
Abstract
Arsenic, an omnipresent environmental contaminant, is regarded as a potent hepatotoxin. Nigella sativa oil (NSO) consumption has been shown to improve hepatic functions in various in vivo models of acute hepatic injury. The present study evaluates the protective efficacy of NSO against sodium arsenate (As)–induced deleterious alterations in the liver. Male Wistar rats were divided into four groups, namely, control, As, NSO, and AsNSO. After pre-treating rats in AsNSO and NSO groups with NSO (2 mL/kg bwt, orally) for 14 days, NSO treatment was further extended for 30 days, with and without As treatment (5 mg/kg bwt, orally), respectively. As induced an upsurge in serum ALT and AST activities indicating liver injury, as also confirmed by the histopathological findings. As caused significant alterations in the activities of membrane marker enzymes and carbohydrate metabolic enzymes, and in the vital components of antioxidant defense system. Marked DNA damage and hepatic arsenic accumulation were also observed in As-treated rats. Oral NSO administration ameliorated these deleterious alterations and improved overall hepatic antioxidant and metabolic status in As-treated rats. Prevention of oxidative damage could be the underlying mechanism of NSO-mediated protective effects. The results suggest that NSO could be a useful dietary supplement in the management of arsenic hepatotoxicity.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Inorganic arsenic, as arsenate (AsV) or arsenite (AsIII), is one of the most potent hazardous substances that exert multiple toxic effects on living organisms. Arsenic toxicity has turned into a global epidemic and is a major concern for public health. Drinking water contaminated with arsenic is the primary source of its exposure, where arsenate is the predominant form (Chung et al. 2014). Arsenic interferes with multiple enzyme catalyzed reactions and affects the entire organ systems in humans and other animals (Adil et al. 2015). Autopsy studies of arsenic-exposed animals have shown that among soft tissues, the liver is the largest repository followed by the kidney (Flora et al. 2008). Various studies have correlated acute and chronic arsenic exposure to the genesis of numerous pathologies including diabetes (Sung et al. 2015), vascular diseases (Prozialeck et al. 2008), blackfoot disease (Tseng 2005), and deleterious effects on the kidney, liver, and nervous and cardiovascular systems (Singh et al. 2011). Analysis at the cellular and molecular levels has revealed that arsenic escalates the formation of free radicals and reactive oxygen species (ROS) that target the cellular biomolecules and cause DNA damage, lipid peroxidation (LPO), and protein oxidation as well as hamper the enzyme activities, eventually resulting in oxidative stress (Jomova et al. 2011). The liver is the primary target organ of arsenic toxicity as it is directly involved in its biotransformation via enzyme cascade reactions (Singh et al. 2011). Arsenic hepatotoxicity is morphologically characterized by hepatocellular injuries, degradation of hepatocytes, hepatomegaly, hepatoportal sclerosis, and progressive fibrosis and cirrhosis (Ghatak et al. 2011). A number of complexing and metal chelating agents like dimercaprol sulfonate and British anti-lewisite have been used as antidotes for the treatment of arsenic toxicity. However, none of these agents is considered safe for clinical use due to the associated side effects (Gupta et al. 2005). Thus, there is a pressing need to identify naturally occurring dietary substances with antioxidant or ROS quenching properties that can serve as effective chemo-protectants against arsenic toxicity. One such source of dietary antioxidants is N. sativa. Since ancient times, the consumption of N. sativa seed and its oil has shown health-promoting effects. These effects have been attributed to the presence of a wide range of well-defined phytochemicals that exhibit anti-inflammatory, anti-carcinogenic, antioxidant, anti-diabetic, hepatoprotective, and nephroprotective properties (Ijaz et al. 2017). Until now, many active components have been isolated from the N. sativa oil (NSO) including thymoquinone, dithymoquinone, thymohydroquinone, thymol, carvacrol, and p-cymene (Ahmad et al. 2013). Additionally, NSO is also a chief source of polyunsaturated fatty acids (PUFA), i.e., ω-3 and ω-6 PUFA present in 1:4 ratio, which is recommended as an excellent dietary PUFA proportion (Sioen et al. 2017). Utilization of ω-3 and ω-6 PUFA in 1:4 ratio has been shown to conquer the pathogenesis of various ailments (Russo 2009). Previously, NSO consumption has been shown to downregulate CCl4-induced nitric oxide synthase mRNA and suppress NO production in rat liver (Ibrahim et al. 2008). Thymoquinone, a major NSO constituent, has been reported to improve plasma and liver antioxidant capacity by upregulating the expression of antioxidant genes in hypercholesterolemic rats (Ismail et al. 2009). Recently, thymoquinone has been shown to ameliorate the hepatotoxicity and neurotoxicity induced by microcystin-LR, a potent hepatotoxin, in mice (Abdel-Daim et al. 2019). Moreover, thymoquinone with diallyl sulfide has been shown its protective potential against fibronil-induced oxidative injury (Abdel-Daim et al. 2018) and malathion-induced toxicity (Abdel-Daim et al. 2020). Thymoquinone has also been reported to attenuate cardiomyopathy in streptozotocin-treated diabetic rats (Atta et al. 2018). In line, several studies have reported the effectiveness of NSO in attenuating toxicities induced by several oxidative stress generating agents including propoxur-induced neurotoxicity (Mohamadin et al. 2010), alcohol-induced gastrointestinal toxicity (Kanter et al. 2005), cyclosporine-induced cardiotoxicity (Ebru et al. 2008), and gentamicin-induced nephrotoxicity (Yaman and Balikci 2010). Recent studies from our lab have shown NSO to confer protection against cisplatin-induced multiple organ toxicity (Farooqui et al. 2017; Farooqui et al. 2016; Shahid et al. 2017). However, the possible preventive/protective potential of NSO in relation to heavy metal toxicity, particularly arsenic-induced hepatotoxicity, remains unexplored. This study aims to investigate sodium arsenate (As)–induced cytotoxic and genotoxic effects on rat liver and possible diminution of these effects by NSO supplementation. We hypothesized that NSO would be able to protect against As-induced noxious effects by strengthening the antioxidant defense and improving the energy metabolism in rat liver.
Material and methods
Chemicals
N. sativa oil was purchased from Mohammedia Products, Red Hills, Nampally, Hyderabad, India. Sodium arsenate was purchased from Sigma-Aldrich Chemical Corp., St. Louis, MO, USA. All other chemicals used were of analytical grade and were obtained either from Sigma Chemical Corp. or SRL (Mumbai, India).
Phytochemistry of N. sativa oil
Bioactive components in the Nigella sativa oil were confirmed by GCMS (gas chromatography mass spectroscopy). Gas chromatography (PerkinElmer, Clarus 680, Elite-5MS, 30 m × 0.25 mm, 0.25 μm) coupled with mass spectrometry technique (PerkinElmer, Clarus SQ 8 T) Column (30 m × 0.25 mm × 0.25 휇m) was employed for all the analysis. The oven temperature was raised to 220 °C from 70 °C at 4 °C min and maintained isothermally for 15 min. The temperatures of the injector and detector were maintained at 220 °C and 240 °C, respectively, with the preparation of 10% of samples in acetonitrile. The split mode ratio of 1:15 was applied for the injection of a 0.5 휇L sample. The carrier gas used was helium at a flow rate of 1 mL/min. Other parameters were kept the same in relation to GC analysis. EI at 70 eV provided the mass spectra with mass scanning done from 40 to 400 u.
Experimental procedure
Adult male Wistar rats, weighing approximately 150 g, were used to conduct the present study. The animals were purchased from National Institute of Biologicals (NIB), New Delhi. Experiments on animals were approved by the Institutional Animal Ethical Committee (IAEC) of the Department of Biochemistry, Faculty of Life Sciences, Aligarh Muslim University, Aligarh, India (714/GO/Re/02/CPCSEA). Prior to use, rats were acclimatized for 7 days in 12 h:12 h light and dark illumination cycles. The animals were kept in polycarbonate cages in a well-ventilated room maintained at 26 ± 2 °C and 50–60% relative humidity and fed standard rodent diet supplied by Aashirwad Industries, Chandigarh, India, with free access to water. Rats were randomized into 4 groups with five rats per group, as mentioned below:
-
Group 1.
