
Abstract
Highly toxic phenol causes a threat to the ecosystem and human body. The development of bioremediation is a crucial issue in environmental protection. Herein, Rhodococcus biphenylivorans B403, which was isolated from the activated sludge of the sewage treatment plant, exhibited a good tolerance and removal efficiency to phenol. The degradation efficiency of phenol increased up to 62.27% in the oligotrophic inorganic medium (MM) containing 500-mg/L phenol at 18 h. R. biphenylivorans B403 cultured in the MM medium showed a higher phenol degradation efficiency than that in the eutrophic LB medium. On the basis of the transcriptomic and proteomic analysis, a total of 799 genes and 123 proteins showed significantly differential expression between two different culture conditions, especially involved in phenol degradation, carbon metabolism, and nitrogen metabolism. R. biphenylivorans B403 could alter the phenol degradation pathway by facing different culture conditions. During the phenol removal in the oligotrophic inorganic medium, muconate cycloisomerase, acetyl-CoA acyltransferase, and catechol 1,2-dioxygenase in the ortho-pathway for phenol degradation showed upregulation compared with those in the eutrophic organic medium. Our study provides novel insights into the possible pathway underlying the response of bacterium to environmental stress for phenol degradation.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Phenol has been extensively used in a variety of industrial application such as wood industry, petrochemical, and leather processing (Zhou et al. 2016). As a highly toxic aromatic pollutant, phenol is listed as the priority pollutant and causes health risk to humans (Duan et al. 2018; Mukherjee and De 2014). Short-term exposure to phenol causes skin diseases and eye burns. Chronic and long-term exposure to this pollutant causes fatigue; damage to the nervous, respiratory, and immune systems; and even cancer (Talaiekhozani et al. 2017; Villegas et al. 2016). To date, bioremediation of phenol attracts a great concern due to their advantages of high degradation efficiencies, low costs, low energy consumption, simple operations, and not creating secondary pollution. Thus, finding new candidate from natural environment for phenol removal is a key on the development of bioremediation.
In nature, bacteria respond to the environmental stress for survival in different environmental conditions. Simultaneously, the aromatic pollutants derived from natural and artificial activities can be degraded by many bacterial species (Gong et al. 2016; Paisio et al. 2012; Wang et al. 2015). Noticeably, Rhodococcus genus is one of the bacterial strains in the natural environment that can effectively degrade phenolic and its derivatives (Arif et al. 2013; Paisio et al. 2012; Zidkova et al. 2010). Further researches focus on the mechanism toward response of Rhodococcus genus to phenol. Szőköl et al. found that the phenol hydroxylase of Rhodococcus erythropolis, a key enzyme in the phenol degradation pathway, was significantly reduced in the transcriptomic analysis when succinic acid and phenol coexisted, while glucose and glycerol had no inhibitory effect on the activity of phenol hydroxylase (Szőköl et al. 2014). Phenol hydroxylase plays an important role in phenol degradation by Rhodococcus ruber (He et al. 2014). The gene cluster coding for enzymes of the phenol degradation in Rhodococcus erythropolis has been verified to increase phenol hydroxylase activity (Zídková et al. 2013). During the phenol catabolism, phenol is firstly decomposed into catechol, which is further cleaved through the ortho- and meta-pathways (Kim et al. 2018). Bacteria are able to degrade phenol through two independent metabolic systems: the ortho-pathway (Lin 2017) and meta-pathway (Basak et al. 2014). Roell et al. used 1-13C phenol labeling and showed that when Rhodococcus PD630 was grown in a medium containing a high concentration of phenol, it simultaneously induced catechol 1,2-dioxygenase and catechol 2,3-dioxygenase (Roell et al. 2019). Margesin et al. detected the activities of 1,2-dioxygenase and catechol 2,3-dioxygenase and showed that the degradation of phenol through the ortho- and meta-pathways occurred in the Rhodococcus NO20-3 strain (Margesin et al. 2005). The nutrition is also important for aromatic pollutant removal by Rhodococcus genus (Suhaila et al. 2013). However, relatively less attention on the phenol removal by Rhodococcus genus was focused in the presence of different culture conditions.
We isolated and identified a bacterium from activated sludge naming as Rhodococcus biphenylivorans B403, which demonstrated capacity to utilize phenol as sole carbon source. The aim of this study was to investigate the effect of different culture conditions on the phenol degradation efficiency and growth characteristics of R. biphenylivorans B403. Moreover, comparative transcriptomic and proteomic analyses were used to elucidate the possible metabolic pathway involving in phenol removal under different culture conditions based on the differentially expressed genes and proteins profiles.
Materials and methods
Bacterial strain and culture media
The bacterial strain R. biphenylivorans B403 (CCTCC NO: M2019087) was isolated and screened from the activated sludge of the sewage treatment plant in Xiaochang County, Hubei Province, China.
A Luria-Bertani bacterial culture medium (LB, L−1) included 5 g of yeast extract, 10 g of peptone, and 10 g of NaCl. An inorganic salt medium (MM, L−1) contained 0.2 g of NaCl, 1 g of NH4NO3, 0.2 g of MgSO4·7H2O, 0.5 g of KH2PO4, 0.5 g of K2HPO4, and a trace amount of FeSO4·7H2O. The pH value of culture medium was adjusted to 6.5–7.0 and then sterilization at 121°C for 30 min. The stock solution of phenol was prepared (5 g/L) and then was added into the cultures of R. biphenylivorans B403 at final concentration of 500 mg/L. All chemicals used in this study were of analytical grade.
