
Abstract
Hydrotalcite-like compounds are a group of layered double hydroxides widely studied as sorbents to remove organic and inorganic contaminants under laboratory conditions. This study is a proof-of-concept of the long-term fate of hydrotalcite compounds under natural environmental conditions, to bridge the gap between laboratory studies and their field application as sorbents. Hydrotalcite (HT) with intercalated carbonate species (HT-CO3) and dodecyl sulphate (HT-DS) were synthesised and placed in two groundwater monitoring wells in Denmark, one contaminated with chlorinated hydrocarbons and another with uncontaminated groundwater. To assess the structural and surface compositional changes of hydrotalcite compounds upon prolonged exposure to groundwater, the material was analysed with powder X-ray diffraction (PXRD), Fourier-transformed infrared spectroscopy (FT-IR), X-ray photoelectron spectroscopy (XPS), and scanning electron microscopy (SEM). The results showed that the stability and dissolution behaviour of hydrotalcite compounds under groundwater conditions depended on the intercalated anion (CO32− > DS) and groundwater dynamics (static flow > dynamic flow), while the hydrotalcite aggregate size only had a minor effect. Groundwater geochemistry influenced the precipitation of insoluble species (CaCO3, and adsorbed sulphate) on the hydrotalcite surface. The instability of hydrotalcite compounds, especially in the case of HT-DS, may constitute a significant limiting factor on their future application as sorbents under dynamic flow conditions.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Groundwater pollution is a worldwide problem, with clean-up often being difficult, costly, and disruptive (Reddy 2008). In recent years, the development and application of nanomaterials for in situ groundwater treatment have provided cost-effective solutions, minimising excavation and reducing operational times (Patil et al. 2015). One example of a nanomaterial that has gained interest is the family of layered double hydroxides (LDH), due to their low production cost, abundance of raw materials in nature, and unique properties such as thermal stability, high anionic exchange capacity, and large specific surface area (Rives 2001; Xu et al. 2015). Of specific interest here are LDH with the general formula [Mg1−xAl x(OH)2]Ax/nn−·m H2O (Miyata 1983), better known as hydrotalcite (HT) compounds. HT compounds consist of positively charged hydroxide layers composed of edge-sharing Mg(OH)6 and Al(OH)6 octahedra, separated by hydrated layers with weakly held inorganic or organic anions (e.g., NO3−, Cl−, SO42−, CO3−2, surfactants) for charge balance (Rives 2001). Hydrotalcite intercalated with carbonate ions (HT-CO3) and dodecyl sulphate ions (HT-DS) are among the most investigated HT compounds for water clean-up (Rojas 2012). For example, HT-CO3 shows high sorption capacities towards metals (Gillman 2006; Park et al. 2007; Zubair et al. 2017) and fluorinated compounds (Rattanaoudom et al. 2012; Hu et al. 2017), while HT-DS shows a high sorption affinity towards phenols (Chuang et al. 2008), pesticides (Bruna et al. 2006; Chaara et al. 2011), and chlorinated hydrocarbons (CHCs, (You et al. 2002; Zhao and Nagy 2004; Alonso-de-Linaje et al. 2019)). While the sorption capacity of HT-CO3 and HT-DS towards various pollutants is well known (Ulibarri and Hermosin 2001; Rojas 2012), including the impact of varying geochemistry (Chuang et al. 2008; Hu et al. 2017; Alonso-de-Linaje et al. 2019) and pollutant loading (Gillman 2006; Zubair et al. 2017), little is known however about the stability of these HT compounds during long-term exposure to natural groundwater, and hence their suitability for groundwater remediation applications.
Currently, our knowledge on the parameters that could impact HT stability in groundwater is derived from laboratory dissolution experiments (e.g., Xu et al. 2015; Jobbágy and Regazzoni 2011). In these studies, it has been clearly shown that pH has a strong control on HT stability, i.e., dissolution behaviour. For example, it was shown that HT-CO3 (Xu et al. 2015) and HT with interlayer chloride (HT-Cl; Jobbágy and Regazzoni 2011) fully dissolve at pH 4, while incongruent dissolution of Mg2+ was observed at higher pH (4–10). This was explained by the solutions being supersaturated with respect to amorphous Al(OH)3, but undersaturated with respect to Mg(OH)2, meaning that while both Mg2+ and Al3+ partially dissolved from the HT surface, Al3+ re-precipitated as Al(OH)3 on the HT surface (Jobbágy and Regazzoni 2011; Xu et al. 2015). Other parameters that impact the HT stability, such as surface area and interlayer composition/bonding strength, were highlighted by Parello et al. (2010). They observed lower dissolution rates of copper-doped HT-CO3 compared with the HT with intercalated ibuprofen species (HT-ibu), which they explained by the higher reactive surface area of HT-ibu (i.e., smaller particles that dissolve faster) and weaker interlayer bonds, compared with HT-CO3. However, other key parameters that could potentially affect the HT stability in field applications, such as varying groundwater chemistry and redox conditions, groundwater flow conditions, prolonged exposures, and microbial activity, have not been assessed, to our knowledge. This information is critical as it will have significant practical implications for the ultimate use of HT compounds as sorbents in contaminated groundwater.
Our study assesses the long-term fate (up to 6 months) of HT-CO3 and HT-DS under natural environmental conditions with the main focus on understanding the structural changes to HT compounds. It is hypothesised that prolonged exposures to dynamic groundwater conditions will impact the surface and bulk composition and structure of HT compounds, eventually altering their ideal sorption properties. Specifically, the objectives are as follows: (i) to perform surface and bulk characterisation of groundwater-exposed samples using powder X-ray diffraction (PXRD), Fourier-transform infrared spectroscopy (FT-IR), X-ray photoelectron spectroscopy (XPS), and scanning electron microscopy (SEM); (ii) to identify potential dissolution/precipitation and/or anion-exchange processes acting on HT-CO3 and HT-DS; and (iii) to determine the effect of HT interlayer anion, post-synthesis HT drying, and groundwater conditions on HT stability, and identify potential challenges associated with HT application for groundwater remediation.