Control (C)
-
Group 2.
Sodium arsenate–treated group (As) was administered As (5 mg/kg bwt, orally) for 30 days. The dose of As was selected based on earlier studies and was much lower than the reported LD50 value of arsenic, i.e., 40 mg/kg bwt (Sumedha and Miltonprabu 2015; Yousef et al. 2008).
-
Group 3.
N. sativa oil–treated group (NSO) was administered NSO (2 mL/kg bwt, orally), once daily during the entire duration of experiment.
-
Group 4.
Sodium arsenate and N. sativa oil–co-treated group (AsNSO) was administered NSO for 14 days prior to and 30 days along with As treatment.
The body weight of animals was noted before and after completion of the experimental protocol. Rats were sacrificed the next day after the last As dose under light ether anesthesia. Blood was collected and the liver was removed from each sacrificed rat and processed for histopathological study and homogenate preparation as described below.
Preparation of liver homogenate
Liver homogenate was prepared by the method of Kaplan and Utiger (1978) with slight modifications. Briefly, Tris-HCl buffer (100 mM, pH 7.5) was used to prepare liver homogenates. Five pulses were passed through liver tissue suspended in the buffer at 4 °C, by a glass–teflon homogenizer (Thomas PA, USA) to obtain a 10% (w/v) homogenate. Then, with a periodic interval of 30 s, three pulses of duration 30 s each were passed using Ultra-Turrex Kunkel homogenizer (TypeT 25). A small volume of liver homogenate was stored at −20 °C for the determination of total –SH groups, GSH levels, and LPO (lipid peroxidation), while the remaining volume was centrifuged at 2000 rpm at 4 °C for 10 min, and the supernatant was collected for the assay of free radical scavenging, hepatocyte membrane, and carbohydrate metabolic enzyme activities. The precision of the analytical methods used was confirmed by repeatability, i.e., intra-day assay as well as an intermediate precision, i.e., inter-day assay. All the experiments were repeated at least 3–5 times to check the reproducibility of the data
Serum parameters
Serum was obtained by centrifuging the blood sample of each rat at 2000 rpm for 10 min at 4 °C. One part of the serum samples was used to determine the activities of transaminases, including ALT (alanine aminotransferase) and AST (aspartate aminotransferase), by using span diagnostics kits based on the method proposed by Reitman and Frankel (1957). Briefly, 50 μL of serum was added to 250 μL substrate buffer (α-ketoglutarate and DL-α-alanine in phosphate buffer at pH 7.4 for ALT and α-ketoglutarate and L-aspartic acid in phosphate buffer at pH 7.4 for AST) and incubated at 37 °C for 30 min. Then, 250 μL of coloring agent (2, 4 dinitrophenyl hydrazine) was added, mixed well, and allowed to stand at room temperature for 20 min. The reaction was stopped by adding 2.5 mL of 4 N NaOH solution and read at 505 nm by using a UV-1700 spectrophotometer (Pharma spec, Shimadzu Corp., Japan). Cholesterol (Chl) levels were determined by the method reported earlier (Farooqui et al. 2016). However, the other part of each serum sample was deproteinated with 3% TCA in 1:3 proportion, and the precipitate obtained after centrifugation was used to quantify the total phospholipids (PLs) (Bartlett 1959).
Carbohydrate metabolic enzymes
Enzyme activities involved in the reduction of NADP or oxidation of NADH were determined in the liver homogenate spectrophotometrically from optical density change at 340 nm. For calculating the concentration of NADPH/NAD+, 6220 M−1 cm−1 was used as the molar extinction constant. The enzyme activities of LDH (lactate dehydrogenase), MDH (malate dehydrogenase), ME (malic enzyme), G6PDH (glucose-6-phosphate dehydrogenase), G6Pase (glucose-6-phosphatase), and FBPase (fructose-1, 6-bisphosphatase) were estimated as reported by Khundmiri et al. (2004). HK (Hexokinase) was estimated as described by Crane and Sols (1953), and the amount of remaining glucose was evaluated as reported by Nelson (1944).
Membrane and lysosomal marker enzymes assay
The activity of ALP (alkaline phosphatase) was assayed by using PNPP as the substrate, by the method reported earlier by Kempson et al. (1979). Briefly, 1.4 mL assay buffer (55 mM glycine; 36 mM NaCl and 45 mM NaOH, pH 10.5) was mixed with 100 μL aliquot of liver homogenate and 15 μL p-nitrophenyl phosphate (final concentration 5.8 mM) and incubated in water bath maintained at 37 °C. Once the yellow color developed, the reaction was stopped by adding 50 μL of 5 N NaOH and the color was read at 405 nm. The activities of GGTase (γ-glutamyl transferase) and LAP (leucine aminopeptidase) were determined by using the substrates γ-glutamyl p-nitroanilide and L-leucine p-nitroanilide respectively, as described by Glossmann and Neville (1972) and Goldmann et al. (1976). Briefly, the reaction was started by adding 100 μL aliquot of liver homogenate to 1.9 mL substrate buffer [(20 mM MgCl2, 2 mM γ- glutamyl-p-nitroanilide, 4 mM glycylglycine, 100 mM Tris-base, pH 8.2 for GGTase) and (50 mM sodium phosphate buffer, 0.33 mM L-leucine p-nitroanilide, pH 7.2 for LAP)] and incubated at 37 °C. Once the yellow color developed, the reaction was stopped by adding 100 μL of 15 M acetic acid and the color was read at 405 nm. The activity of ACPase (acid phosphatase), a lysosomal marker enzyme, was estimated as reported previously (Farooq et al. 2004). Briefly, the reaction mixture was prepared by mixing 2.4 mL acetate buffer (0.05 M sodium acetate, pH 4.5) with 100 μL aliquot of liver homogenate. The reaction was started by adding 0.5 mL p-nitrophenyl phosphate (final concentration 0.8 mM) and incubated for 15 min at 37 °C. The reaction was stopped by adding 2 mL of 2 N NaOH. The yellow color developed was read at 405 nm.
Evaluation of kinetic parameters (KM and Vmax) of membrane enzymes
Kinetic parameters of hepatocyte membrane enzymes (ALP, LAP, and GGTase) were determined by assaying these enzymes at varying substrate concentrations in the liver homogenate. The data was analyzed by double reciprocal Lineweaver-Burk plots.
Free radical scavenging enzyme assays
The activity of SOD (superoxide dismutase) was determined by inhibiting the auto-oxidation of pyrogallol (Marklund and Marklund 1974) and CAT (catalase) by the decomposition of hydrogen peroxide (H2O2) to water (H2O) (Giri et al. 1996). GSH-Px (glutathione peroxidase) activity was assayed by following the oxidation of NADPH to NADP+ in the presence of oxidized glutathione, as reported earlier (Flohé and Günzler 1984). The activity of GR (glutathione reductase) was determined by following the conversion of NADPH to NADP+ (Carlberg and Mannervik 1985). Activities of GST (glutathione-S-transferase) and TR (thioredoxin reductase) were determined by using 1-chloro-2-4-dinitrobenzene and 5,5-dithiobis-2-nitrobenzoic acid as substrates, respectively (Habig et al. 1974; Tamura and Stadtman 1996).