The growth conditions of bacterial strain
R. biphenylivorans B403 was initially activated in LB medium at 28°C with shaking at 200 rpm for 48 h. The activated bacterial strain was inoculated into phenol containing LB and MM medium, respectively. The initial optical density at 600 nm was adjusted to 0.1. The LB and MM media without the addition of phenol were set as the control. The supernatants were harvested each 3 h. The growth was spectrophotometrically determined at 600 nm after centrifugation (UV-1000 spectrophotometer). The growth curves of R. biphenylivorans B403 were drawn based on the measurement of OD600 values.
Analysis of phenol degradation by high-performance liquid chromatography
The samples collected from bacterial culture at different time interval were centrifuged for 5 min at 12000 rpm. Fifty-fold dilution of the supernatant with double distilled water was used for phenol determination by high-performance liquid chromatography (HPLC, Agilent 1200 HPCL System, Agilent, Santa Clara, CA) equipped with an Agilent ZORBAX Eclipse XDB-C18 column (5 μm, 4.5 × 150 mm). The mobile phase was composed of 50% methanol:50% water (v/v). The flow rate was 0.5 mL/min and the injection volume was 10 μL. Phenol was determined at 270 nm at 25°C temperature. Phenol degradation (%) was calculated with the following equation: phenol degradation (%)=(C0-Ct)/C0×100, where C0 was the initial concentration of phenol and Ct was the concentration of phenol determined along the time. All experiments were performed in triplicate.
RNA extraction
R. biphenylivorans B403 was cultured in both LB and MM media which contain 500-mg/L phenol for 14 h, respectively. The bacterial cells were collected by centrifugation at 4°C and washing three times with phosphate buffered saline (PBS). The quick-freezing of the bacterial cells was carried out by using liquid nitrogen. The RNA of each sample was extracted using the Ribo-offTM rRNA depletion kit (Bacteria). Both two RNA samples with different treatment in LB and MM medium were prepared in triplicate.
Transcriptomic sequencing
The Qubit and Agilent 2100 systems were used to detect the concentration and integrity of the extracted total RNA of the samples, followed by the use of Ribo-Zero™ rRNA removal kit to remove the ribosomal RNA (rRNA) and the addition of fragmentation buffer for fragmentation. The fragmented mRNA samples were used as templates for the synthesis of the first cDNA strand and further synthesize the second cDNA strand. The cDNA library was constructed by PCR enrichment. The cDNA library was used for prokaryotic transcriptomic sequencing. The sequencing was performed using Illumina HiSeq 2500 system. L1, L2, and L3 were triplicate for bacterial sample harvested from LB medium containing 500 mg/L of phenol. W1, W2, and W3 were triplicate for bacterial sample harvested from MM medium containing 500 mg/L of phenol.
Differential expression analysis
The clean data was obtained from the raw data by quality control. Then the clean data was assembled into transcripts using Rockhopper with the reference genome (McClure et al. 2013). All sequence of the transcripts was extracted and further annotated based on NCBI non-redundant protein sequences (Nr) database. Thereafter, the analyses of the differentially expressed genes (DEGs) were performed by Gene Ontology (GO) enrichment analysis and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis with a fold change ≥ 2 and a false discovery rate (FDR) ≤ 0.01.
Protein preparation, digestion, and labeling
The protein obtained from the bacterial grown on the differential media supplemented with phenol was used for comparative proteomic analysis. The bacterial pellets were mixed with lysis buffer containing 100-mM Tris-HCl (pH 8.0) and 2.5% (w/v) SDS. The samples were ultrasonicated on ice for 10 min. The supernatants obtained after centrifugation at 12, 000 g for 15 min were further precipitated by acetone. Consequently, the precipitated protein was dissolved and incubated in 100-mM Tris-HCl (pH 8.0) containing 8-M urea and 10-mM dithiothreitol at 37 °C for 1 h. And then 40-mM iodoacetamide was added. The quantitative analysis of protein was performed by the Bradford method (Ontañon et al. 2018). The samples were diluted by 100-mM Tris-HCl (pH 8.0) until the urea concentration decreased to 2 M. Proteins were digested by trypsin at 37 °C overnight. The digestion was terminated by adding trifluoroacetic acid, and the pH was adjusted to 6.0. After centrifugation at 12, 000 g for 15 min, the desalination of supernatant was performed via Sep-Pak C18. The peptide digests were concentrated and over-dried before labeling. Tandem Mass Tag (TMT) labeling was conducted using TMT 10plexTM Isobaric Label Reagent Set (Thermo Scientific, USA) according to the instructions of the manufacturer (McAlister et al. 2012). High pH reverse phase fractionation was finally carried out. Each sample was performed in triplicates.
MS analysis
The LC-MS/MS analysis was carried out on Thermo Scientific Q ExactiveTM HF-X mass spectrometer. The peptides were trapped and separated through trapping column and analytical column (75 μm × 250 mm, 3 μm particle size, 100 Å pore size), respectively. The mobile phase A was composed of 0.1% formic acid in water, and the mobile phase B was composed of 0.1% formic acid in 80% acetonitrile. The flow rate was 300 nL/min. MS spectra were obtained across the scan of m/z ranging from 350 to 1800 in a resolution of 60,000 using an injection time of 50 ms. The twenty most abundant precursors were selected for fragmentation in a resolution of 30,000 using an injection time of 100 ms.
Bioinformatics analysis of proteomic analysis
Protein identification was carried out using MaxQuant (Version 1.6.6.0) together with its integrated search engine, Andromeda (Tyanova et al. 2016), which further searched against Uniprot Rhodococcus biphenylivorans reference proteomic database. The main search parameters were set as oxidation (M) and acetyl (Protein N-term) as the variable modifications, carbamidomethyl (C) as the fixed modification, and trypsin as the digestion. The identified proteins were analyzed with a FDR ≤ 0.01. The differential expressed proteins were screened with a threshold of p ≤ 0.05 and fold change ≥ 1.5 or ≤ 0.667. Based on the identified differential expressed proteins, the annotation, classification, and enrichment analysis were performed using GO, KEGG, and COG (Clusters of Orthologous Groups of proteins).
