borst P, Evers R, Kool M, Wijnholds J (2000) A family of drug transporters: the multidrug resistance-associated proteins. J Natl Cancer Inst 92:1295–1302
Bortfeld M, Rius M, Konig J, Herold-Mende C, Nies AT, Keppler D (2006) Human multidrug resistance protein 8 (MRP8/ABCC11), an apical efflux pump for steroid sulfates, is an axonal protein of the CNS and peripheral nervous system. Neuroscience 137:1247–1257
Bourichi S, Misbahi H, Rodi YK, Chahdi FO, Essassi EM, Szabó S, Szalontai B, Gajdács M, Molnár J, Spengler G (2018) In vitro evaluation of the multidrug resistance reversing activity of novel imidazo[4,5-b]pyridine derivatives. Anticancer Res 38:3999–4003
Boyerinas B, Park SM, Murmann AE, Gwin K, Montag AG, Zillhardt M, Hua YJ, Lengyel E, Peter ME (2012) Let-7 modulates acquired resistance of ovarian cancer to taxanes via IMP-1-mediated stabilization of multidrug resistance 1. Int J Cancer 130:1787–1797
Brozik A, Hegedus C, Erdei Z, Hegedus T, Ozvegy-Laczka C, Szakacs G, Sarkadi B (2011) Tyrosine kinase inhibitors as modulators of ATP binding cassette multidrug transporters: substrates, chemosensitizers or inducers of acquired multidrug resistance? Expert Opin Drug Metab Toxicol 7:623–642
Carozzo A, Yaneff A, Gomez N, Di Siervi N, Sahores A, Diez F, Attorresi AI, Rodriguez-Gonzalez A, Monczor F, Fernandez N, Abba M, Shayo C, Davio C (2019) Identification of MRP4/ABCC4 as a target for reducing the proliferation of pancreatic ductal adenocarcinoma cells by modulating the cAMP efflux. Mol Pharmacol 96:13–25
Ceballos MP, Decandido G, Quiroga AD, Comanzo CG, Livore VI, Lorenzetti F, Lambertucci F, Chazarreta-Cifre L, Banchio C, Alvarez ML, Mottino AD, Carrillo MC (2018) Inhibition of sirtuins 1 and 2 impairs cell survival and migration and modulates the expression of P-glycoprotein and MRP3 in hepatocellular carcinoma cell lines. Toxicol Lett 289:63–74
Cermak R, Wolffram S (2006) The potential of flavonoids to influence drug metabolism and pharmacokinetics by local gastrointestinal mechanisms. Curr Drug Metab 7:729–744
Chahine S, Seabrooke S, O'Donnell MJ (2012) Effects of genetic knock-down of organic anion transporter genes on secretion of fluorescent organic ions by Malpighian tubules of Drosophila melanogaster. Arch Insect Biochem Physiol 81:228–240
Chan E, Chiorean EG, O'Dwyer PJ, Gabrail NY, Alcindor T, Potvin D, Chao R, Hurwitz H (2018) Phase I/II study of mocetinostat in combination with gemcitabine for patients with advanced pancreatic cancer and other advanced solid tumors. Cancer Chemother Pharmacol 81:355–364
Chang XB (2007) A molecular understanding of ATP-dependent solute transport by multidrug resistance-associated protein MRP1. Cancer Metastasis Rev 26:15–37
Chang A (2011) Chemotherapy, chemoresistance and the changing treatment landscape for NSCLC. Lung Cancer (Amsterdam, Netherlands) 71:3–10
Chang HH, Hemberg M, Barahona M, Ingber DE, Huang S (2008) Transcriptome-wide noise controls lineage choice in mammalian progenitor cells. Nature 453:544–547
Chearwae W, Wu CP, Chu HY, Lee TR, Ambudkar SV, Limtrakul P (2006) Curcuminoids purified from turmeric powder modulate the function of human multidrug resistance protein 1 (ABCC1). Cancer Chemother Pharmacol 57:376–388
Chen ZS, Lee K, Kruh GD (2001) Transport of cyclic nucleotides and estradiol 17-beta-D-glucuronide by multidrug resistance protein 4. Resistance to 6-mercaptopurine and 6-thioguanine. J Biol Chem 276:33747–33754
Chen ZS, Lee K, Walther S, Raftogianis RB, Kuwano M, Zeng H, Kruh GD (2002) Analysis of methotrexate and folate transport by multidrug resistance protein 4 (ABCC4): MRP4 is a component of the methotrexate efflux system. Cancer Res 62:3144–3150
Chen ZS, Robey RW, Belinsky MG, Shchaveleva I, Ren XQ, Sugimoto Y, Ross DD, Bates SE, Kruh GD (2003) Transport of methotrexate, methotrexate polyglutamates, and 17beta-estradiol 17-(beta-D-glucuronide) by ABCG2: effects of acquired mutations at R482 on methotrexate transport. Cancer Res 63:4048–4054
Chen LM, Liang YJ, Ruan JW, Ding Y, Wang XW, Shi Z, Gu LQ, Yang XP, Fu LW (2004) Reversal of P-gp mediated multidrug resistance in-vitro and in-vivo by FG020318. J Pharm Pharmacol 56:1061–1066
Chen J, Tian W, Cai H, He H, Deng Y (2012) Down-regulation of microRNA-200c is associated with drug resistance in human breast cancer. Med Oncol 29:2527–2534
Chen N, Kong Y, Wu Y, Gao Q, Fu J, Sun X, Geng Q (2019) CAC1 knockdown reverses drug resistance through the downregulation of P-gp and MRP-1 expression in colorectal cancer. PLoS One 14:e0222035
Cheng Y, He C, Wang M, Ma X, Mo F, Yang S, Han J, Wei X (2019) Targeting epigenetic regulators for cancer therapy: mechanisms and advances in clinical trials. Signal Transduct Target Ther 4:62
Chu XY, Strauss JR, Mariano MA, Li J, Newton DJ, Cai X, Wang RW, Yabut J, Hartley DP, Evans DC, Evers R (2006) Characterization of mice lacking the multidrug resistance protein MRP2 (ABCC2). J Pharmacol Exp Ther 317:579–589
Cizkova D, Morky J, Micuda S, Osterreicher J, Martinkova J (2005) Expression of MRP2 and MDR1 transporters and other hepatic markers in rat and human liver and in WRL 68 cell line. Physiol Res 54:419–428