Estimation of LPO, protein carbonyl content, total -SH, GSH and H2O2 levels
Lipid peroxidation (LPO) was measured from the level of TBARS (thiobarbituric acid reactive substances) (Ohkawa et al. 1979), and GSH levels and total-SH (sulfhydryl) were determined by the yellow color formed after their reaction with 5,5′-dithiobis-2-nitrobenzoic acid as reported by Jollow et al. (1974) and Sedlak and Lindsay (1968) respectively. Protein oxidation was determined by measuring the levels of protein carbonyls, as reported by Levine et al. (1990). Protein carbonyl groups upon reaction with DNPH produce hydrazone measured at 360 nm by using 22,000 M−1 cm−1 as the molar extinction coefficient. The levels of H2O2 were analyzed by using xylenol orange as a color reagent in the presence of 100 mM sorbitol, as reported earlier (Gay and Gebicki 2000).
Estimation of DNA damage
The liver was collected immediately after sacrificing the rats and placed into the RPMI-1640 medium. A single-cell suspension was obtained by mincing the hepatic tissue in the same medium. Comet assay was then carried out following the protocol reported by Singh et al. (1988) with slight modifications. Cells were mixed with 2% LMPA (low melting point agarose) and pipetted over the frosted slides already coated with 1% NMPA (normal melting point agarose), and covered with the coverslip. The agarose layer was then allowed to solidify. After solidification, the coverslips were removed and slides were submerged in cold lysis buffer for 2 h at 4 °C in the dark. After lysis, the DNA was subjected to unwinding in an alkaline electrophoretic buffer for 30 min, and then electrophoresis was done at 300 mA current and 0.7 V/cm field strength. After electrophoresis, neutralization was performed by neutralizing buffer (0.4 M Tris, pH 7.5), followed by staining with ethidium bromide (20 g/mL). Visualization and scoring of slides were done by using an image analysis system (Komet 5.5, Kinetic Imaging, Liverpool, UK) attached to a CX41 fluorescent microscope (Olympus, Japan) equipped with CC camera. Scoring of comets was done at × 100. The images of 50 cells (25 cells from each replicate slide) were analyzed. Comet tail length (DNA migration from the nucleus in μm) was the selected parameter for evaluating nuclear DNA damage.
Estimation of arsenic accumulation
Dry liver samples (100 mg) from each group were placed separately in the digestion vessel, and 2 mL of nitric acid (69%) was added to each vessel. The mixture of liver and acid was then placed in a water bath at 90 °C for 2 h. After completion of the incubation time, the mixture was cooled at room temperature and 2 mL H2O2 (30%) was added and left overnight at 50 °C. The mixture turned into a colorless, watery solution indicating the complete digestion of the liver sample (Khan et al. 2013). The sample was then filtered through the Whatman filter paper, and the level of arsenic was estimated by ICP-MS (inductively coupled plasma–mass spectroscopy) using iCAP Q (Thermo Fisher Scientific, Germany).
Histopathological examination
Fresh pieces of liver tissue were fixed for a week in Karnovsky fixative (immersion fixation). The fixed tissues were then processed for paraffin embedding. Using a rotary microtome, liver sections of thickness 3 μm were obtained and then subjected to hematoxylin-eosin (HE) staining and examined under the light trinocular microscope at × 400 magnification (Elcombe et al. 1985). The scoring was done to determine the severity of damage as (−) none, (+) mild, (++) moderate, and (+++) severe.
Statistical analysis
All data is expressed as mean ± SEM for at least 3 different preparations. Statistical analysis was performed by one-way ANOVA using origin 8 software followed by Student-Newman-Keuls and Dunnets multiple comparison post hoc tests. A probability level of p<0.05 was taken for indicating the statistical significance. Most of the changes among various groups were compared with the values of the control group for better understanding and clarity. However, specific differences and statistical significance among other groups such as As vs. AsNSO were expressed separately.
Results
GS-MS analysis
In GC-MS analysis, the chromatogram was calibrated at 60-min retention time because all major components of NSO were found within that time. Thymoquinone and thymol were identified with the highest peaks at 11.06 min and 18.87 min, respectively. The ratio of thymoquinone to the rest of the components was found to be 14.19%. Besides, ten other constituents, including 1,3,8-p-menthatriene, cis-4-methoxythujane, limonene, longifolene, caryophyllene, methyltetradecanoic acid, hexadecenoic acid, ascorbic acid, linoleic acid, and cyclopropane butanoic acid, were identified in NSO within the 60-min retention time (Fig. 1).

Chromatogram showing constituents of N. sativa oil characterized by GC-MS analysis
Effect of NSO on As-elicited alterations in serum parameters
As-elicited hepatotoxicity was noted by significant elevation of ALT and AST levels in serum. Administration of As resulted in profound increase in serum ALT (+89.14%), AST (+65.62%), Chl (+30.33%), and PLs (+25.57%) (Table 1). However, supplementation of NSO before and along with the As treatment protected against these As-induced adverse alterations in serum parameters. However, NSO administration alone had no significant effect on these parameters.
Effect of NSO on As-elicited alterations in carbohydrate metabolic enzymes
As shown in Table 2, the treatment of rats with As profoundly elevated the activity of HK (+37.83%) and LDH (+64.02%) while the activity of MDH (−68.69%) declined significantly in the liver. In contrast, supplementation of NSO before and simultaneously with As treatment ameliorated the As-elicited elevation of HK and LDH activities in the hepatic tissue. Moreover, MDH activity was also markedly improved upon NSO supplementation. Notable reduction in the activities of G6Pase (−32.14%) and FBPase (−38.55%) was also observed in the As-treated group. The activity of G6PDH was significantly decreased (−62.5%), while the ME activity was profoundly elevated (+81.28%) in the liver homogenate. However, As-induced alterations in G6Pase and FBPase as well as in G6PDH and ME activities were significantly alleviated upon NSO administration. However, no significant alteration in the carbohydrate metabolic enzymes was observed in the rats treated with NSO alone.
Effect of NSO on As-elicited alterations in the membrane and the lysosomal biomarker enzyme
To evaluate the structural integrity of certain cellular organelles like plasma membrane and lysosomes, the effect of As alone and in combination with NSO was estimated on their marker enzymes. As shown in Fig. 2, As treatment resulted in a significant reduction in membrane marker enzyme activities, viz., ALP (−42.08%), GGTase (−40.36%), and LAP (−38.86%) in liver homogenate. Supplementation of NSO to As-administered rats alleviated the As-induced decrease in membrane enzyme activities. Furthermore, the specific activity of the lysosomal marker enzyme, viz., ACPase, was elevated (+49.03%) upon As treatment. In contrast, supplementation of NSO before and simultaneously with As treatment attenuated the As-induced increase in the activity of ACPase.

Effect of NSO on As-induced alterations in the activities of: a ALP, alkaline phosphatase; b GGTase, γ-glutamyl transferase; c LAP, leucine aminopeptidase; and d ACPase, acid phosphatase, in five different preparations of liver homogenate. As, sodium arsenate treated; NSO, N. sativa oil administered; AsNSO, sodium arsenate + N. sativa oil treated. *, significantly different at p < 0.05 from control. ǂ, significantly different at p < 0.05 from As-treated group
Effect of NSO on As-elicited alterations in kinetic parameters of membrane enzymes
As shown in Table 3, oral administration of As for 30 days led to a significant decline in Vmax values of all the membrane marker enzymes. In contrast, supplementation of NSO prior to and simultaneously with As treatment caused a significant improvement in Vmax values. Administration of NSO alone did not alter the values of Vmax of any membrane-bound enzymes. However, the KM values of the membrane enzymes were not significantly changed in any of the four experimental groups.
Effect of NSO on As-elicited alterations in antioxidant defense parameters
Antioxidant defense system comprising of enzymatic and non-enzymatic components plays a vital role in averting or slowing down the progression of many ailments. Since enhanced ROS generation has been reported as the major cause of As-induced hepatocellular toxicity, hence to evaluate the ameliorative potential of NSO against As-induced hepatotoxicity, the effect of As and NSO alone and in combination (AsNSO) was determined on various enzymatic and non-enzymatic parameters of the antioxidant defense system.