Real-time PCR analysis
The samples of bacterial cultures were collected on hour 14. Based on the results of transcriptomic and proteomic analyses, the key gene expression in phenol degradation and carbon metabolism was validated via quantitative real-time PCR (qPCR). The primer sequences used in this study were listed in Table S1. The program of qPCR was carried out using a Bio-Rad CFX96 system associate with TB Green™ Premix Ex Taq™ (Takara). The parameters were set as follows: 95 °C for 30 s, followed by 40 cycles at 95 °C for 5 s, and 60 °C for 30 s. The melting curve procedure was detected followed by 95 °C for 10 s, 65 °C for 5 s, and 95 °C for 5 s. The relative expression fold in selected genes was calculated using the 2−ΔΔCt method with 16S rRNA normalized Ct value.
Results and discussions
Growth characteristics and phenol degradation of R. biphenylivorans B403
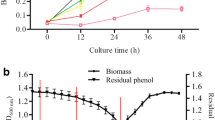
The growth characteristics of R. biphenylivorans B403 in the MM medium and LB medium were assessed in this study, as well as the degradation of phenol. As shown in Fig. 1a, as expected, R. biphenylivorans B403 showed much higher growth in eutrophic medium (LB) than oligotrophic medium (MM). However, R. biphenylivorans B403 which incubated in MM medium still entered the exponential phase from 6 to 27 h and further reached the stationary phase after 36 h. This result implied that it might be used as the sole carbon source by R. biphenylivorans B403, even in the oligotrophic environment. The efficiency of R. biphenylivorans B403 on phenol degradation was shown in Fig. 1b. R. biphenylivorans B403 which is incubated in LB medium showed faster degradation efficiency of phenol than that of the culture in MM medium. The phenol degradation efficiency increased up to 85.87% in the LB medium and up to 35.26% in the MM medium at 15 h, respectively. After 18 h of treatment, the phenol showed almost complete elimination (99.67%) in the LB medium and further degrades up to 62.27% in the MM medium. The biomass of R. biphenylivorans B403 in LB medium showed increasing 13.2-fold higher than that of the culture in the MM medium. However, compared with the treatment in the MM medium, only 1.6-fold increase in phenol degradation by R. biphenylivorans B403 in the LB medium was observed. Moreover, R. biphenylivorans B403 which incubated in MM medium exhibited complete removal of 500 mg/L phenol on 33 h.

Time-course analysis of a growth curves of R. biphenylivorans B403 and b degradation curves of phenol under different culture conditions
Phenol is a well-known hazardous aromatic pollutant for human and environment, which is widely distributed in man-made ecosystems (Zhou et al. 2016). To date, a variety of Gram-positive bacteria are able to aerobically degrade aromatic pollutants (Ye et al. 2020). In our study, R. biphenylivorans B403, which is screened from the activated sludge of the sewage treatment plant, has a potential on degradation of aromatic pollutant. R. biphenylivorans play an important role in environmental bioremediation (Su et al. 2015b). Previous reports also demonstrate that R. biphenylivorans is able to degrade polychlorinated biphenyls (Su et al. 2015a; Ye et al. 2020). Therefore, these results indicated that R. biphenylivorans B403 not only could utilize phenol as sole carbon source but also promote phenol removal in the medium.
Impact of different nutrition on the gene expression profiles of R. biphenylivorans B403 during the phenol degradation
To investigate the possible molecular mechanism toward phenol removal by R. biphenylivorans B403, comparative transcriptomic analysis was performed. The good correlation among the three biological replicates in both two treatment groups was observed (Figure S1). Compared with the bacterium growth in the eutrophic medium (LB), a total of 799 genes are differentially expressed in R. biphenylivorans B403 on oligotrophic medium (MM). Accordingly, 409 genes and 390 genes were upregulated and downregulated in comparison with the sample from the eutrophic medium (LB) (Fig. 2).

Heatmap of the normalized fold change of all differentially expressed gene clusters between two groups with different nutrition. The x-axis represents the sample name and its clustering result, and the y-axis represents the differential gene and its clustering result
The functional classification of these DEGs was performed by GO analysis. These DEGs were successfully enriched with GO terms and further classified into three major GO categories, including molecular function (red), cell component (blue), and biological process (green). The DEGs were divided into 32 subcategories (Fig. 3). Among these 409 upregulated DEGs, genes related to the catalytic activity comprised the highest percentage (176 genes, 43%), whereas genes related to metabolic processes (171 genes) comprised 41.8% of all the upregulated genes. Thereafter, the upregulated DEGs were enriched in cellular processes (103 genes, 25.2%), binding (129 genes, 31.5%), single-organism processes (113 genes, 27.6%), cellular processes (103 genes, 25.2%), membranes (91 genes, 22.3%), and membrane parts (87 genes, 21.3%), respectively. Among these 390 downregulated DEGs, the most important proportions were related to metabolic processes and catalytic activity (159 genes, 40.8%), followed by binding (115 genes, 29.5%), single-organism processes (104 genes, 26.7%), cell process (98 genes, 25.1%), membranes (65 genes, 16.7%), and membrane parts (65 genes, 16.7%).