Materials and methods
Synthesis of HT
Reagent grade metal nitrates (≥ 98%) (Mg(NO3)2·6H2O and Al(NO3)3·9H2O), Na2CO3 (≥ 99.5%), NaOH (≥ 98%), and the anionic surfactant sodium dodecyl sulphate (C12H25SO4, SDS) (≥ 98.5%) were purchased from Sigma-Aldrich and used without further purification. All glassware was cleaned in 3 M HCl solution overnight, thoroughly rinsed with deionised water (MilliQ water, resistivity > 18 Ω cm), and dried at 75 °C before use. All solutions described below were prepared with MilliQ water. In the case of HT-DS synthesis, solutions were prepared with CO2-free MilliQ, obtained by overnight sparging with N2.
Two HT compounds with a Mg/Al molar ratio of 3:1 were synthesised via the coprecipitation method (Cavani et al. 1991; Clearfield et al. 1991; Newman and Jones 1998) by incorporating carbonate ions (HT-CO3) and dodecyl sulphate ions (HT-DS), respectively. This Mg/Al molar ratio is representative of ratios found in natural HTs collected at Snarum, Norway (Roy et al. 1953).
For the synthesis, 50 mL of the metal cations solution (0.75 M Mg(NO3)2·6H2O and 0.25 M Al(NO3)3·9H2O) was added dropwise (1–2 mL/min) to 100 mL of the interlayer anion solution (0.25 M of Na2CO3 or NaC12H25SO4), under vigorous stirring. The pH was maintained at 9–10 by the addition of 1 M NaOH solution using a titrator system (Metrohohm, Tritando 809). The syntheses were performed at room temperature. During HT-DS synthesis, the mixed solutions were continuously sparged with N2 to minimise CO2 absorption.
After titration, the slurries were aged for another hour, then centrifuged, and the supernatant decanted. The wet paste was washed 4–5 times with MilliQ water until the supernatant was free of NO3− (Mohr salt-H2SO4 test). For most exposure experiments (“HT exposure experiments in uncontaminated and contaminated groundwater wells”), the wet paste was used directly, except for one batch of HT-DS that was oven dried at 75 °C for 48 h. The dried HT-DS was ground using an agate mortar and pestle and stored in plastic tubes until use.
HT exposure experiments in uncontaminated and contaminated groundwater wells
About 200–400 mg of HT solids were placed in dialysis tubing cellulose membrane bags obtained from Sigma-Aldrich (diameter = 10 mm; molecular mass cut-off = 14,000). The dialysis tubing was cut into pieces (6–8 cm length), soaked in MilliQ water overnight, and then filled with HT compounds (solid/liquid ratio of 40–80 g/L) before sealing the ends with nylon rope. Individual dialysis bags were transported inside MilliQ water filled zip-lock bags (kept at 20 ± 0.5 °C) to and from the field site to maintain sample integrity. At the field site, the dialysis bags were attached to a nylon rope, with a lead weight at the bottom-end (Fig. 1) and placed into the saturated zone of two separate wells. At selected times (7 days; 1, 2, 4, and 6 months), individual bags were removed from the line and immediately shipped to the laboratory for characterisation (“HT characterisation”). In the laboratory, the content of the dialysis bag was emptied into a tube, centrifuged, and then washed twice with MilliQ water, before the solids were isolated using 20-μm filter centrifuge tubes. The solids were dried at room temperature inside a glass desiccator.

(a) Scheme and (b) photograph of the field experimental set-up. Each rope held 15 dialysis bags: 5 with wet paste HT-CO3 (represented by red stars), 5 with wet paste HT-DS (represented by light blue stars), and 5 with dried HT-DS (represented by dark blue stars) fixed along a length of ~ 1.5 m
Field site characteristics
The test site was located underneath an old dry-cleaning facility in Skovlunde, Denmark, where uncontrolled release of approximately 2 tons of tetrachloroethylene (PCE) occurred over more than 20 years (Schiefler et al. 2018), leading to severe contamination of the local aquifer (Figure SM-1 and SM-2). The top 6–8 m are composed of a saturated clay layer (perched groundwater level), where the horizontal hydraulic conductivity is minimal, i.e., static flow (~ 10−6 m/s). Underneath lies a well-sorted alluvial sandy aquifer, with a groundwater flow of 2·10−5 m/s (Schiefler et al. 2018). In this study, one rope with HT-filled dialysis bags (Fig. 1) was placed within the heavily contaminated clay layer (well MW-1; 4 m depth) and another set was placed within the uncontaminated aquifer (well MW-2; 17 m depth) (Figure SM-1). The geochemical characteristics and contaminant concentration of the two well waters are given in Table SM-1 and SM-2. Note that pH (7.6 ± 0.2) in MW-1 is slightly higher than pH (7.1 ± 0.2) in MW-2. The HCO3− concentration in MW-1 (642 mg/L) is twice the concentration in MW-2 (316 mg/L), while Zn2+ and SO42− concentrations are higher in MW-2 (160 mg/L and 130 mg/L) than in MW-1 (both 47 mg/L). Experiments were conducted from May 2018 to November 2018.
HT characterisation
Changes in the HT structure were determined by PXRD using a Bruker D8 Discover apparatus (CuKα radiation: λ = 1.5405 Å; 2θ = 2.75–70°; 0.02° step). Powder samples were mounted on a Si wafer, and a Si standard was added to allow for corrections of peak shifts and broadening. PXRD patterns were processed using EVA software. The compositional changes of the HT were evaluated by FT-IR spectroscopy using a Perkin-Elmer Spectrum One FT-IR spectrometer and the KBr pellet technique (sample/KBr mass ratio of 1/150). The morphology and size of the HT was evaluated by SEM (FEI Quanta 3D dual beam), run in low vacuum mode (60 Pa), using 10.0 kV and 23.3 pA, with a low vacuum secondary electron detector. Semi-quantitative element analyses were collected using an Oxford X-Max Energy dispersive X-ray spectroscopy (EDXS) detector. The dry samples were mounted on sticky carbon tape and analysed without coating. XPS was used to determine the HT surface (< 10 nm) element composition and speciation. Note, however, that the basal spacing of HT-CO3 and HT-DS is < 10 nm (Miyata 1983). This means that in our experiments, the X-ray beam and subsequent photoelectrons could potentially penetrate multiple HT layers, providing a bulk-like composition (Coenen et al. 2019). XPS spectra were recorded using a Kratos Axis UltraDLD apparatus with Al monochromatic Kα radiation (1486.6 eV; 150 W, ~ 10−9 Torr chamber pressure) and a charge neutraliser to counterbalance charging effects. High-resolution XPS spectra of the HT constituent elements (Al2p, Mg2s, S2p3/2, C1s, O1s, and Zn2p3/2) were obtained to determine the atomic % (at%) and surface Mg/Al and S/Al ratios. The data were calibrated and fitted using commercial CasaXPS software (version 2.3.12). A Shirley background was subtracted, the spectra were energy calibrated to the hydrocarbon C1s peak at 285.0 eV, and the photoelectron peaks were fit using 30–70% Gaussian-Lorentzian contribution, respectively (Stipp and Hochella 1991; Nesbitt 2002). An Ultima 2 (Jovin Yvon Horiba) induced couple plasma optical emission spectroscopy (ICP-OES) instrument was used to determine the bulk chemical composition (i.e., amount of Mg, Al, and S) in acid-digested solid samples (0.05 g in 100 ml 16.5 M HNO3) of unexposed HT-CO3 and HT-DS. The groundwater chemistry was analysed by an external commercial laboratory (Eurofins Miljø, Denmark).