Effect of NSO on As-elicited alterations in enzymatic antioxidants
The antioxidant defense enzyme activities in the hepatic tissue of rats treated with As were altered significantly. As shown in Table 4, significant reductions in activities of SOD (− 33.99%), CAT (− 58.39%), GSH-Px (− 40.00%), GR (− 41.03%), TR (− 53.42%), and GST (− 70.31%) were observed in the liver of As-intoxicated rats. NSO supplementation before and simultaneously with the As treatment significantly alleviated the As-elicited reduction in the activities of SOD, CAT, GSH-Px, GR, TR, and GST. In contrast, the antioxidant defense enzymes’ activities were not altered significantly in rats treated with NSO alone.
Effect of NSO on As-elicited alterations in non-enzymatic antioxidants/parameters of oxidative stress
Elevated generation of ROS leads to increase in LPO, H2O2 levels, and oxidation of protein as well as the sulfhydryl groups (total SH and GSH). LPO was determined by measuring the levels of malondialdehyde (MDA), which gives pink-colored product upon reaction with TBARS. Oxidation of protein leads to the elevation of carbonyl content, which was determined by the reaction with DNPH. As shown in Table 5, treatment of rats with As resulted in a significant increase in LPO (+78.57%) and protein oxidation (+112.83%), while the levels of total SH (−22.09%) and GSH (−69.64%) were significantly declined in the liver homogenate. Furthermore, the level of H2O2 was also increased (+90.85%) in the hepatic tissue of As-treated animals. In contrast, As-induced alterations in LPO, protein oxidation, total SH, GSH, and H2O2 levels were significantly ameliorated upon NSO administration.
Histopathological observations
Histopathological examination of liver tissue of As-treated animals was suggestive of considerable damage to the liver. As treatment resulted in hepatocellular degradation, vacuolation, and infiltration pyknosis (Fig. 3b and Table 6). However, there was no significant histoarchitectural damage to the liver when the rats were treated with NSO alone (Fig. 3c). Supplementation of NSO prior to and simultaneously with the As treatment (Fig. 3d) caused marked attenuation of liver damage elicited by As treatment.

Representative histological images of liver sections stained with hematoxylin and eosin. Control rat liver (a) shows lobules with hepatocytes and central vein and clear sinusoid area whereas As-treated group (b) reveals lobules with necrotic hepatocytes and voluminous sinusoids. The NSO alone group (c) shows the well-preserved hepatic structure similar to that in the control group while the co-treated group, AsNSO (d), shows restored morphology without any obvious necrosis and voluminous sinusoids while the central vein was occupied with secretory material
Effect of NSO on As-elicited DNA damage
Administration of As alone resulted in significant DNA damage in hepatocytes compared to the untreated control (Fig. 4a). This was represented by elongated tail length, indicating an elevated level of breaks in the DNA strand as well as alkali labile sites in hepatocyte DNA (Fig. 4b). Administration of NSO prior to and simultaneously with As reduced the extent of As-induced DNA damage, as evident by the decline in comet tail length (Fig. 4). In contrast, supplementation of NSO alone did not cause damage to the hepatocyte DNA, and the tail length remained the same as that of the untreated group.

a Representative images showing the effect of NSO on As-induced DNA breakage in hepatocytes as evaluated by single-cell gel electrophoresis (comet assay), b The results of comet assay represented as mean values of tail lengths. As, sodium arsenate treated; NSO, N. sativa oil administered; AsNSO, sodium arsenate + N. sativa oil treated. *Significantly different at p < 0.05 from control. ǂ, significantly different at p < 0.05 from As-treated group
+
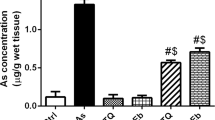
Effect of NSO on arsenic accumulation in the liver
Exposure to As for 30 days caused a significant elevation of arsenic concentration in the hepatic tissue. However, NSO supplementation prior to and along with the As treatment profoundly reduced the accumulation of arsenic in the hepatic tissue (Fig. 5).

Effect of NSO on arsenic accumulation in the hepatic tissue. Results are mean ± SEM for three different samples (n = 3) in each group. As, sodium arsenate treated; NSO, N. sativa oil administered; AsNSO, sodium arsenate + N. sativa oil treated. *Significantly different at p < 0.05 from control. ǂ, significantly different at p < 0.05 from As-treated group
Discussion
The liver has already been recognized as a primary target organ of arsenic toxicity (Liu and Waalkes 2008). Arsenic is known to exert its toxicity through oxidative stress by generating reactive oxygen species (Muthumani and Prabu 2012b). A number of synthetic substances, including metal chelators, have long been used to alleviate arsenic toxicity; however, most of them exhibit toxic manifestations and that stress the need to explore alternative strategies. (Flora and Pachauri 2010; Susan et al. 2019; Mehrandish et al. 2019). In view of the involvement of oxidative stress in arsenic toxicity, an important strategy could be the use of naturally occurring antioxidants and one such dietary source of antioxidants is N. sativa, which has long been revered for its health promoting benefits in physiological as well as pathological conditions (Salem 2005). A number of studies have documented the protective role of N. sativa oil (NSO) against various drug and chemical-induced toxicities (Yaman and Balikci 2010; Mohamadin et al. 2010; Abdel-Daim and Ghazy 2015). However, the ameliorative potential of NSO against arsenic-induced hepatotoxicity, in particular, has not yet been evaluated. In the current study, the effect of NSO against arsenic-induced hepatotoxicity was investigated to identify its potential mechanism of action.
Repeated exposure of rats to sodium arsenate (As), an As (V) compound, for 30 days, caused notable changes in the serum parameters. Serum levels of ALT and AST, the classical diagnostic indicators of hepatotoxicity, were significantly elevated by As intoxication. Serum Chl and PL levels were also elevated significantly in As-treated animals. In contrast, supplementation of NSO prior to and simultaneously with As treatment caused a marked reduction in As-induced enhancement of transaminase activities. Likewise, NSO profoundly lowered the PLs and Chl levels in serum when administered either alone or together with As treatment. It might be ascribed to the hypocholesterolemic effect of NSO, as has been reported earlier (Asgary et al. 2013).
To assess the effect of arsenic on hepatic energy metabolism and examine if NSO administration could restore the metabolic capacity of the liver in As-intoxicated rats, the activities of various enzymes involved in glucose catabolism and its production/synthesis were determined in the homogenates prepared from various experimental groups. LDH, an enzyme of anaerobic glycolysis, catalyzes the inter-conversion of pyruvate and lactate. Production of lactate from pyruvate by LDH generates NAD+, which is required for the continuation of glycolysis under anaerobic conditions. MDH reversibly catalyzes the oxidation of malate to oxaloacetate, an intermediate of the TCA cycle that leads to energy production through aerobic mode. A marked increase in LDH activity with concomitant decrease in MDH activity suggests a shift in hepatic energy production from aerobic mode alternatively to anaerobic glycolysis, most likely due to mitochondrial dysfunction (Pace et al. 2017). The first step in glucose catabolism, whether occurring by aerobic, anaerobic, or pentose phosphate pathway, is catalyzed by hexokinase. Thus, a profound increase in HK activity in the As-treated group indicates an adaptive hepatocellular response to the increased energy demands. The liver is the major site of gluconeogenesis, and therefore, it plays a key role in the homeostatic regulation of blood glucose levels (Tsuneki et al. 2015). As treatment caused a marked decrease in the activities of G6Pase and FBPase, indicating the reduced ability of the liver to synthesize glucose via gluconeogenesis. G6Pase is an endoplasmic reticulum membrane–associated enzyme that generates glucose from glucose-6-phosphate and helps in the release of glucose from hepatocytes into the blood. Membrane disruption and oxidative modification could be the possible reasons for the observed effect of As on these enzymes as reported previously for other toxicants (Ali et al. 2017). As treatment seems to exert a reciprocal effect on the activities of NADPH generating enzymes, G6PDH, and ME. G6PDH, the first enzyme of the pentose phosphate pathway, catalyzes the oxidation of glucose-6-phosphate to 6-phosphogluconate and generates NADPH, which serves as a reductant in biosynthetic reactions and also protects the cell from oxidative damage. Thus, the As-induced decrease in G6PDH activity would have rendered the cells more susceptible to oxidant attack. Increased ME activity could be a compensatory effect to supply NADPH by an alternative mechanism involving ME. However, the As-induced changes in hepatic energy metabolism were greatly restored towards normal by prolonged NSO administration. Taken together, these findings suggest that NSO acts by lowering the number of damaged mitochondria and affected macromolecules or by increasing the number of normally active organelles and macromolecules as reported earlier for other toxicants (Ahmad et al. 2013; Mollazadeh and Hosseinzadeh 2014).