Statistics of GO differential classification between two groups with different nutrition. a Upregulated and b downregulated genes in the MM medium. The x-axis represents the number of genes, and the y-axis represents the classification of the gene function
A total of 260 DEGs were annotated in the KEGG database (Fig. 4), and these DEGs were classified into 5 categories, including cellular processes, environmental information processing, genetic information processing, metabolism, and organismal systems. Most of the annotated DEGs were involved in “metabolism,” implying that the lack of nutrition significantly impacts the gene expression profile of R. biphenylivorans B403. The “metabolism” was closely related to phenol catabolism, such as amino acid metabolism, energy metabolism, carbohydrate metabolism, xenobiotics biodegradation and metabolism, lipid metabolism, and nucleotide metabolism. The phenol catabolic pathway of R. biphenylivorans B403 might be activated in the MM medium more efficiently than that in the LB medium.

KEGG classification statistics of differentially expressed genes between two groups with different nutrition. a Upregulated and b downregulated genes in the MM medium. The y-axis represents the name of the KEGG metabolic pathway, and the x-axis represents the number of genes annotated under the pathway
Effect of different nutrition on phenol degradation pathways of R. biphenylivorans B403
To elucidate the possible mechanism on the effect of changing nutrition for metabolic pathways, especially phenol degradation, carbon metabolism, and nitrogen metabolism, DEGs related to these three pathways are listed in Table S2. The phenol degradation pathway (ko00362) shown in the KEGG database belongs to a branch of the sodium benzoate degradation pathway (Yergeau et al. 2018). In our study, the phenol degradation pathway was obviously impacted by changing nutrition from organic components to inorganic components. Phenol hydroxylase, which involved in the first step of phenol biodegradation, did not show significantly differential expression between two different culture conditions. The intermediate product of phenol degradation, catechol, is able to catabolize to pyruvate, acetyl-CoA, and succinyl-CoA through several DEGs, including dmpB (catechol 2,3-dioxygenase), mhpE (4-hydroxy 2-oxovalerate aldolase), catB (muconate cycloisomerase), fadA (acetyl-CoA acyltransferase), and pcaL (3-oxoadipate enol-lactonase) in the meta- and ortho-pathways (Fig. 5). The normalized fold change of dmpB and mhpE in the meta-pathway and the fadA in the ortho-pathway for phenol degradation were downregulated by 1.23-, 1.21-, and 1.19-fold compared to those in the eutrophic medium (LB), respectively. However, in the oligotrophic environment (MM), the normalized fold change of catB and pcaL in the ortho-pathway for phenol degradation were upregulated by 1.01- and 1.36-fold compared to those in the eutrophic medium (LB), respectively.

Annotation of phenol degradation pathway. Enzymes marked with red, blue, and green arrows in the figure were related to upregulated differential genes, up- and downregulated genes, and downregulated differential genes, respectively
With the addition of inorganic sources, the genes involved in the ortho-pathway for phenol degradation were downregulated. However, the genes involved in the meta-pathway for phenol degradation were upregulated in the presence of inorganic sources. The muconate cycloisomerase and 3-oxoadipate enol-lactonase in the ortho-pathway for phenol degradation were highly expressed, implying that R. biphenylivorans B403 degraded phenol mainly through the ortho-pathway. The needs of carbon source for growth were enhanced when only inorganic sources were supplied to R. biphenylivorans B403. This condition may promote the catabolism of phenol by R. biphenylivorans B403. Thus, the change of nutrition from organic to inorganic source not only benefits to phenol degradation but also alters the degradation pathway of phenol by R. biphenylivorans B403.
Effect of different nutrition on carbon metabolism of R. biphenylivorans B403
The sufficient carbon sources play a key role in bioremediation because the carbon metabolism provides energy for cell growth and pollutant removal (Song et al. 2009). In our study, 8 DEGs related to carbon metabolism of R. biphenylivorans B403 exhibited downregulation in oligotrophic-treated sample (MM) compared with the eutrophic-treated sample (LB), including fdoG (formate dehydrogenase major subunit), gcvP (glycine dehydrogenase), serB (phosphoserine phosphatase), gapA (glyceraldehyde 3-phosphate dehydrogenase), aceF (pyruvate dehydrogenase), mmsA (semialdehyde dehydrogenase), acnA (aconitate hydratase), and fumA (fumarate hydratase). However, cysK (cysteine synthase), maeA (malate dehydrogenase), aceA (isocitrate lyase), atoB (acetyl-CoA C-acetyltransferase), and fadJ (3-hydroxyacyl-CoA dehydrogenase) were upregulated by 1.58-, 1.47-, 2.00-, 1.16-, and 1.09-fold, respectively.
Bacterial species are widely distributed to contaminated sites and enable use of the aromatic pollutants as growth substrates (Djokic et al. 2013; Gu et al. 2018). The carbon metabolism plays a central role in microbial activity. The aceA and atoB are able to change the carbon source flow in the metabolic pathway, which was beneficial to the accumulation of carbon sources (Bringaud et al. 2006). When the bacterial strain suffered phenol in the oligotrophic environment, these upregulated genes might be beneficial for the use of phenol by R. biphenylivorans B403. Among the R. biphenylivorans B403 cultured in the LB medium, most of genes involved in carbon metabolism were upregulated. In nature, bacteria can adapt their catabolic metabolism in response to their nutritional situation. The carbon catabolite repression may be enhanced by changing the culture medium from oligotrophic (MM) to eutrophic condition (LB), which may further influence gene expression related to carbon metabolism. Generally, the induction of stress by microorganisms occurs when they are grown in stress condition or primary food-deficient condition. Previous study demonstrate that the induction of stress obviously improved the removal of aromatic pollutants by microorganisms (Liu et al. 2020). In our study, not only the nutrition-deficient condition but also the phenol stress may enforce R. biphenylivorans B403 to utilize phenol as carbon source, which may further improve the phenol degradation capacity of R. biphenylivorans B403.