Colavita JPM, Todaro JS, de Sousa M, May M, Gomez N, Yaneff A, Di Siervi N, Aguirre MV, Guijas C, Ferrini L, Davio C, Rodriguez JP (2020) Multidrug resistance protein 4 (MRP4/ABCC4) is overexpressed in clear cell renal cell carcinoma (ccRCC) and is essential to regulate cell proliferation. Int J Biol Macromol 161:836–847
Cole SP (2014) Targeting multidrug resistance protein 1 (MRP1, ABCC1): past, present, and future. Annu Rev Pharmacol Toxicol 54:95–117
Cole SP, Bhardwaj G, Gerlach JH, Mackie JE, Grant CE, Almquist KC, Stewart AJ, Kurz EU, Duncan AM, Deeley RG (1992) Overexpression of a transporter gene in a multidrug-resistant human lung cancer cell line. Science (New York, NY) 258:1650–1654
Corbo C, Molinaro R, Parodi A, Toledano Furman NE, Salvatore F, Tasciotti E (2016) The impact of nanoparticle protein corona on cytotoxicity, immunotoxicity and target drug delivery. Nanomedicine (London, England) 11:81–100
Danza K, Silvestris N, Simone G, Signorile M, Saragoni L, Brunetti O, Monti M, Mazzotta A, De Summa S, Mangia A, Tommasi S (2016) Role of miR-27a, miR-181a and miR-20b in gastric cancer hypoxia-induced chemoresistance. Cancer Biol Ther 17:400–406
Dean M, Allikmets R (2001) Complete characterization of the human ABC gene family. J Bioenerg Biomembr 33:475–479
Deeley RG, Westlake C, Cole SP (2006) Transmembrane transport of endo- and xenobiotics by mammalian ATP-binding cassette multidrug resistance proteins. Physiol Rev 86:849–899
Deng W, Dai CL, Chen JJ, Kathawala RJ, Sun YL, Chen HF, Fu LW, Chen ZS (2013) Tandutinib (MLN518) reverses multidrug resistance by inhibiting the efflux activity of the multidrug resistance protein 7 (ABCC10). Oncol Rep 29:2479–2485
Dhayat SA, Abdeen B, Kohler G, Senninger N, Haier J, Mardin WA (2015) MicroRNA-100 and microRNA-21 as markers of survival and chemotherapy response in pancreatic ductal adenocarcinoma UICC stage II. Clin Epigenetics 7:132
Draper MP, Martell RL, Levy SB (1997) Indomethacin-mediated reversal of multidrug resistance and drug efflux in human and murine cell lines overexpressing MRP, but not P-glycoprotein. Br J Cancer 75:810–815
Du X, Liu B, Luan X, Cui Q, Li L (2018) miR-30 decreases multidrug resistance in human gastric cancer cells by modulating cell autophagy. Exp Ther Med 15:599–605
Elbadawy M, Usui T, Yamawaki H, Sasaki K (2018a) Development of an experimental model for analyzing drug resistance in colorectal cancer. Cancers 10:164
Elbadawy M, Usui T, Yamawaki H, Sasaki K (2018b) Novel functions of death-associated protein kinases through mitogen-activated protein kinase-related signals. Int J Mol Sci 19:3031
Elbadawy M, Usui T, Mori T, Tsunedomi R, Hazama S, Nabeta R, Uchide T, Fukushima R, Yoshida T, Shibutani M, Tanaka T, Masuda S, Okada R, Ichikawa R, Omatsu T, Mizutani T, Katayama Y, Noguchi S, Iwai S, Nakagawa T, Shinohara Y, Kaneda M, Yamawaki H, Sasaki K (2019a) Establishment of a novel experimental model for muscle-invasive bladder cancer using a dog bladder cancer organoid culture. Cancer Sci 110:2806–2821
Elbadawy M, Usui T, Yamawaki H, Sasaki K (2019b) Emerging roles of C-Myc in cancer stem cell-related signaling and resistance to cancer chemotherapy: a potential therapeutic target against colorectal cancer. Int J Mol Sci 20:2340
Elbadawy M, Sato Y, Mori T, Goto Y, Hayashi K, Yamanaka M, Azakami D, Uchide T, Fukushima R, Yoshida T, Shibutani M, Kobayashi M, Shinohara Y, Abugomaa A, Kaneda M, Yamawaki H, Usui T, Sasaki K (2021) Anti-tumor effect of trametinib in bladder cancer organoid and the underlying mechanism. Cancer Biol Ther. https://doi.org/10.1080/15384047.2021.1919004
Feng DD, Zhang H, Zhang P, Zheng YS, Zhang XJ, Han BW, Luo XQ, Xu L, Zhou H, Qu LH, Chen YQ (2011) Down-regulated miR-331-5p and miR-27a are associated with chemotherapy resistance and relapse in leukaemia. J Cell Mol Med 15:2164–2175
Fernandes J, Gattass CR (2009) Topological polar surface area defines substrate transport by multidrug resistance associated protein 1 (MRP1/ABCC1). J Med Chem 52:1214–1218
Fernandez SB, Hollo Z, Kern A, Bakos E, Fischer PA, Borst P, Evers R (2002) Role of the N-terminal transmembrane region of the multidrug resistance protein MRP2 in routing to the apical membrane in MDCKII cells. J Biol Chem 277:31048–31055
Ferraro A (2016) Altered primary chromatin structures and their implications in cancer development. Cell Oncol (Dordr) 39:195–210
Funazo T, Tsuji T, Ozasa H, Furugaki K, Yoshimura Y, Oguri T, Ajimizu H, Yasuda Y, Nomizo T, Sakamori Y, Yoshida H, Kim YH, Hirai T (2020) Acquired resistance to alectinib in ALK-rearranged lung cancer due to ABCC11/MRP8 overexpression in a clinically paired resistance model. Mol Cancer Ther 19:1320–1327
García-Domínguez DJ, Hontecillas-Prieto L, Rodríguez-Núñez P, Pascual-Pasto G, Vila-Ubach M, García-Mejías R, José Robles M, Tirado OM, Mora J, Carcaboso AM, de Álava E (2018) The combination of epigenetic drugs SAHA and HCI-2509 synergistically inhibits EWS-FLI1 and tumor growth in Ewing sarcoma. Oncotarget 9:31397–31410
Garrido W, Munoz M, San Martin R, Quezada C (2011) FK506 confers chemosensitivity to anticancer drugs in glioblastoma multiforme cells by decreasing the expression of the multiple resistance-associated protein-1. Biochem Biophys Res Commun 411:62–68