The polyunsaturated fatty acid constituents of membrane phospholipids are highly vulnerable to free radical attack that triggers the self-propagating LPO reactions resulting in impaired membrane structure and function, due to the loss and/or inactivation of membrane-bound enzymes (Shahid et al. 2018). To assess the damage to hepatocyte membrane, the activities of various membrane marker enzymes, i.e., ALP, LAP, and GGTase, were determined in various experimental groups. The specific activities of all the three marker enzymes were significantly declined in the As-treated group (Fig. 2). The observed decline might be attributed to the oxidative modification and consequent inactivation of these enzymes by As-generated ROS. In contrast, prolonged NSO administration to As-exposed rats prevented As-induced decline in the activities of all these enzymes. Marked reduction in lipid peroxidation and protein oxidation/oxidative modification of hepatocyte membrane enzymes by NSO, owing to its inherent antioxidant properties, could have resulted in the noticeable improvement in membrane enzyme activities in As-treated NSO-fed rats. Being an excellent source of essential fatty acid, NSO could have substituted the damaged PUFA of membrane phospholipids that were targeted by As-induced ROS, thereby facilitating the repair process and preventing the loss of membrane enzymes (Farooqui et al. 2016). Furthermore, the kinetic studies showed that decrease in the activities of these membrane enzymes was due to decline in their Vmax value, while the value of KM remained unaltered by As treatment (Table 3). Thus, the affinity of membrane enzymes for their substrates remains the same upon administration of NSO, As or As plus NSO (Robinson 2015).
In agreement with the earlier studies, activity of ACPase in the hepatic tissue was profoundly increased by repeated As exposure (Rizwan et al. 2014). It has been documented that the size and number of lysosomes in the cell increase upon arsenic intoxication (Bolt et al. 2010). In contrast, pre and co-treatment with NSO ameliorated the As-induced elevation in ACPase activity.
Increased production of ROS has been shown to correlate with the severity of toxic manifestations of arsenic toxicity (Jomova et al. 2011). Cellular metabolism of arsenic leads to the generation of ROS including hydrogen peroxide (H2O2), the superoxide anion (O2−), and the hydroxyl radical (.OH) (Jomova et al. 2011). The higher levels of ROS cause wide ranging damage to the cellular macromolecules, such as proteins, lipids, and nucleic acids, and eventually damage the cell membrane, mitochondria, microsomes, and lysosomes in various tissues including the liver (Muthumani and Prabu 2012a). As-intoxication significantly decreased the total thiols and enhanced LPO, evidenced by overwhelmingly high levels of MDA, increased protein carbonyls, an index of protein oxidation and induced DNA damage in the hepatic tissue suggesting induction of oxidative stress. The decrease in total thiol content can be attributed to the high reactivity of arsenic towards –SH groups and/or to the oxidation of –SH groups by As-induced ROS. The level of H2O2, a non-radical ROS, was also significantly elevated by As treatment. H2O2 is itself a strong oxidant; however, upon reaction with transition metal ions like iron in the cell, it generates the more damaging hydroxyl radicals (Collin 2019). DNA is also potentially modified and/or damaged by As-induced ROS. The genotoxic potential of arsenic has been reported previously (Singh et al. 2008). Oral administration of As induced DNA degradation in the hepatocytes as evident by a significantly higher mean tail length in As-exposed hepatocytes as compared to the cells from control rat liver. Besides direct chemical modification by free radicals, DNA could have also been damaged by lipid oxidation products like unsaturated aldehydes and MDA that can bind to DNA to generate mutagenic lesions (Gentile et al. 2017).
GSH is the most abundant non-enzymatic antioxidant in the mammalian liver, where its levels can be readily modulated by regulating the biosynthetic pathways involved (Lu 2013). Sodium arsenate treatment caused a significant decline in the hepatic GSH levels. Arsenic has been shown to deplete cellular GSH levels by utilizing GSH as an electron donor in the reductive conversion of pentavalent arsenate to trivalent arsenite and as a cofactor in the subsequent oxidative methylation reactions. In addition, arsenic is known to directly bind to the sulfhydryl groups of GSH or oxidize GSH via enhanced free radical generation (Rubino 2015). However, pre- and co-treatment with NSO greatly lowered the As-induced oxidative stress as evidenced by the significantly normalized values of LPO and protein carbonyls along with total –SH levels. The level of As-induced DNA damage was also markedly reduced, probably due to the antioxidant and anti-mutagenic actions of NSO (Majdalawieh and Fayyad 2016). As-induced alterations in H2O2 and GSH levels were also alleviated by NSO administration. Similar protective effects of NSO on liver damage by other toxicants have been reported earlier by other investigators (Farooqui et al. 2016).
To combat oxidative stress and maintain a proper reducing environment, the liver is well equipped with robust enzymatic antioxidants that play an important role in the metabolism and neutralization of ROS (Marí et al. 2010). The activities of all major antioxidant defense enzymes were found to decrease in As-administered rats in comparison to the control group. SOD and catalase are known to be inactivated by ROS and free radicals (Ighodaro and Akinloye 2018). Furthermore, SOD, catalase, and GSH-Px being metalloproteins depend on various essential trace elements, i.e., Cu, Fe, and Se, respectively, for their catalytic activities and are thus the potential targets of arsenic toxicity (Birben et al. 2012). Arsenic forms complexes with these trace elements and can therefore directly interact with the enzymes and inhibit their activities (Birben et al. 2012). Thus, the increase in LPO as observed in As-exposed rats could be a consequence of increased levels of superoxide anions and H2O2, as the activities of the enzymes that utilize them as substrates (i.e., SOD and catalase) were decreased significantly by As intoxication. TR, a central component of thioredoxin system, and GR help in maintaining redox homeostasis by catalyzing thiol-disulfide exchange reactions and thus function in defense against oxidative stress (Mukwevho et al. 2014). Reduced NADPH supply due to marked lowering of G6PDH activity in As-treated rats could have adversely affected the activities of NADPH-dependent enzymes, GR and TR. A significant decline in GR activity in As-intoxicated rats would have further lowered the hepatic GSH levels. Reduced levels of GSH and total thiols along with lowered activities of SOD, CAT, GSH-Px, GR, and TR indicate that As exerted an exhaustive effect on antioxidant reserves, and that confirms the involvement of ROS in As hepatotoxicity, as reported earlier (Singh et al. 2011).
In contrast, NSO administration conferred significant protection against As-induced inhibitory effect on all the major antioxidant enzymes, thus restoring the antioxidant power of the hepatic tissue to resist As-generated free radical attack. Furthermore, various NSO constituents namely p-cymene, carvacrol, t-anethole, and in particular thymoquinone, owing to their intrinsic free radical scavenging activities, would have acted synergistically to neutralize/quench As-generated free radicals before they could reach their cellular targets (Salem 2005). These effects would have, therefore, substantially decreased the oxidative damage to cellular macromolecules and organelles, resulting in mitigation of the adverse effects of As on the liver. This is in agreement with previous reports that have shown NSO or its constituents to protect against various pathologies where antioxidant status is severely compromised (Salem 2005).