Results and discussion
HT-CO3 (inorganic-HT)
HT-CO3 material before exposure to groundwater
The PXRD pattern of the initial unexposed HT-CO3 material (Fig. 2a) exhibited the characteristic reflections of a well-crystallised HT with symmetric and consecutive reflections at 2θ = 11.41° (003), 22.82° (006), 34.61° (009), a d(003) basal spacing of 7.74 Å, and a d(110) spacing of 1.53 Å, consistent with previous studies on 3:1 HT-CO3 (Miyata 1983; Drits and Bookin 2011). The FT-IR results showed the characteristic absorption bands expected for HT-CO3 (Fig. 2b). We have given the same band assignments as found in Kloprogge et al. (2002), with the structural hydroxyl stretching mode at ~ 3500 cm−1, OH stretching mode of interlayer H2O-CO3 bridging at 3000–3100 cm−1, H2O bending mode at 1635 cm−1, ʋ2 and ʋ3 CO3 modes at 857 cm−1 and 1365 cm−1, and Mg-O/Al-O lattice vibrations between 800 and 500 cm−1 (Fig. 2b). The high-resolution XPS Al2p, Mg2s, and O1s spectra showed peak maxima at B.E. 74.4 eV, 88.7 eV, and 531.9 eV, respectively (Figure SM-3, SM-4), consistent with previous XPS studies on HT compounds (Table SM-3) (Dupin et al. 2004). The O1s peak could be divided into two components at 531.5 and 532.5 eV, which correspond to carbonate interlayer species and hydroxyl species from the hydroxide sheets (i.e., Mg(OH)2 and Al(OH)3 groups, Figure SM-4e,pre) (Sommer 1975; Dupin et al. 2004). The presence of carbonate interlayer species was also seen in the C1s spectra at B.E. 289.2 eV (Table SM-4). The bulk chemical composition calculated from ICP-OES showed a Mg/Al molar ratio of 3:1 (Table SM-5) which is very similar to the surface Mg/Al ratio derived from XPS (Table SM-3, Fig. SM-3). SEM analyses of unexposed HT-CO3 showed dense particle aggregates, where individual HT particles were difficult to distinguish (Fig. 3a).

(a) PXRD and (b) FT-IR of initial, unexposed HT-CO3. In PXRD patterns, peaks are labelled with Miller indices, and symbol ( ) denotes reflections from the added silicon standard
) denotes reflections from the added silicon standard

SEM images of initial, unexposed and exposed HT samples. (a) Initial HT-CO3; (b) HT-CO3 after 4 months in well MW-1 and (c) well MW-2; (d) initial HT-DS; (e) HT-DS after 4 months in well MW-1; (f) detail of two aggregates of HT-DS after 4 months in well MW-2; yellow arrows point the location of Fe-rich precipitates found between HT-DS aggregates. Unfortunately, the 6-month MW-2-exposed HT-CO3 could not be recovered
HT-CO3 exposure to contaminated groundwater well; MW-1
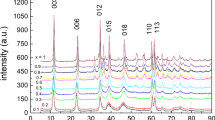
The PXRD patterns of all HT-CO3 samples retrieved from well MW-1 during the 6-month exposure showed very similar peak locations compared with the unexposed, initial material (“HT-CO3 material before exposure to groundwater”) (Fig. 4a, Table SM-6). For instance, the positions of reflections (003) and (110) did not change significantly (σ = 0.023 Å) indicating that the HT-CO3 interlayer composition and Mg/Al molar ratio did not change (Cavani et al. 1991). Noteworthy that the HT-CO3 peak intensities increased with exposure time (Fig. 4a), particularly after 1-month exposure. A change in PXRD peak intensity can be related to many factors (Waseda et al. 2011). Here we hypothesise that the increase in crystallinity observed is attributed to HT-CO3 crystallinity increased with prolonged. All samples, but particularly the 7-day and 1-month exposed samples, exhibited additional new peaks (Fig. 4a), characteristic of calcite (CaCO3) (Kontoyannis and Vagenas 2000).

HT-CO3 before (initial) and after exposure to contaminated groundwater in MW-1 (the colour gradient moves from green to blue with increased exposure time): (a) PXRD patterns, (b) FT-IR spectra, and (c) XPS Mg/Al ratios. HT-CO3 before (initial) and after exposure to uncontaminated groundwater in MW-2: (d) PXRD patterns, (e) FT-IR spectra, (f) XPS Mg/Al ratios. In the PXRD patterns (a, c), peaks are labelled with Miller indices, and symbols (▲) and ( ) denote reflections for calcite and the added silicon standard. In graph (f), (♦) links to Mg/Al axis and (
) denote reflections for calcite and the added silicon standard. In graph (f), (♦) links to Mg/Al axis and ( ) to Al/S axis. Unfortunately, the 6-month MW-2 HT-CO3 sample could not be recovered
) to Al/S axis. Unfortunately, the 6-month MW-2 HT-CO3 sample could not be recovered
Similarly to PXRD, FT-IR spectra did not show much changes upon increased exposure to MW-1 (Fig. 4b). Only the sample retrieved after 7 days showed a minor new absorption band at 1114 cm−1, which could indicate the presence of a minor amount of sulphate (Kloprogge et al. 2002). The presence of calcite is difficult to identify in FT-IR spectra because it induces similar CO3 vibrations as observed for the HT interlayer carbonate (Rodriguez-Blanco et al. 2011).