Giri AK, Aittokallio T (2019) DNMT inhibitors increase methylation in the cancer genome. Front Pharmacol 10:385
Grant CE, Kurz EU, Cole SP, Deeley RG (1997) Analysis of the intron-exon organization of the human multidrug-resistance protein gene (MRP) and alternative splicing of its mRNA. Genomics 45:368–378
Grasse S, Lienhard M, Frese S, Kerick M, Steinbach A, Grimm C, Hussong M, Rolff J, Becker M, Dreher F, Schirmer U, Boerno S, Ramisch A, Leschber G, Timmermann B, Grohé C, Lüders H, Vingron M, Fichtner I, Klein S, Odenthal M, Büttner R, Lehrach H, Sültmann H, Herwig R, Schweiger MR (2018) Epigenomic profiling of non-small cell lung cancer xenografts uncover LRP12 DNA methylation as predictive biomarker for carboplatin resistance. Genome Med 10:55
He SM, Li R, Kanwar JR, Zhou SF (2011) Structural and functional properties of human multidrug resistance protein 1 (MRP1/ABCC1). Curr Med Chem 18:439–481
Hipfner DR, Gauldie SD, Deeley RG, Cole SP (1994) Detection of the M(r) 190,000 multidrug resistance protein, MRP, with monoclonal antibodies. Cancer Res 54:5788–5792
Hjorth CF, Nielsen AS, Sorensen HT, Lash TL, Damkier P, Hamilton-Dutoit S, Cronin-Fenton D (2019) Multi-drug resistance protein 2 (MRP2) expression, adjuvant tamoxifen therapy, and risk of breast cancer recurrence: a Danish population-based nested case-control study. Acta Oncol (Stockholm, Sweden) 58:168–174
Hooijberg JH, Broxterman HJ, Kool M, Assaraf YG, Peters GJ, Noordhuis P, Scheper RJ, Borst P, Pinedo HM, Jansen G (1999) Antifolate resistance mediated by the multidrug resistance proteins MRP1 and MRP2. Cancer Res 59:2532–2535
Huang H, Li J, Shen J, Lin L, Wu X, Xiang S, Li Y, Xu Y, Zhao Q, Zhao Y, Kaboli PJ, Li M, Li X, Wang W, Wen Q, Xiao Z (2020) Increased ABCC4 expression induced by ERRalpha leads to docetaxel resistance via efflux of docetaxel in prostate cancer. Front Oncol 10:1474
Huisman MT, Chhatta AA, van Tellingen O, Beijnen JH, Schinkel AH (2005) MRP2 (ABCC2) transports taxanes and confers paclitaxel resistance and both processes are stimulated by probenecid. Int J Cancer 116:824–829
Iram SH, Cole SP (2011) Expression and function of human MRP1 (ABCC1) is dependent on amino acids in cytoplasmic loop 5 and its interface with nucleotide binding domain 2. J Biol Chem 286:7202–7213
Ivnitski-Steele I, Larson RS, Lovato DM, Khawaja HM, Winter SS, Oprea TI, Sklar LA, Edwards BS (2008) High-throughput flow cytometry to detect selective inhibitors of ABCB1, ABCC1, and ABCG2 transporters. Assay Drug Dev Technol 6:263–276
Jaramillo AC, Cloos J, Lemos C, Stam RW, Kaspers GJL, Jansen G, Peters GJ (2019) Ex vivo resistance in childhood acute lymphoblastic leukemia: correlations between BCRP, MRP1, MRP4 and MRP5 ABC transporter expression and intracellular methotrexate polyglutamate accumulation. Leuk Res 79:45–51
Jedlitschky G, Leier I, Buchholz U, Barnouin K, Center M, Keppler D (1994) ATP-dependent glutathione conjugate transport in HL60 cells overexpressing the multidrug resistance associated protein (MRP): 3. Anti-Cancer Drugs 5:4
Jedlitschky G, Leier I, Buchholz U, Barnouin K, Kurz G, Keppler D (1996) Transport of glutathione, glucuronate, and sulfate conjugates by the MRP gene-encoded conjugate export pump. Cancer Res 56:988–994
Jedlitschky G, Leier I, Buchholz U, Hummel-Eisenbeiss J, Burchell B, Keppler D (1997) ATP-dependent transport of bilirubin glucuronides by the multidrug resistance protein MRP1 and its hepatocyte canalicular isoform MRP2. Biochem J 327(Pt 1):305–310
Jung M, Gao J, Cheung L, Bongers A, Somers K, Clifton M, Ramsay EE, Russell AJ, Valli E, Gifford AJ, George J, Kennedy CJ, Wakefield MJ, Topp M, Ho GY, Australian Ovarian Cancer Study, Scott CL, Bowtell DD, deFazio A, Norris MD, Haber M, Henderson MJ (2020) ABCC4/MRP4 contributes to the aggressiveness of Myc-associated epithelial ovarian cancer. Int J Cancer 147:2225–2238
Kagohara LT, Stein-O'Brien GL, Kelley D, Flam E, Wick HC, Danilova LV, Easwaran H, Favorov AV, Qian J, Gaykalova DA, Fertig EJ (2018) Epigenetic regulation of gene expression in cancer: techniques, resources and analysis. Brief Funct Genomics 17:49–63
Kao HH, Chang MS, Cheng JF, Huang JD (2003) Genomic structure, gene expression, and promoter analysis of human multidrug resistance-associated protein 7. J Biomed Sci 10:98–110
Karasik A, Varadi A, Szeri F (2018) In vitro transport of methotrexate by Drosophila multidrug resistance-associated protein. PLoS One 13:e0205657
Karla PK, Quinn TL, Herndon BL, Thomas P, Pal D, Mitra A (2009) Expression of multidrug resistance associated protein 5 (MRP5) on cornea and its role in drug efflux. J Ocular Pharmacol Ther : the official journal of the Association for Ocular Pharmacology and Therapeutics 25:121–132
Kawabe T, Chen ZS, Wada M, Uchiumi T, Ono M, Akiyama S, Kuwano M (1999) Enhanced transport of anticancer agents and leukotriene C4 by the human canalicular multispecific organic anion transporter (cMOAT/MRP2). FEBS Lett 456:327–331
Keppler D (2011) Multidrug resistance proteins (MRPs, ABCCs): importance for pathophysiology and drug therapy. Handb Exp Pharmacol 299-323(201):299–323