Histopathological observations of the liver strongly corroborate the biochemical results. The liver from As-treated animals showed extensive damage marked by necrotic hepatocytes, voluminous sinusoids, disorganized hepatic cords, and hepatocellular degeneration, as depicted in Fig. 3b. These As-induced changes were greatly alleviated by prolonged administration of NSO, which could have neutralized the overproduced ROS, thereby reducing the morphological and cellular damage (Fig. 3d).
Another interesting finding of the present study is the observed ability of NSO to reduce arsenic accumulation in the hepatic tissue, and that might have played an important role in protecting the liver from arsenic-induced deleterious alterations. GSH-dependent efflux of arsenic from the cells has been shown to confer protection to human cells against arsenic, while depletion of GSH enhances the cellular retention of arsenic and consequent susceptibility of cells to arsenic (Carew et al. 2011; Leslie 2012). Oral administration of NSO prior to and along with As treatment was found to restore GSH levels in the liver, and that might have played a crucial role in increasing the efflux of arsenic and reducing the tissue arsenic accumulation, as observed in the co-treated group. The recovery of GSH levels in NSO-fed As-treated rats would have in turn restored the activity of GST, an important biotransformation enzyme that catalyzes GSH conjugation to xenobiotics to enhance their hydrophilicity and consequent elimination from the body (Allocati et al. 2018).
In conclusion, our results show that oxidative stress induced by As contributes to the development of hepatotoxicity, manifested as dramatic alterations in various cellular/physiological processes. Prolonged NSO treatment, however, greatly reversed the As-induced hepatotoxicity suggesting prevention of oxidative damage as the underlying mechanism of NSO-mediated protective effects on liver functions. Thus, NSO can be considered a potential chemoprotective agent against liver damage caused by long-term arsenic exposure.
Availability of data and materials
Data and materials can be made available on reasonable request.
References
Abdel-Daim MM, Abushouk AI, Bungău SG, Bin-Jumah M, el-kott AF, Shati AA, Aleya L, Alkahtani S (2020) Protective effects of thymoquinone and diallyl sulphide against malathion-induced toxicity in rats. Environ Sci Pollut Res 27:10228–10235
Abdel-Daim MM, Ghazy EW (2015) Effects of Nigella sativa oil and ascorbic acid against oxytetracycline-induced hepato-renal toxicity in rabbits. Iran J Basic Med Sci 18:221–227
Abdel-Daim MM, Sayed AA, Abdeen A, Aleya L, Ali D, Alkahtane AA, Alarifi S, Alkahtani S (2019) Piperine enhances the antioxidant and anti-inflammatory activities of thymoquinone against microcystin-LR-induced hepatotoxicity and neurotoxicity in mice. Oxidative Med Cell Longev 2019:1–10. https://doi.org/10.1155/2019/1309175
Abdel-Daim MM, Shaheen HM, Abushouk AI, Toraih EA, Fawzy MS, Alansari WS, Aleya L, Bungau S (2018) Thymoquinone and diallyl sulfide protect against fipronil-induced oxidative injury in rats. Environ Sci Pollut Res 25:23909–23916
Adil M, Kandhare AD, Visnagri A, Bodhankar SL (2015) Naringin ameliorates sodium arsenite-induced renal and hepatic toxicity in rats: decisive role of KIM-1, Caspase-3, TGF-β, and TNF-α. Ren Fail 37:1396–1407
Ahmad A, Husain A, Mujeeb M, Khan SA, Najmi AK, Siddique NA, Damanhouri ZA, Anwar F (2013) A review on therapeutic potential of Nigella sativa: a miracle herb. Asian Pac J Trop Biomed 3:337–352. https://doi.org/10.1016/S2221-1691(13)60075-1
Ali SN, Ansari FA, Arif H, Mahmood R (2017) Sodium chlorate induces DNA damage and DNA-protein cross-linking in rat intestine: a dose dependent study. Chemosphere 177:311–316
Allocati N, Masulli M, Di Ilio C, Federici L (2018) Glutathione transferases: substrates, inihibitors and pro-drugs in cancer and neurodegenerative diseases. Oncogenesis 7:8. https://doi.org/10.1038/s41389-017-0025-3
Asgary S, Ghannadi A, Dashti G, Helalat A, Sahebkar A, Najafi S (2013) Nigella sativa L. improves lipid profile and prevents atherosclerosis: evidence from an experimental study on hypercholesterolemic rabbits. J Funct Foods 5:228–234. https://doi.org/10.1016/j.jff.2012.10.011
Atta MS, El-Far AH, Farrag FA et al (2018) Thymoquinone attenuates cardiomyopathy in streptozotocin-treated diabetic rats. Oxidative Med Cell Longev:2018
Bartlett GR (1959) Colorimetric assay methods for free and phosphorylated glyceric acids. J Biol Chem 234:469–471
Birben E, Sahiner UM, Sackesen C, Erzurum S, Kalayci O (2012) Oxidative stress and antioxidant defense. World Allergy Organ J 5:9–19. https://doi.org/10.1097/WOX.0b013e3182439613
Bolt AM, Douglas RM, Klimecki WT (2010) Arsenite exposure in human lymphoblastoid cell lines induces autophagy and coordinated induction of lysosomal genes. Toxicol Lett 199:153–159. https://doi.org/10.1016/j.toxlet.2010.08.017
Carew MW, Naranmandura H, Shukalek CB, le XC, Leslie EM (2011) Monomethylarsenic diglutathione transport by the human multidrug resistance protein 1 (MRP1/ABCC1). Drug Metab Dispos 39:2298–2304. https://doi.org/10.1124/dmd.111.041673
Carlberg I, Mannervik B (1985) [59] Glutathione reductase. In: Methods in Enzymology. Academic Press, pp 484–490
Chung J-Y, Yu S-D, Hong Y-S (2014) Environmental source of arsenic exposure. J Prev Med Public Health 47:253–257
Collin F (2019) Chemical basis of reactive oxygen species reactivity and involvement in neurodegenerative diseases. Int J Mol Sci 20:2407
Crane RK, Sols A (1953) The association of hexokinase with particulate fractions of brain and other tissue homogenates. J Biol Chem 203:273–292
Ebru U, Burak U, Yusuf S, Reyhan B, Arif K, Faruk TH, Emin M, Aydın K, lhan Atilla, Semsettin S, Kemal E (2008) Cardioprotective effects of Nigella sativa oil on cyclosporine a-induced cardiotoxicity in rats. Basic Clin Pharmacol Toxicol 103:574–580. https://doi.org/10.1111/j.1742-7843.2008.00313.x
Elcombe CR, Rose MS, Pratt IS (1985) Biochemical, histological, and ultrastructural changes in rat and mouse liver following the administration of trichloroethylene: possible relevance to species differences in hepatocarcinogenicity. Toxicol Appl Pharmacol 79:365–376
Farooq N, Yusufi ANK, Mahmood R (2004) Effect of fasting on enzymes of carbohydrate metabolism and brush border membrane in rat intestine. Nutr Res 24:407–416. https://doi.org/10.1016/j.nutres.2004.01.004
Farooqui Z, Afsar M, Rizwan S, Khan AA, Khan F (2016) Oral administration of Nigella sativa oil ameliorates the effect of cisplatin on membrane enzymes, carbohydrate metabolism and oxidative damage in rat liver. Toxicol Rep 3:328–335. https://doi.org/10.1016/j.toxrep.2016.02.004
Farooqui Z, Ahmed F, Rizwan S, Shahid F, Khan AA, Khan F (2017) Protective effect of Nigella sativa oil on cisplatin induced nephrotoxicity and oxidative damage in rat kidney. Biomed Pharmacother 85:7–15. https://doi.org/10.1016/j.biopha.2016.11.110