Effect of the nutrition on nitrogen metabolism of R. biphenylivorans B403
Generally, phenol significantly affects the metabolism of microbial cells via the interaction with cell membranes and intracellular macromolecules (Putrinš et al. 2010). The nitrogen metabolism–related genes in R. biphenylivorans B403 were also regulated by nutrition. The nirD (nitrite reductase) and gltB (glutamate synthase) in oligotrophic-treated sample (MM) increased by 2.5- and 1.09-fold, respectively. On the contrary, gdhA (glutamate dehydrogenase), an enzyme central to glutamate metabolism, was downregulated in the oligotrophic environment (Log2 fold change = −5.70). Previous literature shows that increasing of phenol concentration leads to suppression of nitrite reductase activity (Xia et al. 2019). However, nirD in R. biphenylivorans B403 could be upregulated in the presence of phenol. Besides, glutamate synthase plays an important role in central nitrogen metabolism (Guillamón et al. 2001), which is closely related to the synthesis of glutathione. Glutamic acid is also the main amino acid for nitrogen metabolism. Upregulation of gdhA in the eutrophic environment benefits to the oxidative deamination of glutamic acid. Therefore, although the partial nitrogen metabolism of R. biphenylivorans B403 was inhibited without sufficient organic sources, the phenol degradation capacity of R. biphenylivorans B403 could be improved by facing harsh environment.
Highly expressed differential genes of R. biphenylivorans B403 in the presence of organic or inorganic sources
Thirty-two DEGs higher than 16-fold differences were classified as highly expressed differential genes (HDEGs) in our study. As shown in Table S3, a total of 32 HDEGs were observed in oligotrophic-treated sample (MM) compared with the eutrophic-treated sample (LB). Twenty-seven of 32 genes were annotated, 17 genes were upregulated, and 10 genes were downregulated in the MM medium compared with the expression in LB medium. The cluster heatmap analysis for HDEGs was carried out (Fig. 6). The expression of sulfonate transport–related genes, including ssuA1 (sulfonate transport system substrate-binding protein), ssuC1 (sulfonate transport system permease protein), ssuB (sulfonate transport system ATP-binding protein), ssuA2 (sulfonate transport system substrate–binding protein), and ssuC2 (sulfonate transport system permease protein) showed significant increase (8.49-, 8.23-, 5.93-, 4.29-, and 4.00-fold, respectively) in the MM medium. The expression of redox reactions–related genes, including oxidoreductase (CEJ39_RS03315), FMN coenzyme (CEJ39_RS03685), oxidoreductase (CEJ39_RS03245), and monooxygenase (CEJ39_RS11875) exhibited significant increase (5.97-, 5.81-, 4.17-, and 4.08-fold, respectively) in the MM medium. Biotin is a coenzyme for many hydroxylases, acts as a CO2 carrier in the hydroxylase reaction, and is an indispensable substrate for lipid and protein metabolisms (Yao et al. 2018). Three biotin synthase genes, including bioB (biotin synthase), bioD (dethiobiotin synthetase), and bioF (8-amino-7-oxononanoate synthase), were upregulated by 6.86-, 4.55-, and 4.33-fold, respectively. The entC (isochorismate synthase), entB (bifunctional isochorismate lyase), and entA (2,3-dihydro-2,3-dihydroxybenzoate dehydrogenase) were upregulated by 4.60-, 4.37-, and 4.29-fold, respectively. In addition, the genes related to amino acid metabolism were upregulated in MM treated R. biphenylivorans B403, including aspartic and ornithine metabolism. The expression of racD and pvdA increased by 4.82-, 4.60-fold, respectively, in MM treated R. biphenylivorans B403.

Heatmap of the normalized fold change of 32 highly differentially expressed gene clusters between two groups with different nutrition. The red and blue colors in cells reflect high and low fold change, respectively
The sulfonate transport–related genes upregulated in our study belong to the ATP-binding cassette (ABC) transporter. Generally, ABC transporter is widely distributed from bacteria to fungi (Cason et al. 2012; González-Guerrero et al. 2010). ABC transporter could be induced in the presence of a variety of environmental stress, especially pollutants. The activated sulfonate transport activity caused by oligotrophic environment might accelerate the exchange between intracellular and extracellular environment, which further improved the phenol removal by R. biphenylivorans B403. Noticeably, the expression of FMN coenzyme–related gene was significantly induced by oligotrophic environment. The rate of oxidative deamination of glutamic acid in R. biphenylivorans B403 might also be accelerated. The entC, entB, and entA generally involves in the synthesis of iron carriers. When the iron carriers contacted the iron-carrier receptor protein on the cell membrane, the iron atoms were released directly into the cytoplasm or the entire iron-carrier complex entered the cell through the ABC transporter (Braun 2005). The environmental stress led to activation of the resistance of R. biphenylivorans B403, which may contribute to rebuild the cellular homeostasis and improve phenol removal by R. biphenylivorans B403. The racD serves as a carrier for K+ and Mg2+ ions to transport electrolytes while reducing oxygen consumption (Ito et al. 2008). The pvdA has a detoxifying effect on the ammonia accumulated in the cells (Alfakih 2014). The upregulation of racD and pvdA in R. biphenylivorans B403 helps the bacterial cells to adapt to the environment when phenol was added. In all, these results indicated that the oligotrophic environment only that contains inorganic sources could obviously evoke the adaptive response of R. biphenylivorans B403 to environmental stress and improve the removal of aromatic pollutant.