Kim WJ, Kakehi Y, Hirai M, Arao S, Hiai H, Fukumoto M, Yoshida O (1995) Multidrug resistance-associated protein-mediated multidrug resistance modulated by cyclosporin A in a human bladder cancer cell line. Jpn J Cancer Res : Gann 86:969–977
Kobayashi M, Funayama R, Ohnuma S, Unno M, Nakayama K (2016) Wnt-beta-catenin signaling regulates ABCC3 (MRP3) transporter expression in colorectal cancer. Cancer Sci 107:1776–1784
Koike K, Kawabe T, Tanaka T, Toh S, Uchiumi T, Wada M, Akiyama S, Ono M, Kuwano M (1997) A canalicular multispecific organic anion transporter (cMOAT) antisense cDNA enhances drug sensitivity in human hepatic cancer cells. Cancer Res 57:5475–5479
Komdeur R, Plaat BE, van der Graaf WT, Hoekstra HJ, Hollema H, van den Berg E, Zwart N, Scheper RJ, Molenaar WM (2003) Expression of multidrug resistance proteins, P-gp, MRP1 and LRP, in soft tissue sarcomas analysed according to their histological type and grade. Eur J Cancer (Oxford, England : 1990) 39:909–916
Kool M, de Haas M, Scheffer GL, Scheper RJ, van Eijk MJ, Juijn JA, Baas F, Borst P (1997) Analysis of expression of cMOAT (MRP2), MRP3, MRP4, and MRP5, homologues of the multidrug resistance-associated protein gene (MRP1), in human cancer cell lines. Cancer Res 57:3537–3547
Kool M, van der Linden M, de Haas M, Scheffer GL, de Vree JM, Smith AJ, Jansen G, Peters GJ, Ponne N, Scheper RJ, Elferink RP, Baas F, Borst P (1999) MRP3, an organic anion transporter able to transport anti-cancer drugs. Proc Natl Acad Sci U S A 96:6914–6919
Kovacs D, Szoke K, Igaz N, Spengler G, Molnar J, Toth T, Madarasz D, Razga Z, Konya Z, Boros IM, Kiricsi M (2016) Silver nanoparticles modulate ABC transporter activity and enhance chemotherapy in multidrug resistant cancer. Nanomed Nanotechnol Biol Med 12:601–610
Kranz J, Hessel S, Aretz J, Seidel A, Petzinger E, Geyer J, Lampen A (2014) The role of the efflux carriers Abcg2 and Abcc2 for the hepatobiliary elimination of benzo[a]pyrene and its metabolites in mice. Chem Biol Interact 224:36–41
Kruh GD, Belinsky MG (2003) The MRP family of drug efflux pumps. Oncogene 22:7537–7552
Kruh GD, Zeng H, Rea PA, Liu G, Chen ZS, Lee K, Belinsky MG (2001) MRP subfamily transporters and resistance to anticancer agents. J Bioenerg Biomembr 33:493–501
Kummar S, Chen HX, Wright J, Holbeck S, Millin MD, Tomaszewski J, Zweibel J, Collins J, Doroshow JH (2010) Utilizing targeted cancer therapeutic agents in combination: novel approaches and urgent requirements. Nat Rev Drug Discov 9:843–856
Lage H, Perlitz C, Abele R, Tampe R, Dietel M, Schadendorf D, Sinha P (2001) Enhanced expression of human ABC-transporter tap is associated with cellular resistance to mitoxantrone. FEBS Lett 503:179–184
Landesman-Milo D, Goldsmith M, Leviatan Ben-Arye S, Witenberg B, Brown E, Leibovitch S, Azriel S, Tabak S, Morad V, Peer D (2013) Hyaluronan grafted lipid-based nanoparticles as RNAi carriers for cancer cells. Cancer Lett 334:221–227
Lee K, Klein-Szanto AJ, Kruh GD (2000) Analysis of the MRP4 drug resistance profile in transfected NIH3T3 cells. J Natl Cancer Inst 92:1934–1940
Leier I, Jedlitschky G, Buchholz U, Cole SP, Deeley RG, Keppler D (1994) The MRP gene encodes an ATP-dependent export pump for leukotriene C4 and structurally related conjugates. J Biol Chem 269:27807–27810
Leslie EM, Mao Q, Oleschuk CJ, Deeley RG, Cole SP (2001) Modulation of multidrug resistance protein 1 (MRP1/ABCC1) transport and atpase activities by interaction with dietary flavonoids. Mol Pharmacol 59:1171–1180
Li Y, Revalde JL, Reid G, Paxton JW (2010) Interactions of dietary phytochemicals with ABC transporters: possible implications for drug disposition and multidrug resistance in cancer. Drug Metab Rev 42:590–611
Li C, Zou J, Zheng G, Chu J (2016a) MiR-30a decreases multidrug resistance (MDR) of gastric cancer cells. Med Sci Monit : international medical journal of experimental and clinical research 0, 0 22:4509–4515
Li F, Zhou X, Zhou H, Jia J, Li L, Zhai S, Yan B (2016b) Reducing both Pgp overexpression and drug efflux with anti-cancer gold-paclitaxel nanoconjugates. PLoS One 11:e0160042
Li W, Zhang H, Assaraf YG, Zhao K, Xu X, Xie J, Yang DH, Chen ZS (2016c) Overcoming ABC transporter-mediated multidrug resistance: molecular mechanisms and novel therapeutic drug strategies. Drug Resist Updat 27:14–29
Li YF, Ji HH, Zhang ZL, Zhang TT, Gan W, Zhang SF (2017) Targeting MRP4 expression by anti-androgen treatment reverses MRP4-mediated docetaxel resistance in castration-resistant prostate cancer. Oncol Lett 14:1748–1756
Limtrakul P, Chearwae W, Shukla S, Phisalphong C, Ambudkar SV (2007) Modulation of function of three ABC drug transporters, P-glycoprotein (ABCB1), mitoxantrone resistance protein (ABCG2) and multidrug resistance protein 1 (ABCC1) by tetrahydrocurcumin, a major metabolite of curcumin. Mol Cell Biochem 296:85–95
Ling V, Thompson LH (1974) Reduced permeability in CHO cells as a mechanism of resistance to colchicine. J Cell Physiol 83:103–116
Liu J, Zhao Y, Guo Q, Wang Z, Wang H, Yang Y, Huang Y (2012) TAT-modified nanosilver for combating multidrug-resistant cancer. Biomaterials 33:6155–6161
Liu H, Wu X, Huang J, Peng J, Guo L (2015) miR-7 modulates chemoresistance of small cell lung cancer by repressing MRP1/ABCC1. Int J Exp Pathol 96:240–247