In terms of compositional surface changes, XPS data showed that the Mg/Al ratio decreased initially with increasing exposure time, to about 2.4 for the 1-month sample but then remained relatively unchanged for the 2-, 4-, and 6-month exposed samples (Fig. 4c, Table SM-3). Seeing that bulk Mg/Al ratios of exposed samples were likely still close to 3:1, as indicated by the unchanged lattice parameter a in the PXRD (Table SM-6), this decrease in surface Mg/Al ratio could indicate an increase in surface Al (Table SM-3). This in turn can be explained by some HT surface dissolution, releasing both Mg2+ and Al3+, and precipitation of released Al3+ as Al(OH)3 on HT surfaces, because Al3+ solubility is extremely low at near neutral pH (< 0.004 mM at MW-1 pH of 7.6 ± 0.2, Table SM-1) (Ksp [Al(OH)3] = 10−27 vs Ksp [Mg(OH)2] = 10−11 in water at 25 °C). A similar process has been argued before in HT-CO3 and HT-Cl dissolution experiments at pHs between 4 and 10 (Jobbágy and Regazzoni 2011; Xu et al. 2015). The observation that the HT-CO3 surface Mg/Al ratio decreased only within the first month of exposure but then remained fairly constant, indicates that a somewhat steady-state was reached, i.e., no further dissolution occurred. This could be due to the Al(OH)3 surface precipitates preventing further HT dissolution as proposed by Jobbágy and Regazzoni (2011) and/or represent a steady state in the well, as the water flow was almost static in the clay layer (i.e., very low hydraulic conductivity). Establishment of such equilibrium conditions in MW-1 could also explain the enhanced HT-CO3 crystallinity observed in PXRD after 1-month exposure (Fig. 4a), allowing for the material to age, i.e., recrystallise. A dissolution-precipitation process would give rise to crystallites with less defects and an improved crystallinity, thus accounting for the intensity increase observed. In contrast to PXRD, calcite was not observed in XPS (i.e., Ca 2p3/2 from CaCO3, B.E = 347.7 ± 0.15 eV) (Stipp and Hochella 1991). This is most likely because the area selected for XPS (~ 700 × 300 μm) did not contain any calcite crystals. XPS results did however reveal the presence of Zn2+ (B.E = 1022.0 ± 0.3 eV, Table SM-3, Figure SM-4), likely in the form of Zn(OH)2 (B.E. = 1021.8 ± 0.2 eV; Deroubaix and Marcus 1992). From the similarity in ionic radii between Zn2+ and Mg2+ (0.88 Å vs 0.86 Å), this could indicate some Zn2+ incorporation and/or sorption onto HT-CO3 surfaces during exposure to MW-1 water. Overall, however, Zn2+ concentrations were very low (~ 0.1 at%), and this likely did not affect HT-CO3 surface properties.
SEM of HT-CO3 retrieved after 4-month exposure to MW-1 showed HT aggregates that were somewhat more rounded compared with the unexposed HT-CO3 (Fig. 3b). This could be due to surface dissolution at HT surfaces, as also suggested by XPS.
Overall, these characterisation results indicate that the HT-CO3 structure remained stable, which is explained by the high affinity of the carbonate ions for the HT interlayer; i.e., carbonate can outcompete most other anions (Rives 2001; Eiby et al. 2016). The comparison of PXRD, FTIR, and XPS results looking for structural changes from HT-CO3 before and after exposure to contaminants (i.e., CHCs and BTEX) in MW-1 showed no concrete evidence of structural degradation. Moreover, the static flow conditions in MW-1 limited dissolution processes, allowing for a steady state to establish after a month.
HT-CO3 exposure to uncontaminated groundwater well; MW-2
The structure of HT-CO3, as probed by PXRD and FT-IR, did not change over the 6-month exposure to the uncontaminated groundwater in MW-2 well (Fig. 4d, e). Calcite was detected in HT-CO3 samples, and FT-IR signals from sulphate were more pronounced when compared with MW-1-exposed HT-CO3 samples (Fig. 4a). Specifically, small FT-IR absorption bands were observed at ~ 980 cm−1, ~1114 cm−1, and ~ 613 cm−1, which are consistent with the ν1, ν3, and ν4 modes of sulphate (Kloprogge and Frost 2001; Kloprogge et al. 2002) (Fig. 4e). Also high-resolution XPS S2p3/2 spectra showed a peak at B.E. 169.9 ± 0.4 eV characteristic of sulphate (B.E. = 169.5 ± 0.2 eV; Peisert et al. 1994) (Table SM-3, Figure SM-4). The presence of sulphate is best explained by adsorption onto HT-CO3 surfaces because there is no indication of sulphate intercalation; i.e., the basal spacing remained unchanged (d003 of HT-SO4 is 8.2 Å compared with 7.78 Å for HT-CO3, Miyata 1983; Table SM-6). The enhanced adsorption of sulphate onto HT-CO3 samples in MW-2 is maybe not unexpected seeing the SO42− concentration in MW-2 well was about three times higher than in MW-1 well (Table SM-1).
The XPS Mg/Al surface ratio of the MW-2-exposed HT-CO3 samples steadily decreased with increased exposure reaching a value close to 1.5 after 4 months (Fig. 4f, Table SM-3). This decrease suggests that the extent of HT-CO3 surface dissolution and consequent surface Al(OH)3 precipitation was higher under MW-2 conditions compared with MW-1. This may be best explained by the slightly lower pH in MW-2 (pH = 7.1 ± 0.2) as well as the dynamic flow conditions in the sandy aquifer, leading to continuous HT-CO3 dissolution. As a result, no equilibrium could be reached between the HT-CO3 surface and the continuously exchanging MW-2 groundwater. SEM images of the 4-month MW-2-exposed HT-CO3 sample (Fig. 3c) showed rounded particle aggregates, very similar to what was observed for the 4-month MW-1-exposed HT-CO3 sample (Fig. 3b).