Correlation between differentially expressed genes, bacterial strain growth, and phenol degradation
In our study, the differentially expressed genes showed obvious difference between two nutrient systems, especially related to phenol degradation, carbon metabolism, and nitrogen metabolism. Meanwhile, the growth and the phenol removal efficiency also exhibited significant difference. The phenol degradation was greatly improved in the LB medium because the biomass of R. biphenylivorans B403 in the LB medium was significantly higher than that in the MM medium. However, the oligotrophic environment preferred induction of stress-related genes in R. biphenylivorans B403. The R. biphenylivorans B403 cultured in the MM medium required almost double the time to completely degrade 500 mg/L of phenol compared with the R. biphenylivorans B403 cultured in the LB medium. The utilization efficiency of the organic source by the bacterial cells was high, resulting in a significant increase in biomass of R. biphenylivorans B403 in the eutrophic medium (LB). However, the unit biomass of R. biphenylivorans B403 cultured in the oligotrophic environment (MM) showed a higher phenol degradation efficiency than that in the LB medium (Figure S2). The degradation efficiency of phenol per unit biomass in the MM medium within 18 h was 63.12%, which was higher than that of the culture in LB medium. Stronger phenol-induced stress enhanced the ability of single cell to degrade phenol. Due to the higher biomass of the R. biphenylivorans B403 in the organic source system, the bacterial community response promoted the higher comprehensive degradation efficiency of phenol by R. biphenylivorans B403.
Verification of the proposed mechanism via proteomic analysis
To confirm whether the change of trophic condition could alter the phenol degradation pathway by R. biphenylivorans B403, a proteomic analysis was carried out. According to the proteomic analysis, a total of 123 differentially expressed proteins were identified. Among them, 61 of the 123 proteins were significantly upregulated in the oligotrophic MM environment (Log2 fold change ≥ 1) (Table S4), while 62 of the 123 proteins were significantly upregulated in the eutrophic LB environment (Log2 fold change ≤ −1) (Table S5). On the basis of GO enrichment analysis, the change of nutrient condition significantly influenced the cellular process, metabolic process, cell, cell part, catalytic activity, and binding in the three major GO categories (Fig. 7). As expected, more proteins related to energy production and conversion were activated in LB medium (A0A2Z4VFX8, A0A2Z4VEW6, A0A2Z4VET2, A0A2Z4VDS4, A0A2Z4VP80, A0A2Z4VRF1, and A0A2Z4VGD1) than that in MM medium (A0A2Z4VL42, A0A2Z4VI69, and A0A2Z4VH72). The genes involved in phenol degradation are also affected by the existence of available substrates (Szőköl et al. 2014). As mentioned above in transcriptomic analysis, the genes in both carbon metabolism and phenol degradation showed significant differential expression under two different culture media. When the carbon catabolite repression was alleviated without the addition of rich nutrient, the ortho-pathway for phenol degradation in R. biphenylivorans B403 may be activated for supporting growth. Consistent with the result of energy production and conversion, 7 of the 11 differentially expressed proteins related to amino acid transport and metabolism were activated in eutrophic environment (A0A2Z4VIE7, A0A2Z4VM02, A0A2Z4VE21, A0A2Z4VNQ3, A0A2Z4VL81, A0A2Z4VCE0, and A0A2Z4VN11). Noticeably, KEGG enrichment analysis demonstrated that the change of nutrition from organic to inorganic source alters the phenol degradation by R. biphenylivorans B403 from meta- to ortho-pathway (Table 1). Catechol 1,2-dioxygenase (catA), which is involved in ortho-pathway for phenol degradation, was upregulated in MM medium. On the contrary, 2-keto-4-pentenoate hydratase, 4-oxalocrotonate decarboxylase, acetaldehyde dehydrogenase, 4-hydroxy-2-oxovalerate aldolase, and 5-car`ymethyl-2-hydroxymuconate semialdehyde dehydrogenase were activated in the presence of eutrophic condition, which involved in meta-pathway for phenol degradation. Although a slight difference was observed between transcriptomic and proteomic analysis, the results still indicated that the change of nutrient condition could switch the metabolic pattern of phenol by R. biphenylivorans B403. Additionally, the upregulation of aceA, catB, and catA in MM medium was validated compared to those in LB medium based on the qPCR analysis (Figure S3), indicating that the change of growth condition could regulate phenol degradation by R. biphenylivorans B403.

GO enrichment analysis of the differentially expressed proteins in MM treatment compared with those in LB treatment
Conclusions
In summary, our study found that R. biphenylivorans B403, a typical Gram-positive bacterium, is able to use phenol as sole carbon source even in the oligotrophic environment. Based on comparative transcriptomic and proteomic analysis, the gene and protein expression profiles of R. biphenylivorans B403 exhibited significant difference between two different mediums, especially related to phenol degradation, carbon metabolism, and nitrogen metabolism. Compared with the eutrophic environment for phenol degradation, R. biphenylivorans B403 could alter the phenol degradation pathway by facing the oligotrophic environment. The induction of stress simultaneously caused by deficient nutrition and xenobiotic pollutant may be beneficial to phenol removal. Thus, a better and deeper understanding of aromatic pollutant removal by a bacterium potentially provides an alternative for bioremediation strategy.