Lorenzi F, Babaei-Jadidi R, Sheard J, Spencer-Dene B, Nateri AS (2016) Fbxw7-associated drug resistance is reversed by induction of terminal differentiation in murine intestinal organoid culture. Mol Ther Methods Clin Dev 3:16024
Low FG, Shabir K, Brown JE, Bill RM, Rothnie AJ (2020) Roles of ABCC1 and ABCC4 in proliferation and migration of breast cancer cell lines. Int J Mol Sci 21:7664
Lozano E, Asensio M, Perez-Silva L, Banales JM, Briz O, Marin JJG (2020) MRP3-mediated chemoresistance in cholangiocarcinoma: target for chemosensitization through restoring SOX17 expression. Hepatology (Baltimore, Md) 72:949–964
Luo Q, Wu X, Zhang Y, Shu T, Ding F, Chen H, Zhao P, Chang W, Zhu X, Liu Z (2018) ARID1A ablation leads to multiple drug resistance in ovarian cancer via transcriptional activation of MRP2. Cancer Lett 427:9–17
Macconaill LE, Garraway LA (2010) Clinical implications of the cancer genome. J Clin Oncol 28:5219–5228
Mackall CL, Merchant MS, Fry TJ (2014) Immune-based therapies for childhood cancer. Nat Rev Clin Oncol 11:693–703
Madon J, Hagenbuch B, Landmann L, Meier PJ, Stieger B (2000) Transport function and hepatocellular localization of mrp6 in rat liver. Mol Pharmacol 57:634–641
Maeng HJ, Lee WJ, Jin QR, Chang JE, Shim WS (2014) Upregulation of COX-2 in the lung cancer promotes overexpression of multidrug resistance protein 4 (MRP4) via PGE2-dependent pathway. Eur J Pharm Sci : official journal of the European Federation for Pharmaceutical Sciences 62:189–196
Mahdizadeh S, Karimi G, Behravan J, Arabzadeh S, Lage H, Kalalinia F (2016) Crocin suppresses multidrug resistance in MRP overexpressing ovarian cancer cell line. Daru: journal of Faculty of Pharmacy, Tehran University of Medical Sciences 24:17
Malinen MM, Ito K, Kang HE, Honkakoski P, Brouwer KLR (2019) Protein expression and function of organic anion transporters in short-term and long-term cultures of Huh7 human hepatoma cells. Eur J Pharm Sci : official journal of the European Federation for Pharmaceutical Sciences 130:186–195
Marbeuf-Gueye C, Salerno M, Quidu P, Garnier-Suillerot A (2000) Inhibition of the P-glycoprotein- and multidrug resistance protein-mediated efflux of anthracyclines and calceinacetoxymethyl ester by PAK-104P. Eur J Pharmacol 391:207–216
Masetto F, Chegaev K, Gazzano E, Mullappilly N, Rolando B, Arpicco S, Fruttero R, Riganti C, Donadelli M (2020) MRP5 nitration by NO-releasing gemcitabine encapsulated in liposomes confers sensitivity in chemoresistant pancreatic adenocarcinoma cells. Biochimica et biophysica acta. Mol Cell Res 1867:118824
Mechetner EB, Roninson IB (1992) Efficient inhibition of P-glycoprotein-mediated multidrug resistance with a monoclonal antibody. Proc Natl Acad Sci U S A 89:5824–5828
Melamed JR, Riley RS, Valcourt DM, Billingsley MM, Kreuzberger NL, Day ES (2017) Quantification of siRNA duplexes bound to gold nanoparticle surfaces. Methods Mol Biol (Clifton, NJ) 1570:1–15
Mercier C, Masseguin C, Roux F, Gabrion J, Scherrmann JM (2004) Expression of P-glycoprotein (ABCB1) and Mrp1 (ABCC1) in adult rat brain: focus on astrocytes. Brain Res 1021:32–40
Michaelis M, Rothweiler F, Klassert D, von Deimling A, Weber K, Fehse B, Kammerer B, Doerr HW, Cinatl J Jr (2009) Reversal of P-glycoprotein-mediated multidrug resistance by the murine double minute 2 antagonist nutlin-3. Cancer Res 69:416–421
Morris ME, Zhang S (2006) Flavonoid-drug interactions: effects of flavonoids on ABC transporters. Life Sci 78:2116–2130
Myint K, Biswas R, Li Y, Jong N, Jamieson S, Liu J, Han C, Squire C, Merien F, Lu J, Nakanishi T, Tamai I, McKeage M (2019) Identification of MRP2 as a targetable factor limiting oxaliplatin accumulation and response in gastrointestinal cancer. Sci Rep 9:2245
Narayana K, Reddy M, Chaluvadi M, Devarakonda K (2001) Bioflavonoids classification, pharmacological, biochemical effects and therapeutic potential. Indian J Pharm 33:2–16
Nezami M (2019) Sequential and dual inhibition of pleiotropic targets in cancer—a novel strategy to sensitize tumor cells to targeted therapies and overcome resistance. J Cancer Ther 10:166–177
Nguyen H, Zhang S, Morris ME (2003) Effect of flavonoids on MRP1-mediated transport in Panc-1 cells. J Pharm Sci 92:250–257
Nieth C, Priebsch A, Stege A, Lage H (2003) Modulation of the classical multidrug resistance (MDR) phenotype by RNA interference (RNAi). FEBS Lett 545:144–150
Nooter K, Westerman AM, Flens MJ, Zaman GJ, Scheper RJ, van Wingerden KE, Burger H, Oostrum R, Boersma T, Sonneveld P et al (1995) Expression of the multidrug resistance-associated protein (MRP) gene in human cancers. Clin Cancer Res: an official journal of the American Association for Cancer Research 1:1301–1310
Nowacka-Zawisza M, Wisnik E (2017) DNA methylation and histone modifications as epigenetic regulation in prostate cancer (Review). Oncol Rep 38:2587–2596
Oguri T, Ozasa H, Uemura T, Bessho Y, Miyazaki M, Maeno K, Maeda H, Sato S, Ueda R (2008) MRP7/ABCC10 expression is a predictive biomarker for the resistance to paclitaxel in non-small cell lung cancer. Mol Cancer Ther 7:1150–1155
Oshima H, Kon J, Ooe H, Hirata K, Mitaka T (2008) Functional expression of organic anion transporters in hepatic organoids reconstructed by rat small hepatocytes. J Cell Biochem 104:68–81