HT-DS (Organo-HT)
HT-DS material before exposure to groundwater
The PXRD pattern of the initial, unexposed HT with intercalated dodecyl sulphate (HT-DS) showed a crystalline structure with symmetric and consecutive reflections at 2θ = 3.29° (003), 6.69° (006), 10.35° (009), a d(003) (interlayer) spacing of 26.86 Å, and a d(110) spacing of 1.53 Å (Fig. 4a), in agreement with previous studies on HT-DS (Clearfield et al. 1991; Alonso-de-Linaje et al. 2019). The FT-IR showed characteristic HT-DS absorption bands: metal-oxide-metal lattice vibration modes at 800–500 cm−1, S=O antisymmetric and symmetric stretching at 1220 and 1065 cm−1, C–H stretching at ~ 3000 cm−1, and C–H bending modes at ~ 1469 cm−1 (Fig. 5b) (Bruna et al. 2006).

(a) PXRD pattern and (b) FT-IR spectra of initial, unexposed HT-DS before. In the PXRD pattern (a), peaks are labelled with Miller indices, and symbol (♦) denotes the reflections for the added silicon standard
The XPS widecan of HT-DS displayed peaks with binding energies consistent with Al2p (75.2, eV), Mg2s (89.4, eV), S2p3/2 (168.8 eV), C1s (285 eV), and O1s (532.2 eV) (Table SM-7, Figure SM-5) (Lin et al. 2018). The high-resolution XPS O1s spectra showed binding energies characteristic of hydroxide species in the HT hydroxide layer, while the S2p3/2 spectra showed the presence of sulphate species from intercalated DS (Table SM-7) (Moulder et al. 1995). The surface Mg/Al molar ratio was 3:1 (Table SM-7), identical to the Mg/Al ratio determined from bulk chemical analysis (Table SM-5). SEM images from unexposed HT-DS showed strongly aggregated HT-DS particles, similar to the unexposed HT-CO3 (“HT-CO3 material before exposure to groundwater”) where individual particles are difficult to distinguish (Fig. 3d).
HT-DS exposure to contaminated groundwater well; MW-1
Overall, the PXRD and FTIR of all HT-DS samples (added as wet paste to dialysis bags) retrieved from MW-1 during the 6-month period showed almost identical patterns compared with the unexposed, initial HT-DS (Fig. 6a, b). These results indicate that the general bulk structure remained unaltered during exposure, and the DS remained in the interlayer. Upon further analysis of the PXRD patterns, some small changes in the basal spacing and crystal lattice parameters were observed (Table SM-8), but they did not seem to correlate with exposure time and generally deviated by less than 5% from values obtained from the unexposed, initial HT-DS. A slight decrease in HT-DS basal spacing (i.e., lattice parameter c) can occur if the interlayer water content decreases and/or the DS interlayer arrangement changed (i.e., tilt of the surfactants molecules with respect to the HT layers) (Table SM-8) (Kopka et al. 1988). In terms of the changing lattice parameter a, small decreases could occur due to slight decreases in the bulk Mg/Al molar ratio. The ionic radius of Mg2+ (0.86 Å) is slightly larger than that of Al3+ (0.67 Å) (Shannon 1976), so a reduction between cation-cation distance could result in a decrease of the a constant (Pausch et al. 1986).

HT-DS before (initial) and after exposure to the contaminated groundwater in MW-1 (the colour gradient moves from green to blue with increased exposure time): (a) PXRD pattern, (b) FT-IR spectra, and (c) XPS Mg/Al ratios. HT-DS before (initial) and after exposure to the uncontaminated groundwater in MW-2: (d) PXRD patterns, (e) FT-IR spectra, and (f) XPS Mg/Al ratios. The high-intensity PXRD peaks observed at low 2θ angles (0–8°) for samples after 2-month exposure in MW-2 were related to a low signal-to-noise ratio in this area of the PXRD pattern. In the PXRD patterns (a, c) peaks are labelled with Miller indices and symbol ( ) denotes reflections of the added silicon standard. In graphs (c) and (f), (♦) links to Mg/Al axis and (
) denotes reflections of the added silicon standard. In graphs (c) and (f), (♦) links to Mg/Al axis and ( ) to Al/S axis
) to Al/S axis
In terms of HT-DS surface compositional changes during exposure to MW-1, XPS showed that the surface Mg/Al and S/Al molar ratios initially decreased and then stabilised at around 1.8 and 0.5, respectively, after 2 months (Fig. 6c Table SM-7). The decrease in these two ratios is caused by surface dissolution of HT-DS and the subsequent precipitation of Al(OH)3. The somewhat steady-state after 2 months is likely due to the almost static flow conditions in MW-1, similar to observations for MW-1-exposed HT-CO3 samples. In XPS, some minor Zn2+ was also detected on HT-DS surfaces after 2-month exposure, similar to MW-1-exposed HT-CO3. Again, this could indicate Zn2+ incorporation and/or adsorption onto HT-DS surfaces (Table SM-7, Figure SM-5). Similar to HT-CO3 PXRD, FTIR, and XPS analysis, results from HT-DS did not show any evidence of structural changes related to the presence of contaminants after exposure to MW-1. Compared with the initial HT-DS (Fig. 3d), SEM images of 4-month MW-1-exposed HT-DS samples (Fig. 3e) showed no significant variation in HT-DS aggregate size and morphology.
HT-DS exposure to uncontaminated groundwater well; MW-2
PXRD of HT-DS samples exposed to the uncontaminated groundwater in MW-2 showed clear changes in the bulk structure with increasing exposure time. The cell lattice parameter a gradually decreased from 3.051 to 3.012 over the first 2 months of exposure (Table SM-8). After 4-month exposure the HT-DS samples lost all characteristic HT PXRD reflections (i.e., (003), (006), and (110); Fig. 6d). These results indicate gradual structural changes, likely due to continuous dissolution processes under the dynamic flow conditions in MW-2, which eventually led to a total loss of the HT-DS crystalline structure and a transformation into an amorphous solid in the time between the 2- and 4-month exposure. For comparison, Pausch et al. (1986) reported that the HT structure loses stability once the cell parameter a has decreased to a value of 3.040 (Figure SM-6). Similarly, Misra and Perrota (1992) reported that HT phases require a minimal a value of 3.038. Here, we observed that HT-DS exposed to the MW-2 groundwater exhibited a values well below 3.040 already after 1-week exposure; thus, the HT-structure likely already became unstable early on (Fig. 6d and Table SM-8).