Flohé L, Günzler WA (1984) [12] Assays of glutathione peroxidase. In: Methods in Enzymology. Academic Press, pp 114–120
Flora SJ, Chouhan S, Kannan GM et al (2008) Combined administration of taurine and monoisoamyl DMSA protects arsenic induced oxidative injury in rats. Oxidative Med Cell Longev 1:39–45
Flora SJS, Pachauri V (2010) Chelation in metal intoxication. Int J Environ Res Public Health 7:2745–2788. https://doi.org/10.3390/ijerph7072745
Gay C, Gebicki JM (2000) A critical evaluation of the effect of sorbitol on the ferric–xylenol orange hydroperoxide assay. Anal Biochem 284:217–220. https://doi.org/10.1006/abio.2000.4696
Gentile F, Arcaro A, Pizzimenti S, Daga M, Paolo Cetrangolo G, Dianzani C, Lepore A, Graf M, R. J. Ames P, Barrera G, 1 Department of Medicine and Health Sciences “V. Tiberio”, University of Molise, Campobasso, Italy, 2 Department of Clinical and Biological Sciences, University of Torino, Torino, Italy, 3 Department of Drug Science and Technology, University of Torino, Torino, Italy, 4 Department of Molecular Medicine and Medical Biotechnologies, University of Naples Federico II, Naples, Italy, 5 CEDOC, NOVA Medical School, Universidade NOVA de Lisboa, Lisboa, Portugal, and Department of Haematology, Dumfries Royal Infirmary, Dumfries, Scotland, UK (2017) DNA damage by lipid peroxidation products: implications in cancer, inflammation and autoimmunity. AIMS Genet 4:103–137. https://doi.org/10.3934/genet.2017.2.103
Ghatak S, Biswas A, Dhali GK, Chowdhury A, Boyer JL, Santra A (2011) Oxidative stress and hepatic stellate cell activation are key events in arsenic induced liver fibrosis in mice. Toxicol Appl Pharmacol 251:59–69. https://doi.org/10.1016/j.taap.2010.11.016
Giri U, Iqbal M, Athar M (1996) Porphyrin-mediated photosensitization has a weak tumor promoting activity in mouse skin: possible role of in situ-generated reactive oxygen species. Carcinogenesis 17:2023–2028. https://doi.org/10.1093/carcin/17.9.2023
Glossmann H, Neville DM (1972) γ-Glutamyltransferase in kidney brush border membranes. FEBS Lett 19:340–344
Goldmann DR, Schlesinger H, Segal S (1976) Isolation and characterization of the brush border fraction from newborn rat renal proximal tubule cells. Biochimica et Biophysica Acta (BBA)-Biomembranes 419:251–260
Gupta R, Kannan GM, Sharma M, S. Flora SJ (2005) Therapeutic effects of Moringa oleifera on arsenic-induced toxicity in rats. Environ Toxicol Pharmacol 20:456–464. https://doi.org/10.1016/j.etap.2005.05.005
Habig WH, Pabst MJ, Jakoby WB (1974) Glutathione S-Transferases: the first enzymatic step in mercapturic acid formation. J Biol Chem 249:7130–7139
Ibrahim ZS, Ishizuka M, Soliman M et al (2008) Protection by Nigella sativa against carbon tetrachloride-induced downregulation of hepatic cytochrome P450 isozymes in rats. Jpn J Vet Res 56:119–128
Ighodaro OM, Akinloye OA (2018) First line defence antioxidants-superoxide dismutase (SOD), catalase (CAT) and glutathione peroxidase (GPX): their fundamental role in the entire antioxidant defence grid. Alexandria J Med 54:287–293. https://doi.org/10.1016/j.ajme.2017.09.001
Ijaz H, Tulain UR, Qureshi J, Danish Z, Musayab S, Akhtar MF, Saleem A, Khan KK, Zaman M, Waheed I, Khan I, Abdel-Daim M (2017) Nigella sativa (Prophetic Medicine): a review. Pak J Pharm Sci 30:229–234
Ismail M, Al-Naqeeb G, Chan KW (2009) Nigella sativa thymoquinone-rich fraction greatly improves plasma antioxidant capacity and expression of antioxidant genes in hypercholesterolemic rats. Free Radic Biol Med 48:664–672. https://doi.org/10.1016/j.freeradbiomed.2009.12.002
Jollow DJ, Mitchell JR, Zampaglione N, Gillette JR (1974) Bromobenzene-induced liver necrosis. Protective role of glutathione and evidence for 3,4-bromobenzene oxide as the hepatotoxic metabolite. PHA 11:151–169. https://doi.org/10.1159/000136485
Jomova K, Jenisova Z, Feszterova M, Baros S, Liska J, Hudecova D, Rhodes CJ, Valko M (2011) Arsenic: toxicity, oxidative stress and human disease. J Appl Toxicol 31:95–107
Kanter M, Demir H, Karakaya C, Ozbek H (2005) Gastroprotective activity of Nigella sativa L oil and its constituent, thymoquinone against acute alcohol-induced gastric mucosal injury in rats. World J Gastroenterol 11:6662–6666. https://doi.org/10.3748/wjg.v11.i42.6662
Kaplan MM, Utiger RD (1978) Iodothyronine metabolism in rat liver homogenates. J Clin Invest 61:459–471
Kempson SA, Kim JK, Northrup TE, Knox FG, Dousa TP (1979) Alkaline phosphatase in adaptation to low dietary phosphate intake. Am J Physiol Endocrinol Metab 237:E465
Khan N, Jeong IS, Hwang IM, Kim JS, Choi SH, Nho EY, Choi JY, Kwak BM, Ahn JH, Yoon T, Kim KS (2013) Method validation for simultaneous determination of chromium, molybdenum and selenium in infant formulas by ICP-OES and ICP-MS. Food Chem 141:3566–3570
Khundmiri SJ, Asghar M, Khan F, Salim S, Yusufi AN (2004) Effect of ischemia and reperfusion on enzymes of carbohydrate metabolism in rat kidney. J Nephrol 17:377–383
Leslie EM (2012) Arsenic–glutathione conjugate transport by the human multidrug resistance proteins (MRPs/ABCCs). J Inorg Biochem 108:141–149. https://doi.org/10.1016/j.jinorgbio.2011.11.009
Levine RL, Garland D, Oliver CN, et al (1990) [49] Determination of carbonyl content in oxidatively modified proteins. In: Methods in Enzymology. Academic Press, pp 464–478
Liu J, Waalkes MP (2008) Liver is a target of arsenic carcinogenesis. Toxicol Sci 105:24–32
Lu SC (2013) GLUTATHIONE SYNTHESIS. Biochim Biophys Acta 1830:3143–3153. https://doi.org/10.1016/j.bbagen.2012.09.008
Majdalawieh AF, Fayyad MW (2016) Recent advances on the anti-cancer properties of Nigella sativa, a widely used food additive. J Ayurveda Integr Med 7:173–180. https://doi.org/10.1016/j.jaim.2016.07.004
Marí M, Colell A, Morales A, von Montfort C, Garcia-Ruiz C, Fernández-Checa JC (2010) Redox control of liver function in health and disease. Antioxid Redox Signal 12:1295–1331. https://doi.org/10.1089/ars.2009.2634
Marklund S, Marklund G (1974) Involvement of the superoxide anion radical in the autoxidation of pyrogallol and a convenient assay for superoxide dismutase. Eur J Biochem 47:469–474. https://doi.org/10.1111/j.1432-1033.1974.tb03714.x
Mehrandish R, Rahimian A, Shahriary A (2019) Heavy metals detoxification, a review of herbal compounds for chelation therapy in heavy metals toxicity. J Herbmed Pharmacol 8:69–77
Mohamadin AM, Sheikh B, Abd El-Aal AA et al (2010) Protective effects of Nigella sativa oil on propoxur-induced toxicity and oxidative stress in rat brain regions. Pestic Biochem Physiol 98:128–134. https://doi.org/10.1016/j.pestbp.2010.05.011
Mollazadeh H, Hosseinzadeh H (2014) The protective effect of Nigella sativa against liver injury: a review. Iran J Basic Med Sci 17:958–966
Mukwevho E, Ferreira Z, Ayeleso A (2014) Potential role of sulfur-containing antioxidant systems in highly oxidative environments. Molecules 19:19376–19389