Palii SS, Van Emburgh BO, Sankpal UT, Brown KD, Robertson KD (2008) DNA methylation inhibitor 5-Aza-2'-deoxycytidine induces reversible genome-wide DNA damage that is distinctly influenced by DNA methyltransferases 1 and 3B. Mol Cell Biol 28:752–771
Patel H, Jadhav H, Ansari I, Pawara R, Surana S (2019) Pyridine and nitro-phenyl linked 1,3,4-thiadiazoles as MDR-TB inhibitors. Eur J Med Chem 167:1–9
Peng Y, Croce CM (2016) The role of MicroRNAs in human cancer. Signal Transduct Target Ther 1:15004
Peterson BG, Tan KW, Osa-Andrews B, Iram SH (2017) High-content screening of clinically tested anticancer drugs identifies novel inhibitors of human MRP1 (ABCC1). Pharmacol Res 119:313–326
Pham AN, Wang J, Fang J, Gao X, Zhang Y, Blower PE, Sadee W, Huang Y (2009) Pharmacogenomics approach reveals MRP1 (ABCC1)-mediated resistance to geldanamycins. Pharm Res 26:936–945
Pratt S, Shepard RL, Kandasamy RA, Johnston PA, Perry W 3rd, Dantzig AH (2005) The multidrug resistance protein 5 (ABCC5) confers resistance to 5-fluorouracil and transports its monophosphorylated metabolites. Mol Cancer Ther 4:855–863
Reid G, Wielinga P, Zelcer N, De Haas M, Van Deemter L, Wijnholds J, Balzarini J, Borst P (2003) Characterization of the transport of nucleoside analog drugs by the human multidrug resistance proteins MRP4 and MRP5. Mol Pharmacol 63:1094–1103
Rejinold NS, Yoo J, Jon S, Kim YC (2018) Curcumin as a novel nanocarrier system for doxorubicin delivery to MDR cancer cells: in vitro and in vivo evaluation. ACS Appl Mater Interfaces 10:28458–28470
Richmond A, Su Y (2008) Mouse xenograft models vs GEM models for human cancer therapeutics. Dis Model Mech 1:78–82
Ritchie TK, Kwon H, Atkins WM (2011) Conformational analysis of human ATP-binding cassette transporter ABCB1 in lipid nanodiscs and inhibition by the antibodies MRK16 and UIC2. J Biol Chem 286:39489–39496
Rosa R, Monteleone F, Zambrano N, Bianco R (2014) In vitro and in vivo models for analysis of resistance to anticancer molecular therapies. Curr Med Chem 21:1595–1606
Ryu S, Kawabe T, Nada S, Yamaguchi A (2000) Identification of basic residues involved in drug export function of human multidrug resistance-associated protein 2. J Biol Chem 275:39617–39624
Saghafinia S, Mina M, Riggi N, Hanahan D, Ciriello G (2018) Pan-cancer landscape of aberrant DNA methylation across human tumors. Cell Rep 25(1066-1080):e8
Sampath J, Adachi M, Hatse S, Naesens L, Balzarini J, Flatley RM, Matherly LH, Schuetz JD (2002) Role of MRP4 and MRP5 in biology and chemotherapy. AAPS PharmSci 4:E14
Schiffmann I, Greve G, Jung M, Lubbert M (2016) Epigenetic therapy approaches in non-small cell lung cancer: update and perspectives. Epigenetics 11:858–870
Schneider BJ, Shah MA, Klute K, Ocean A, Popa E, Altorki N, Lieberman M, Schreiner A, Yantiss R, Christos PJ, Palmer R, You D, Viale A, Kermani P, Scandura JM (2017) Phase I study of epigenetic priming with azacitidine prior to standard neoadjuvant chemotherapy for patients with resectable gastric and esophageal adenocarcinoma: evidence of tumor hypomethylation as an indicator of major histopathologic response. Clin Cancer Res : an official journal of the American Association for Cancer Research 23:2673–2680
Sharma A, Vatapalli R, Abdelfatah E, Wyatt McMahon K, Kerner Z, Guzzetta AA, Singh J, Zahnow C, Baylin BS, Yerram S, Hu Y, Azad N, Ahuja N (2017) Hypomethylating agents synergize with irinotecan to improve response to chemotherapy in colorectal cancer cells. PLoS One 12:e0176139
Shen T, Kuang YH, Ashby CR, Lei Y, Chen A, Zhou Y, Chen X, Tiwari AK, Hopper-Borge E, Ouyang J, Chen ZS (2009) Imatinib and nilotinib reverse multidrug resistance in cancer cells by inhibiting the efflux activity of the MRP7 (ABCC10). PLoS One 4:e7520
Shi J, Kantoff PW, Wooster R, Farokhzad OC (2017) Cancer nanomedicine: progress, challenges and opportunities. Nat Rev Cancer 17:20–37
Shi B, Xu FF, Xiang CP, Jia R, Yan CH, Ma SQ, Wang N, Wang AJ, Fan P (2020) Effect of sodium butyrate on ABC transporters in lung cancer A549 and colorectal cancer HCT116 cells. Oncol Lett 20:148
Shishodia S, Singh T, Chaturvedi MM (2007) Modulation of transcription factors by curcumin. Adv Exp Med Biol 595:127–148
Shoemaker RH, Scudiero DA, Melillo G, Currens MJ, Monks AP, Rabow AA, Covell DG, Sausville EA (2002) Application of high-throughput, molecular-targeted screening to anticancer drug discovery. Curr Top Med Chem 2:229–246
Slot AJ, Wise DD, Deeley RG, Monks TJ, Cole SP (2008) Modulation of human multidrug resistance protein (MRP) 1 (ABCC1) and MRP2 (ABCC2) transport activities by endogenous and exogenous glutathione-conjugated catechol metabolites. Drug Metab Dispos 36:552–560
Slot AJ, Molinski SV, Cole SP (2011) Mammalian multidrug-resistance proteins (MRPs). Essays Biochem 50:179–207
Smeets PH, van Aubel RA, Wouterse AC, van den Heuvel JJ, Russel FG (2004) Contribution of multidrug resistance protein 2 (MRP2/ABCC2) to the renal excretion of p-aminohippurate (PAH) and identification of MRP4 (ABCC4) as a novel PAH transporter. J Am Soc Nephrol : JASN 15:2828–2835
Song L, Li Y, Li W, Wu S, Li Z (2014) miR-495 enhances the sensitivity of non-small cell lung cancer cells to platinum by modulation of copper-transporting P-type adenosine triphosphatase A (ATP7A). J Cell Biochem 115:1234–1242