This gradual structural loss is also shown by the FT-IR data. Specifically, a progressive decrease in major FT-IR band intensities was observed with increasing exposure time, and ultimately the loss of specific bands (e.g., C–H bend at 1469 cm−1 and S=O antisymmetric and symmetric stretching at 1220 and 1065 cm−1) after 4-month exposure to MW-2 (Fig. 6e).
The XPS data showed a steady decrease in the surface Mg/Al and S/Al molar ratios with increasing exposure time, yielding values of 0.8 and 0.1, respectively, after 4 months (Fig. 6f, Table SM-7), reaffirming the continuous loss of HT-DS structure as observed with PXRD and FTIR. XPS further showed the presence of some silicates or hydrated silicates (e.g., magnesium silicates) (Table SM-7, Figure SM-5) (NIST, 2000) on HT-DS surfaces after 2-month exposure. Compared with unexposed HT-DS (Fig. 3d), SEM images of the 6-month MW-2-exposed HT-DS sample showed more compacted aggregates where individual particles were even more difficult to spot (Fig. 3f). EDXS mapping further revealed the presence of some Fe-rich precipitates in between HT-DS aggregates (yellow arrows in Fig. 3f, Fig. SM-7), but these compounds were not detected with any of the other techniques.
Effect of post-synthesis drying treatment on HT-DS
Our recent study showed that drying of HT-DS following synthesis (oven dried at 75 °C for 48 h) enhanced the formation of large aggregates, which in turn reduced its sorption capacity towards CHC (Alonso-de-Linaje et al. 2019). We also study the effect of post-synthesis drying on HT-DS stability under aquifer conditions here. The PXRD, FT-IR, and XPS results showed that HT-DS stability was not affected by post-synthesis treatment because trends for both HT-DS types (whether added as wet paste or dry solid) with exposure time were almost identical (wet paste: Fig. 6, Table SM-7, SM-8; dry solid: Figure SM-8, Table SM-9, SM-10). The surface of both HT-DS samples reached stability after 2 months of exposure to MW-1, while faster dissolution rates were observed in MW-2, with the eventual loss of the HT-DS structure after 2 months of exposure. This implies that the size of HT-DS aggregates has a minor effect on the dissolution behaviour of HT-DS surfaces, while other geochemical factors (i.e., pH, water chemistry, groundwater flow) play a more substantial role.
Comparison of the long-term fate of HT-CO3 vs HT-DS under natural environmental conditions
For the first time, the long-term stability of HT-CO3 and HT-DS was tested under real aquifer conditions, where MW-1 was heavily contaminated with chlorinated hydrocarbons while MW-2 was not. Results showed that the presence of chlorinated hydrocarbons and BTEX did not significantly affect the stability of HT compounds in MW-1. On the other hand, aquifer flow conditions and the type of intercalated anion affected HT stability in these settings. Under almost static groundwater conditions (MW-1), both HT-CO3 and HT-DS partly dissolved over the first 2 months, and then stabilised. Overall, HT-DS dissolved more than HT-CO3 as indicated by the lower surface Mg/Al ratio. When exposed to dynamic groundwater flow conditions (MW-2), HT-CO3 and HT-DS dissolution were dramatically enhanced and did not stabilise over the monitored 6-month period. This ultimately led to complete disintegration of HT-DS into a new amorphous phase between months 2 and 4, while HT-CO3 was able to retain its primary structure up until month 6. The enhanced stability of HT-CO3 is explained by the higher affinity of CO32− (i.e., cation binding strength) towards the interlayer (Parello et al. 2010). The progressive dissolution of surface exposed hydroxide layers of HT-CO3 and HT-DS was enabled by the protonation of surface hydroxyl groups as observed in previous laboratory studies (Parello et al. 2010; Xu et al. 2015). Overall, our results indicate that groundwater dynamics and geochemistry influenced HT long-term fate, while the presence of chlorinated hydrocarbons and BTEX or aggregate size has a minor effect on the HT stability.
Perspectives on the HT suitability for remediation applications
So far, only a few studies have focused on the stability of HT compounds in aqueous solutions, but these were all based on laboratory dissolution experiments (Parello et al. 2010; Jobbágy and Regazzoni 2011; Xu et al. 2015) and ignored key environmental factors like groundwater flow. Most groundwater remediation systems are subjected to dynamic water flow, and static flow is only observed under very particular conditions (i.e., in very low hydraulic conductivity media). For instance, groundwater is constantly passing through in situ remediation systems such as permeable reactive barriers, while for ex situ systems (e.g., pump and treat) or wastewater treatment plants, a constant flow is needed to pump the contaminated groundwater into the treatment facility.
From a remediation perspective, the instability of HT compounds especially under dynamic flow (i.e., high hydraulic conductivity) constitutes an important limiting factor. This consideration is particularly important in the case of HT-DS, while HT-CO3 displayed enhanced stability even under dynamic flow conditions. With time, the partial dissolution of HT-CO3 and HT-DS particles might release the previously sorbed contaminants, irrespective of the sorption mechanism (e.g., anion intercalation or surface adsorption). Additionally, surface adsorbates (i.e., Al(OH)3, adsorbed sulphate and Zn2+ species, or CaCO3 crystals) might act as a protective layer, reducing the contact between groundwater and the sorbent, thus reducing sorption capacity, something we observed in MW-1. A further point to consider is biological activity which could also affect the long-term fate of HT, especially in the case of HT-DS, as anaerobic biodegradation of DS by denitrifying bacteria was previously recognised (Paulo et al. 2013). This could not be assessed here due to the experimental design (i.e., sealed dialysis bags prevented contact between bacteria and HT compounds), but should be investigated in future studies.
The current study demonstrated that exposure to dynamic aquifer conditions clearly compromised HT stability, even for the most stable HT, i.e., HT-CO3. This proves the need for further HT stability analyses under real groundwater conditions as stability is clearly affected by groundwater flow conditions and HT type.