Muthumani M, Prabu SM (2012a) Silibinin potentially protects arsenic-induced oxidative hepatic dysfunction in rats. Toxicol Mech Methods 22:277–288. https://doi.org/10.3109/15376516.2011.647113
Muthumani M, Prabu SM (2012) Arsenic induced oxidative stress and its possible reversal by chelation therapy. Research and Reviews: A Journal of Toxicology (RRJoT) 2(2):16–37
Nelson N (1944) A photometric adaptation of the Somogyi method for the determination of glucose. J Biol Chem 153:375–380
Ohkawa H, Ohishi N, Yagi K (1979) Assay for lipid peroxides in animal tissues by thiobarbituric acid reaction. Anal Biochem 95:351–358. https://doi.org/10.1016/0003-2697(79)90738-3
Pace C, Dagda R, Angermann J (2017) Antioxidants protect against arsenic induced mitochondrial cardio-toxicity. Toxics 5:38
Prozialeck WC, Edwards JR, Nebert DW, Woods JM, Barchowsky A, Atchison WD (2008) The vascular system as a target of metal toxicity. Toxicol Sci 102:207–218. https://doi.org/10.1093/toxsci/kfm263
Reitman S, Frankel S (1957) A colorimetric method for the determination of serum glutamic oxalacetic and glutamic pyruvic transaminases. Am J Clin Pathol 28:56–63
Rizwan S, Naqshbandi A, Farooqui Z, Khan AA, Khan F (2014) Protective effect of dietary flaxseed oil on arsenic-induced nephrotoxicity and oxidative damage in rat kidney. Food Chem Toxicol 68:99–107. https://doi.org/10.1016/j.fct.2014.03.011
Robinson PK (2015) Enzymes: principles and biotechnological applications. Essays Biochem 59:1–41
Rubino FM (2015) Toxicity of glutathione-binding metals: a review of targets and mechanisms. Toxics 3:20–62. https://doi.org/10.3390/toxics3010020
Russo GL (2009) Dietary n−6 and n−3 polyunsaturated fatty acids: from biochemistry to clinical implications in cardiovascular prevention. Biochem Pharmacol 77:937–946. https://doi.org/10.1016/j.bcp.2008.10.020
Salem ML (2005) Immunomodulatory and therapeutic properties of the Nigella sativa L. seed. Int Immunopharmacol 5:1749–1770. https://doi.org/10.1016/j.intimp.2005.06.008
Sedlak J, Lindsay RH (1968) Estimation of total, protein-bound, and nonprotein sulfhydryl groups in tissue with Ellman’s reagent. Anal Biochem 25:192–205. https://doi.org/10.1016/0003-2697(68)90092-4
Shahid F, Farooqui Z, Rizwan S, Abidi S, Parwez I, Khan F (2017) Oral administration of Nigella sativa oil ameliorates the effect of cisplatin on brush border membrane enzymes, carbohydrate metabolism and antioxidant system in rat intestine. Exp Toxicol Pathol 69:299–306. https://doi.org/10.1016/j.etp.2017.02.001
Shahid F, Farooqui Z, Khan F (2018) Cisplatin-induced gastrointestinal toxicity: an update on possible mechanisms and on available gastroprotective strategies. Eur J Pharmacol 827:49–57
Singh AP, Goel RK, Kaur T (2011) Mechanisms pertaining to arsenic toxicity. Toxicol Int 18:87
Singh N, Kumar D, Raisuddin S, Sahu AP (2008) Genotoxic effects of arsenic: prevention by functional food-jaggery. Cancer Lett 268:325–330. https://doi.org/10.1016/j.canlet.2008.04.011
Singh NP, McCoy MT, Tice RR, Schneider EL (1988) A simple technique for quantitation of low levels of DNA damage in individual cells. Exp Cell Res 175:184–191. https://doi.org/10.1016/0014-4827(88)90265-0
Sioen I, van Lieshout L, Eilander A, Fleith M, Lohner S, Szommer A, Petisca C, Eussen S, Forsyth S, Calder PC, Campoy C, Mensink RP (2017) Systematic review on n-3 and n-6 polyunsaturated fatty acid intake in European countries in light of the current recommendations-Focus on specific population groups. Ann Nutr Metab 70:39–50
Sumedha NC, Miltonprabu S (2015) Diallyl trisulfide ameliorates arsenic-induced hepatotoxicity by abrogation of oxidative stress, inflammation, and apoptosis in rats. Hum Exp Toxicol 34:506–525
Susan A, Rajendran K, Sathyasivam K, Krishnan UM (2019) An overview of plant-based interventions to ameliorate arsenic toxicity. Biomed Pharmacother 109:838–852
Sung T-C, Huang J-W, Guo H-R (2015) Association between arsenic exposure and diabetes: a meta-analysis. In: BioMed Research International. https://www.hindawi.com/journals/bmri/2015/368087/. Accessed 7 Oct 2019
Tamura T, Stadtman TC (1996) A new selenoprotein from human lung adenocarcinoma cells: purification, properties, and thioredoxin reductase activity. PNAS 93:1006–1011. https://doi.org/10.1073/pnas.93.3.1006
Tseng C-H (2005) Blackfoot disease and arsenic: a never-ending story. J Environ Sci Health C 23:55–74. https://doi.org/10.1081/GNC-200051860
Tsuneki H, Tokai E, Nakamura Y, Takahashi K, Fujita M, Asaoka T, Kon K, Anzawa Y, Wada T, Takasaki I, Kimura K, Inoue H, Yanagisawa M, Sakurai T, Sasaoka T (2015) Hypothalamic orexin prevents hepatic insulin resistance via daily bidirectional regulation of autonomic nervous system in mice. Diabetes 64:459–470
Yaman İ, Balikci E (2010) Protective effects of nigella sativa against gentamicin-induced nephrotoxicity in rats. Exp Toxicol Pathol 62:183–190. https://doi.org/10.1016/j.etp.2009.03.006
Yousef MI, El-Demerdash FM, Radwan FM (2008) Sodium arsenite induced biochemical perturbations in rats: ameliorating effect of curcumin. Food Chem Toxicol 46:3506–3511
Acknowledgements
Financial support to the Department of Biochemistry from the UGC-SAP-DRS III, DST-FIST, and DBT-PURSE programs is gratefully acknowledged. TA is the recipient of a senior research fellowship from DBT (Department of Biotechnology), Government of India. CIL, Central University of Punjab, Bhatinda, is acknowledged for providing the facilities for ICP-MS studies. Department of Chemistry, Aligarh Muslim University, Aligarh, is also acknowledged for providing the facilities for GC-MS studies.
Author information
Authors and Affiliations
Contributions
Conceived and designed the experiments: FK and TA; performed the experiments: TA and ZF; performed the histological experiment: IP and SA; analyzed the data: FK, TA, and SR; wrote the manuscript: TA; revised the manuscript: FK
Corresponding author
Ethics declarations
Ethical approval
Animal experiments were permitted by the Ministry of Environment, Forests and Climate Change, Government of India, under registration no. 714/GO/Re/02/CPCSEA issued by the Committee for the Purpose of Control and Supervision of Experiments on Animals (CPCSEA) dated 29th October 2014 and approved by the Institutional Animal Ethics Committee (IAEC) of the Department of Biochemistry, Faculty of Life Sciences, Aligarh Muslim University, Aligarh, India.
Consent to participate
Not applicable
Consent to publish
Not applicable
Conflict of interest
The authors declare no competing interests.
Additional information
Responsible Editor: Mohamed M. Abdel-Daim
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Alam, T., Rizwan, S., Farooqui, Z. et al. Oral Nigella sativa oil administration alleviates arsenic-induced redox imbalance, DNA damage, and metabolic and histological alterations in rat liver. Environ Sci Pollut Res 28, 41464–41478 (2021). https://doi.org/10.1007/s11356-021-13493-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11356-021-13493-6