Spitzwieser M, Pirker C, Koblmuller B, Pfeiler G, Hacker S, Berger W, Heffeter P, Cichna-Markl M (2016) Promoter methylation patterns of ABCB1, ABCC1 and ABCG2 in human cancer cell lines, multidrug-resistant cell models and tumor, tumor-adjacent and tumor-distant tissues from breast cancer patients. Oncotarget 7:73347–73369
Sui H, Cai GX, Pan SF, Deng WL, Wang YW, Chen ZS, Cai SJ, Zhu HR, Li Q (2014) miR200c attenuates P-gp-mediated MDR and metastasis by targeting JNK2/c-Jun signaling pathway in colorectal cancer. Mol Cancer Ther 13:3137–3151
Sun C, Li N, Yang Z, Zhou B, He Y, Weng D, Fang Y, Wu P, Chen P, Yang X, Ma D, Zhou J, Chen G (2013) miR-9 regulation of BRCA1 and ovarian cancer sensitivity to cisplatin and PARP inhibition. J Natl Cancer Inst 105:1750–1758
Takayanagi S, Kataoka T, Ohara O, Oishi M, Kuo MT, Ishikawa T (2004) Human ATP-binding cassette transporter ABCC10: expression profile and p53-dependent upregulation. J Exp Ther Oncol 4:239–246
Tatosian DA, Shuler ML (2009) A novel system for evaluation of drug mixtures for potential efficacy in treating multidrug resistant cancers. Biotechnol Bioeng 103:187–198
Tian J, Xu YY, Li L, Hao Q (2017) MiR-490-3p sensitizes ovarian cancer cells to cisplatin by directly targeting ABCC2. Am J Transl Res 9:1127–1138
To KK, Leung WW, Ng SS (2015) Exploiting a novel miR-519c-HuR-ABCG2 regulatory pathway to overcome chemoresistance in colorectal cancer. Exp Cell Res 338:222–231
Tsuruo T, Iida H, Nojiri M, Tsukagoshi S, Sakurai Y (1983) Potentiation of chemotherapeutic effect of vincristine in vincristine resistant tumor bearing mice by calmodulin inhibitor clomipramine. Aust J Pharm 6:145–147
Uemura T, Oguri T, Ozasa H, Takakuwa O, Miyazaki M, Maeno K, Sato S, Ueda R (2010) ABCC11/MRP8 confers pemetrexed resistance in lung cancer. Cancer Sci 101:2404–2410
Uemura T, Oguri T, Maeno K, Sone K, Takeuchi A, Fukuda S, Kunii E, Takakuwa O, Kanemitsu Y, Ohkubo H, Takemura M, Ito Y, Niimi A (2019) ABCC11 gene polymorphism as a potential predictive biomarker for an oral 5-fluorouracil derivative drug S-1 treatment in non-small cell lung cancer. Cancer Chemother Pharmacol 84:1229–1239
Ulrich AB, Schmied BM, Standop J, Schneider MB, Pour PM (2002) Pancreatic cell lines: a review. Pancreas 24:111–120
Usui T, Elbadawy M, Sasaki K (2018a) Session 6: advances in anticancer therapy. J Vet Pharmacol Ther 41:28–30
Usui T, Sakurai M, Umata K, Elbadawy M, Ohama T, Yamawaki H, Hazama S, Takenouchi H, Nakajima M, Tsunedomi R, Suzuki N, Nagano H, Sato K, Kaneda M, Sasaki K (2018b) hedgehog signals mediate anti-cancer drug resistance in three-dimensional primary colorectal cancer organoid culture. Int J Mol Sci 19:1098
Valinezhad Sani F, Palizban A, Mosaffa F, Jamialahmadi K (2020) Glucosamine reverses drug resistance in MRP2 overexpressing ovarian cancer cells. Eur J Pharmacol 868:172883
van Zanden JJ, Wortelboer HM, Bijlsma S, Punt A, Usta M, Bladeren PJ, Rietjens IM, Cnubben NH (2005) Quantitative structure activity relationship studies on the flavonoid mediated inhibition of multidrug resistance proteins 1 and 2. Biochem Pharmacol 69:699–708
Vanhoefer U, Cao S, Minderman H, Toth K, Scheper RJ, Slovak ML, Rustum YM (1996) PAK-104P, a pyridine analogue, reverses paclitaxel and doxorubicin resistance in cell lines and nude mice bearing xenografts that overexpress the multidrug resistance protein. Clin Cancer Res: an official journal of the American Association for Cancer Research 2:369–377
Versantvoort CH, Broxterman HJ, Lankelma J, Feller N, Pinedo HM (1994) Competitive inhibition by genistein and ATP dependence of daunorubicin transport in intact MRP overexpressing human small cell lung cancer cells. Biochem Pharmacol 48:1129–1136
Wang EJ, Johnson WW (2003) The farnesyl protein transferase inhibitor lonafarnib (SCH66336) is an inhibitor of multidrug resistance proteins 1 and 2. Chemotherapy 49:303–308
Wang H, Chi CH, Zhang Y, Shi B, Jia R, Wang BJ (2019) Effects of histone deacetylase inhibitors on ATP-binding cassette transporters in lung cancer A549 and colorectal cancer HCT116 cells. Oncol Lett 18:63–71
Werner HM, Mills GB, Ram PT (2014) Cancer systems biology: a peek into the future of patient care? Nature reviews. Clin Oncol 11:167–176
Wielinga PR, Reid G, Challa EE, van der Heijden I, van Deemter L, de Haas M, Mol C, Kuil AJ, Groeneveld E, Schuetz JD, Brouwer C, De Abreu RA, Wijnholds J, Beijnen JH, Borst P (2002) Thiopurine metabolism and identification of the thiopurine metabolites transported by MRP4 and MRP5 overexpressed in human embryonic kidney cells. Mol Pharmacol 62:1321–1331
Wijnholds J, Mol CA, van Deemter L, de Haas M, Scheffer GL, Baas F, Beijnen JH, Scheper RJ, Hatse S, De Clercq E, Balzarini J, Borst P (2000) Multidrug-resistance protein 5 is a multispecific organic anion transporter able to transport nucleotide analogs. Proc Natl Acad Sci U S A 97:7476–7481
Wilting RH, Dannenberg JH (2012) Epigenetic mechanisms in tumorigenesis, tumor cell heterogeneity and drug resistance. Drug Resist Updat 15:21–38
Xin Y, Huang M, Guo WW, Huang Q, Zhang LZ, Jiang G (2017) Nano-based delivery of RNAi in cancer therapy. Mol Cancer 16:134