Conclusions
This study provides, for the first time, experimental data and analysis on the long-term fate (6 months) of synthetic HT-CO3 and HT-DS (oven dried and wet paste) under natural groundwater conditions. Various characterisation techniques were combined to bridge the gap between laboratory observations and field applications. The experiments were conducted at a site in Denmark, deploying HT compounds in two wells, one with CHC contaminated groundwater and one well with uncontaminated groundwater. The stability of HT compounds in groundwater is primarily affected by intercalated anion (CO3 > DS) and groundwater dynamics (static flow > dynamic flow). Dissolution of HT-CO3 and HT-DS exposed to groundwater occurred at the solid-liquid interface affecting particle surface and progressively dissolving the HT aggregates if steady-state is not reached. Post-synthesis drying treatment of the HT-DS (wet vs oven dried) did not significantly affect the dissolution kinetics.
References
Alonso-de-Linaje V, Mangayayam MC, Tobler DJ et al (2019) Sorption of chlorinated hydrocarbons from synthetic and natural groundwater by organo-hydrotalcites : towards their applications as remediation nanoparticles. Chemosphere In press. https://doi.org/10.1016/j.chemosphere.2019.124369
Bruna F, Pavlovic I, Barriga C et al (2006) Adsorption of pesticides carbetamide and metamitron on organohydrotalcite. Appl Clay Sci 33:116–124. https://doi.org/10.1016/j.clay.2006.04.004
Cavani F, Trifiró F, Vaccari A (1991) Hydrotalcite-type anionic clays: preparation, properties and applications. Catal Today 11:173–301. https://doi.org/10.1016/0920-5861(91)80068-K
Chaara D, Bruna F, Ulibarri MA, Draoui K, Barriga C, Pavlovic I (2011) Organo/layered double hydroxide nanohybrids used to remove non ionic pesticides. J Hazard Mater 196:350–359. https://doi.org/10.1016/j.jhazmat.2011.09.034
Chuang YH, Tzou YM, Wang MK et al (2008) Removal of 2-chlorophenol from aqueous solution by Mg/Al layered double hydroxide (LDH) and modified LDH. Ind Eng Chem Res 47:3813–3819. https://doi.org/10.1021/ie071508e
Clearfield A, Kieke M, Kwan J et al (1991) Intercalation of dodecyl sulfate into layered double hydroxides. J Incl Phenom Mol Recognit Chem 11:361–378. https://doi.org/10.1007/BF01041414
Coenen K, Gallucci F, Mezari B et al (2019) Investigating the role of the different metals in hydrotalcite Mg / Al-based adsorbents and their interaction with acidic sorbate species. Chem Eng Sci 200:138–146. https://doi.org/10.1016/j.ces.2019.01.046
Deroubaix G, Marcus P (1992) X-ray photoelectron spectroscopy analysis of copper and zinc oxides and sulphides. Surf Interface Anal 18:39–46. https://doi.org/10.1002/sia.740180107
Drits VA, Bookin A (2011) Crystral structure and X-ray indentification of layared double hydroxides. In: Rives V (ed) Layered double hydroxides: present and future. Nova Science Publishers, Inc., New York, pp 41–100
Dupin JC, Martinez H, Guimon C et al (2004) Intercalation compounds of Mg-Al layered double hydroxides with dichlophenac: different methods of preparation and physico-chemical characterization. Appl Clay Sci 27:95–106. https://doi.org/10.1016/j.clay.2004.03.001
Eiby SHJ, Tobler DJ, Nedel S et al (2016) Competition between chloride and sulphate during the reformation of calcined hydrotalcite. Appl Clay Sci 132–133:650–659. https://doi.org/10.1016/j.clay.2016.08.017
Gillman GP (2006) A simple technology for arsenic removal from drinking water using hydrotalcite. Sci Total Environ 366:926–931. https://doi.org/10.1016/j.scitotenv.2006.01.036
Hu Z, Song X, Wei C, Liu J (2017) Behavior and mechanisms for sorptive removal of perfluorooctane sulfonate by layered double hydroxides. Chemosphere 187:196–205. https://doi.org/10.1016/j.chemosphere.2017.08.082
Jobbágy M, Regazzoni AE (2011) Dissolution of nano-size Mg – Al – Cl hydrotalcite in aqueous media. Appl Clay Sci 51:366–369. https://doi.org/10.1016/j.clay.2010.11.027
Kloprogge JT, Frost RL (2001) Infrared and Raman spectroscopic studies of layered double hydroxides (LDHS). In: Rives V (ed) Layered double hydroxides: present and future. Nova Science Publishers, Inc., New York, pp 153–215
Kloprogge JT, Wharton D, Hickel L, Frost RL (2002) Infrared and Raman study of interlayer anions CO3 2– , NO3 – , SO4 2– and ClO4 – in Mg/Al- hydrotalcite. Am Mineral 87:623–629. https://doi.org/10.2138/am-2002-5-604
Kontoyannis CG, Vagenas NV (2000) Calcium carbonate phase analysis using XRD and FT-Raman spectroscopy. Analyst 125:251–255. https://doi.org/10.1039/a908609i
Kopka H, Beneke K, Lagaly G (1988) Anionic surfactants between double metal hydroxide layers. J Colloid Interface Sci 123:427–436. https://doi.org/10.1016/0021-9797(88)90263-9
Lin ST, Tran HN, Chao HP, Lee JF (2018) Layered double hydroxides intercalated with sulfur-containing organic solutes for efficient removal of cationic and oxyanionic metal ions. Appl Clay Sci 162:443–453. https://doi.org/10.1016/j.clay.2018.06.011
Misra C, Perrota J (1992) Compositon and properties of synthetic hydrotalcites. Clay Clay Miner 40:145–150. https://doi.org/10.1346/CCMN.1992.0400202
Miyata S (1983) Anion-exchange properties of hydrotalcite-like compounds. Clay Clay Miner 31:305–311. https://doi.org/10.1346/CCMN.1983.0310409
Moulder JF, Stickle WF, Sobol PE et al (eds) (1995) Handbook of X-ray photoelectron spectroscopy. Perkin-Elmer Corporation, Minnesota
Nesbitt HW (2002) Chapter 6: Interpretation of X-ray photoelectron spectra with applications to mineralogy and geochemistry. In: Hederson GS, Baker DS (eds) Synchrotron radiation, earth, Evironmental and materials science applications. Mineral Association of Canada Short Course 30, pp 131–158