References
Alfakih AA (2014) Overview on the fungal metabolites involved in mycopathy. Open Journal of Medical Microbiology 4:38–63
Arif NM, Ahmad SA, Syed MA, Shukor MY (2013) Isolation and characterization of a phenol-degrading Rhodococcus sp. strain AQ5NOL 2 KCTC 11961BP. J. Basic Microbiol. 53:9–19
Basak SP, Sarkar P, Pal P (2014) Isolation and characterization of phenol utilizing bacteria from industrial effluent-contaminated soil and kinetic evaluation of their biodegradation potential. J. Environ. Sci. Heal. A 49:67–77
Braun V (2005) Bacterial iron transport related to virulence. Contrib. Microbiol. 12:210–233
Bringaud F, Riviere L, Coustou V (2006) Energy metabolism of trypanosomatids: adaptation to available carbon sources. Mol. Biochem. Parasitol. 149:1–9
Cason ED, Piater LA, Ev H (2012) Reduction of U(VI) by the deep subsurface bacterium, Thermus scotoductus SA-01, and the involvement of the ABC transporter protein. Chemosphere 86:572–577
Djokic L, Narancic T, Biocanin M, Saljnikov E, Casey E, Vasiljevic B, Nikodinovic-Runic J (2013) Phenol removal from four different natural soil types by Bacillus sp. PS11. Appl. Soil Ecol. 70:1–8
Duan W, Meng F, Cui H, Lin Y, Wang G, Wu J (2018) Ecotoxicity of phenol and cresols to aquatic organisms: a review. Ecotoxicol. Environ. Saf. 157:441–456
Gong B, Wu P, Huang Z, Li Y, Dang Z, Ruan B, Kang C, Zhu N (2016) Enhanced degradation of phenol by Sphingomonas sp. GY2B with resistance towards suboptimal environment through adsorption on kaolinite. Chemosphere 148:388–394
González-Guerrero M, Benabdellah K, Valderas A, Azcón-Aguilar C, Ferrol N (2010) GintABC1 encodes a putative ABC transporter of the MRP subfamily induced by Cu, Cd, and oxidative stress in Glomus intraradices. Mycorrhiza 20:137–146
Gu QH, Wu QP, Zhang JM, Guo WP, Ding Y, Wang J, Wu HQ, Sun M, Hou LF, Wei XH, Zhang YX (2018) Isolation and transcriptome analysis of phenol-degrading bacterium from carbon-sand filters in a full-scale drinking water treatment plant. Front. Microbiol. 9:16
Guillamón JM, van Riel NAW, Giuseppin MLF, Verrips CT (2001) The glutamate synthase (GOGAT) of Saccharomyces cerevisiae plays an important role in central nitrogen metabolism. FEMS Yeast Res. 1:169–175
He Z, Niu C, Lu Z (2014) Individual or synchronous biodegradation of di-n-butyl phthalate and phenol by Rhodococcus ruber strain DP-2. J. Hazard. Mater. 273:104–109
Ito T, Hemmi H, Kataoka K, Mukai Y, Yoshimura T (2008) A novel zinc-dependent D-serine dehydratase from Saccharomyces cerevisiae. Biochem. J 409:399–406
Kim D, Choi KY, Yoo M, Zylstra GJ, Kim E (2018) Biotechnological potential of Rhodococcus biodegradative pathways. J. Microbiol. Biotechnol. 28:1037–1051
Lin J (2017) Stress responses of Acinetobacter strain Y during phenol degradation. Arch. Microbiol. 199:365–375
Liu J, Liu F, Ding C, Ma F, Yu H, Shi Y, Zhang X (2020) Response of Trametes hirsuta to hexavalent chromium promotes laccase-mediated decolorization of reactive black 5. Ecotoxicol. Environ. Saf. 205:111134
Margesin R, Fonteyne P, Redl B (2005) Low-temperature biodegradation of high amounts of phenol by Rhodococcus spp. and basidiomycetous yeasts. Res. Microbiol. 156:68–75
McAlister GC, Huttlin EL, Haas W, Ting L, Jedrychowski MP, Rogers JC, Kuhn K, Pike I, Grothe RA, Blethrow JD, Gygi SP (2012) Increasing the multiplexing capacity of TMTs using reporter ion isotopologues with isobaric masses. Anal. Chem. 84:7469–7478
McClure R, Balasubramanian D, Sun Y, Bobrovskyy M, Sumby P, Genco CA, Vanderpool CK, Tjaden B (2013) Computational analysis of bacterial RNA-Seq data. Nucleic Acids Res. 41:e140–e140
Mukherjee R, De S (2014) Adsorptive removal of phenolic compounds using cellulose acetate phthalate–alumina nanoparticle mixed matrix membrane. J. Hazard. Mater. 265:8–19
Ontañon OM, Landi C, Carleo A, Gagliardi A, Bianchi L, González PS, Agostini E, Bini L (2018) What makes A. guillouiae SFC 500-1A able to co-metabolize phenol and Cr(VI)? A proteomic approach. J. Hazard. Mater. 354:215–224
Paisio CE, Talano MA, González PS, Busto VD, Talou JR, Agostini E (2012) Isolation and characterization of a Rhodococcus strain with phenol-degrading ability and its potential use for tannery effluent biotreatment. Environ. Sci. Pollut. R. 19:3430–3439
Putrinš M, Ilves H, Lilje L, Kivisaar M, Hõrak R (2010) The impact of ColRS two-component system and TtgABC efflux pump on phenol tolerance of Pseudomonas putida becomes evident only in growing bacteria. BMC Microbiol. 10:110
Roell GW, Carr RR, Campbell TP, Shang Z, Henson WR, Czajka JJ, Martin HG, Zhang F, Foston M, Dantas G (2019) A concerted systems biology analysis of phenol metabolism in Rhodococcus opacus PD630. Metab. Eng. 55:120–130
Song H, Liu Y, Xu W, Zeng G, Aibibu N, Xu L, Chen B (2009) Simultaneous Cr(VI) reduction and phenol degradation in pure cultures of Pseudomonas aeruginosa CCTCC AB91095. Bioresour. Technol. 100:5079–5084