Yadav AK, Srikrishna S, Gupta SC (2016) Cancer drug development using Drosophila as an in vivo tool: from bedside to bench and back. Trends Pharmacol Sci 37:789–806
Yamada A, Ishikawa T, Ota I, Kimura M, Shimizu D, Tanabe M, Chishima T, Sasaki T, Ichikawa Y, Morita S, Yoshiura K, Takabe K, Endo I (2013) High expression of ATP-binding cassette transporter ABCC11 in breast tumors is associated with aggressive subtypes and low disease-free survival. Breast Cancer Res Treat 137:773–782
Yang G, Wu D, Zhu J, Jiang O, Shi Q, Tian J, Weng Y (2013) Upregulation of miR-195 increases the sensitivity of breast cancer cells to adriamycin treatment through inhibition of Raf-1. Oncol Rep 30:877–889
Yang J, Song P, Zhou G (2019) A study on the correlations of MRP-1 expression with the pathogenesis and prognosis of colorectal cancer. J BUON : official journal of the Balkan Union of Oncology 24:84–90
Yang J, Sontag D, Gong Y, Minuk GY (2021) Enhanced gemcitabine cytotoxicity with knockdown of multidrug resistance protein genes in human cholangiocarcinoma cell lines. J Gastroenterol Hepatol 36:1103–1109
Yano S, Hanibuchi M, Nishioka Y, Nokihara H, Nishimura N, Tsuruo T, Sone S (1999) Combined therapy with anti-P-glycoprotein antibody and macrophage colony-stimulating factor gene transduction for multiorgan metastases of multidrug-resistant human small cell lung cancer in NK cell-depleted SCID mice. Int J Cancer 82:105–111
Yonetomi Y, Sekioka T, Kadode M, Kitamine T, Kamiya A, Matsumura N, Fujita M, Kawabata K (2015) Leukotriene C4 induces bronchoconstriction and airway vascular hyperpermeability via the cysteinyl leukotriene receptor 2 in S-hexyl glutathione-treated guinea pigs. Eur J Pharmacol 754:98–104
Yuan YH, Wang HY, Lai Y, Zhong W, Liang WL, Yan FD, Yu Z, Chen JK, Lin Y (2019) Epigenetic inactivation of HOXD10 is associated with human colon cancer via inhibiting the RHOC/AKT/MAPK signaling pathway. Cell Commun Signal : CCS 17:9
Zeng H, Bain LJ, Belinsky MG, Kruh GD (1999) Expression of multidrug resistance protein-3 (multispecific organic anion transporter-D) in human embryonic kidney 293 cells confers resistance to anticancer agents. Cancer Res 59:5964–5967
Zhan M, Zhao X, Wang H, Chen W, Xu S, Wang W, Shen H, Huang S, Wang J (2016) miR-145 sensitizes gallbladder cancer to cisplatin by regulating multidrug resistance associated protein 1. Tumour Biol 37:10553–10562
Zhang H, Patel A, Ma SL, Li XJ, Zhang YK, Yang PQ, Kathawala RJ, Wang YJ, Anreddy N, Fu LW, Chen ZS (2014) In vitro, in vivo and ex vivo characterization of ibrutinib: a potent inhibitor of the efflux function of the transporter MRP1. Br J Pharmacol 171:5845–5857
Zhang L, Zhao J, Liang C, Liu M, Xu F, Wang X (2017a) A novel biosensor based on intestinal 3D organoids for detecting the function of BCRP. Drug Deliv 24:1453–1459
Zhang Y, Mei Q, Liu Y, Li X, Brock MV, Chen M, Dong L, Shi L, Wang Y, Guo M, Nie J, Han W (2017b) The safety, efficacy, and treatment outcomes of a combination of low-dose decitabine treatment in patients with recurrent ovarian cancer. Oncoimmunology 6:e1323619
Zhang L, Liang C, Xu P, Liu M, Xu F, Wang X (2019a) Characterization of in vitro Mrp2 transporter model based on intestinal organoids. Regul Toxicol Pharmacol : RTP 108:104449
Zhang Y, Tao L, Fan LX, Huang K, Luo HM, Ge H, Wang X, Wang Q (2019b) Cx32 mediates cisplatin resistance in human ovarian cancer cells by affecting drug efflux transporter expression and activating the EGFRAkt pathway. Mol Med Rep 19:2287–2296
Zheng LS, Wang F, Li YH, Zhang X, Chen LM, Liang YJ, Dai CL, Yan YY, Tao LY, Mi YJ, Yang AK, To KK, Fu LW (2009) Vandetanib (Zactima, ZD6474) antagonizes ABCC1- and ABCG2-mediated multidrug resistance by inhibition of their transport function. PLoS One 4:e5172
Zhu Y, Yu F, Jiao Y, Feng J, Tang W, Yao H, Gong C, Chen J, Su F, Zhang Y, Song E (2011) Reduced miR-128 in breast tumor-initiating cells induces chemotherapeutic resistance via Bmi-1 and ABCC5. Clin Cancer Res: an official journal of the American Association for Cancer Research 17:7105–7115
Author information
Authors and Affiliations
Contributions
Study conception and design: ME, ARE, and HME. Acquisition of data: ME, ARE, HME, RFR, EAM, MA, HS, ASM, AE, AHA, and AHE. Writing and editing of the manuscript: ARE, HME, ME, AA, EAM, RFR, and AHE. Critically reviewed the article: ME, HME, and ARE. All authors have approved the final version of the manuscript.
Corresponding author
Ethics declarations
Ethics approval
Not applicable
Consent for publication
• The manuscript is not previously published in the same or very similar form in another journal.
• The manuscript is not under consideration in other journals.
Conflict of interest
The authors declare no competing interests.
Additional information
Responsible Editor: Lotfi Aleya
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Elfadadny, ., El-Husseiny, H.M., Abugomaa, A. et al. Role of multidrug resistance-associated proteins in cancer therapeutics: past, present, and future perspectives. Environ Sci Pollut Res 28, 49447–49466 (2021). https://doi.org/10.1007/s11356-021-15759-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11356-021-15759-5