Newman SP, Jones W (1998) Synthesis, characterization and applications of layered double hydroxides containing organic guests. New J Chem 22:105–115. https://doi.org/10.1039/a708319j
Parello ML, Rojas R, Giacomelli CE (2010) Dissolution kinetics and mechanism of Mg-Al layered double hydroxides: a simple approach to describe drug release in acid media. J Colloid Interface Sci 351:134–139. https://doi.org/10.1016/j.jcis.2010.07.053
Park M, Choi CL, Seo YJ et al (2007) Reactions of Cu 2+ and Pb 2+ with Mg/Al layered double hydroxide. Appl Clay Sci 37:143–148. https://doi.org/10.1016/j.clay.2006.12.006
Patil SS, Shedbalkar UU, Truskewycz A et al (2015) Nanoparticles for environmental clean-up: a review of potential risks and emerging solutions. Environ Technol Innov 5:10–21. https://doi.org/10.1016/j.eti.2015.11.001
Paulo AMSMS, Plugge CMM, García-Encina PAA, Stams AJMJM (2013) Anaerobic degradation of sodium dodecyl sulfate (SDS) by denitrifying bacteria. Int Biodeterior Biodegrad 84:14–20. https://doi.org/10.1016/j.ibiod.2013.05.027
Pausch I, Lohse H-H, Schürmann K, Allmann R (1986) Synthesis of disordered and Al-rich hydrotalcite-like compounds. Clay Clay Miner 34:507–510. https://doi.org/10.1346/CCMN.1986.0340502
Peisert H, Chassé T, Streubel P et al (1994) Relaxation energies in XPS and XAES of solid sulfur compounds. J Electron Spectros Relat Phenomena 68:321–328. https://doi.org/10.1016/0368-2048(94)02129-5
Rattanaoudom R, Visvanathan C, Boontanon SK (2012) Removal of concentrated PFOS and PFOA in synthetic industrial wastewater by powder activated carbon and hydrotalcite. J Water Sustain 2:245–258. https://doi.org/10.11912/jws.2.4.245-258
Reddy KR (2008) Physical and chemical groundwater remediation technologies. In: Darnault CJG (ed) Overexploitation and contamination of shared groundwater resources. Springer, Dordrecht, pp 257–288
Rives V (2001) Layered double hydroxides: present and future. Nova Science Publishers, Inc., New York
Rodriguez-Blanco JD, Shaw S, Benning LG (2011) The kinetics and mechanisms of amorphous calcium carbonate (ACC) crystallization to calcite, via vaterite. Nanoscale 3:265–271. https://doi.org/10.1039/c0nr00589d
Rojas R (2012) Layered double hydroxides application as sorbents for environmental remediation. In: Calixto Carillo A, Analiz Griego D (eds) Hydroxides: synthesis, Types and Applications. Nova Science Publishers, Inc., pp 39–71
Roy DM, Roy R, Osborn EF (1953) The system MgO-Al2O3-H2O and influence of carbonate and nitrate anions on the phase equilibria. Am J Sci 251:337–361
Schiefler AA, Tobler DJ, Overheu ND, Tuxen N (2018) Extent of natural attenuation of chlorinated ethenes at a contaminated site in Denmark. Energy Procedia 146:188–193. https://doi.org/10.1016/j.egypro.2018.07.024
Shannon RD (1976) Revised effective ionic radii and systematic studies of interatomic distances in halides and chalcogenides. Acta Crystallogr Sect A 32:751–767. https://doi.org/10.1107/S0567739476001551
Sommer S (1975) X-ray photoelectron spectra of C1S and O1S in carbonate minerals. Am Mineral 60:483–484
Stipp SL, Hochella MF (1991) Structure and bonding environments at the calcite surface as observed with X-ray photoelectron spectroscopy (XPS) and low energy electron diffraction (LEED). Geochim Cosmochim Acta 55:1723–1736. https://doi.org/10.1016/0016-7037(91)90142-R
Ulibarri MA, Hermosin MC (2001) Layered double hydroxides in water decontamination. In: Rives V (ed) Layered double hydroxides: present and future. Nova Science Publishers, Inc., New York, pp 251–284
Waseda Y, Matsubara E, Shinoda K (2011) X-ray diffraction crystalography. Springer, Berlin Heidelberg
Xu S, Liao MC, Zeng HY et al (2015) Surface modification and dissolution behavior of Mg-Al hydrotalcite particles. J Taiwan Inst Chem Eng 56:174–180. https://doi.org/10.1016/j.jtice.2015.04.013
You Y, Zhao H, Vance GF (2002) Surfactant-enhanced adsorption of organic compounds by layered double hydroxides. Colloids Surf A Physicochem Eng Asp 205:161–172. https://doi.org/10.1016/S0927-7757(01)01137-2
Zhao H, Nagy KL (2004) Dodecyl sulfate-hydrotalcite nanocomposites for trapping chlorinated organic pollutants in water. J Colloid Interface Sci 274:613–624. https://doi.org/10.1016/j.jcis.2004.03.055
Zubair M, Daud M, McKay G et al (2017) Recent progress in layered double hydroxides (LDH)-containing hybrids as adsorbents for water remediation. Appl Clay Sci 143:279–292. https://doi.org/10.1016/j.clay.2017.04.002
Acknowledgements
The authors would like to thank Markus Reischer (Niras, Denmark), Adrian Schiefler (Capital Region of Denmark), Clara Ribagorda (AECOM, Spain), and Arantxa Martinez (AECOM, Spain) for their help in the field. Marco C. Mangayayam and Denis Okhrimenko (University of Copenhagen, Denmark) are acknowledged for their help with XPS data collection.
Funding
This study was funded by Metal-Aid Innovative Training Network (ITN), supported by a grant from the European Commission’s Marie Skɫowdowska Curie Actions program (No. 675219).
Author information
Authors and Affiliations
Corresponding author
Additional information
Responsible Editor: Tito Roberto Cadaval Jr
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
ESM 1
(DOCX 2020 kb).
Rights and permissions
About this article

Cite this article
Alonso-de-Linaje, V., Tobler, D.J., Espinosa, R. et al. Hydrotalcite stability during long-term exposure to natural environmental conditions. Environ Sci Pollut Res 27, 23801–23811 (2020). https://doi.org/10.1007/s11356-020-08460-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11356-020-08460-6