Su X, Liu Y, Hashmi MZ, Hu J, Ding L, Wu M, Shen C (2015a) Rhodococcus biphenylivorans sp. nov., a polychlorinated biphenyl-degrading bacterium. Antonie Van Leeuwenhoek 107:55–63
Su X, Sun F, Wang Y, Hashmi MZ, Guo L, Ding L, Shen C (2015b) Identification, characterization and molecular analysis of the viable but nonculturable Rhodococcus biphenylivorans. Sci. Rep. 5:18590
Suhaila YN, Rosfarizan M, Ahmad SA, Abdul Latif I, Ariff AB (2013) Nutrients and culture conditions requirements for the degradation of phenol by Rhodococcus UKMP-5M. J. Environ. Biol. 34:635–643
Szőköl J, Rucká L, Šimčíková M, Halada P, Ne?Vera J, Pátek M (2014): Induction and carbon catabolite repression of phenol degradation genes in Rhodococcus erythropolis and Rhodococcus jostii. Appl. Microbiol. Biotechnol. 98, 8267-8279
Talaiekhozani A, Talaei MR, Rezania S (2017) An overview on production and application of Ferrate (VI) for chemical oxidation, coagulation and disinfection of water and wastewater. J. Environ. Chem. Eng. 5:1828–1842
Tyanova S, Temu T, Cox J (2016) The MaxQuant computational platform for mass spectrometry-based shotgun proteomics. Nat. Protoc. 11:2301–2319
Villegas LGC, Mashhadi N, Chen M, Mukherjee D, Taylor KE, Biswas N (2016) A short review of techniques for phenol removal from wastewater. Curr. Pollut. Rep. 2:157–167
Wang Q, Li Y, Li J, Wang Y, Wang C, Wang P (2015) Experimental and kinetic study on the cometabolic biodegradation of phenol and 4-chlorophenol by psychrotrophic Pseudomonas putida LY1. Environ. Sci. Pollut. R. 22:565–573
Xia T, Xie M, Chen D, Xiao Z (2019) Impact of phenol on the performance, kinetics, microbial communities and functional genes of an autotrophic denitrification system. Bioprocess Biosystems Eng. 42:1105–1114
Yao C, Chou J, Wang T, Zhao H, Zhang B (2018) Pantothenic acid, vitamin c, and biotin play important roles in the growth of Lactobacillus helveticus. Front. Microbiol. 9
Ye Z, Li H, Jia Y, Fan J, Wan J, Guo L, Su X, Zhang Y, Wu W-M, Shen C (2020) Supplementing resuscitation-promoting factor (Rpf) enhanced biodegradation of polychlorinated biphenyls (PCBs) by Rhodococcus biphenylivorans strain TG9(T). Environ. Pollut. 263:114488
Yergeau E, Tremblay J, Joly S, Labrecque M, Maynard C, Pitre FE, Starnaud M, Greer CW (2018) Soil contamination alters the willow root and rhizosphere metatranscriptome and the root–rhizosphere interactome. ISME J. 12:869–884
Zhou W, Guo W, Zhou H, Chen X (2016) Phenol degradation by Sulfobacillus acidophilus TPY via the meta-pathway. Microbiol. Res. 190:37–45
Zidkova L, Otevrel M, Wimmerova L, Patek M (2010) Continuous phenol degradation in wastewater by Rhodococcus jostii rha1; model batch test and continuous verification in a biofilter. J. Biotechnol. 150:215
Zídková L, Szőköl J, Rucká L, Pátek M, Nešvera J (2013) Biodegradation of phenol using recombinant plasmid-carrying Rhodococcus erythropolis strains. Int. Biodeterior. Biodegrad. 84:179–184
Acknowledgements
This work was supported by China National Key R&D Program (2020YFA0908400; 2019YFA090500), the Science and Technology Innovation Program of Hubei Province (2020BBA056; 2019ABA117), the Central Committee Guides Local Science and Technology Development Projects (2018ZYYD034), and the Wuhan Science and Technology Plan (2019020701011496).
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Funding
China National Key R&D Program (2020YFA0908400)
China National Key R&D Program (2019YFA090500)
Science and Technology Innovation Program of Hubei Province (2020BBA056)
Science and Technology Innovation Program of Hubei Province (2019ABA117)
Central Committee Guides Local Science and Technology Development Projects (2018ZYYD034)
Wuhan Science and Technology Plan (2019020701011496)
Author information
Authors and Affiliations
Contributions
Conceptualization, Zhengbing Jiang and Huiting Song; data curation, Xiaohang Xie and Jiashu Liu; formal analysis, Xiaohang Xie; funding acquisition, Zhengbing Jiang; investigation, Xiaohang Xie and Hong Pan; methodology, Xiaohang Xie, Huanan Li, and Meng Ye; project administration, Zhengbing Jiang and Huiting Song; resources, Huiting Song; software, Jiashu Liu and Jingwei Zhu; supervision, Huiting Song; validation, Huanan Li; writing—original draft, Jiashu Liu; writing—review and editing, Jiashu Liu.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable
Consent for publication
Not applicable
Competing interests
The authors declare no competing interests.
Additional information
Responsible Editor: Diane Purchase
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
ESM 1
(DOCX 513 kb)
Rights and permissions
About this article

Cite this article
Xie, X., Liu, J., Jiang, Z. et al. The conversion of the nutrient condition alter the phenol degradation pathway by Rhodococcus biphenylivorans B403: A comparative transcriptomic and proteomic approach. Environ Sci Pollut Res 28, 56152–56163 (2021). https://doi.org/10.1007/s11356-021-14374-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11356-021-14374-8