
Abstract
In this study, a new magnetic adsorbent based on magnetite-sporopollenin/graphene oxide (Fe3O4-SP/GO) was successfully developed. The adsorbent was applied for magnetic solid phase extraction (MSPE) of three selected polar organophosphorus pesticides (OPPs), namely, dimethoate, phenthoate, and phosphamidon, prior to gas chromatography analysis with electron capture detection (GC-μECD). The Fe3O4-SP/GO adsorbent combines the advantages of superior adsorption capability of the modified sporopollenin (SP) with graphene oxide (GO) and magnetite (Fe3O4) for easy isolation from sample solution. Several MSPE parameters were optimized. Under optimized conditions, excellent linearity (R2 ≥ 0.9994) was achieved using matrix match calibration in the range of 0.1 to 500 ng mL−1. The limit of detection (LOD) method (S/N = 3) was from 0.02 to 0.05 ng mL−1. The developed Fe3O4-SP/GO MSPE method was successfully applied for the determination of these three polar OPPs in cucumber, long beans, bell pepper, and tomato samples. Good recoveries (81.0–120.0%) and good relative standard deviation (RSD) (1.4–7.8%, n = 3) were obtained for the spiked OPPs (1 ng mL−1) from real samples. This study is beneficial for adsorptive removal of toxic pesticide compounds from vegetable samples.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Pesticides are applied in agriculture to combat pests that destroy crops; hence, they improve the quality and quantity of the products. Due to their relatively low cost and high chemical activity, the use of pesticides eventually leads to high-quality agricultural products (Mahpishanian et al. 2015; Lucchi and Benelli 2018). Generally, more than 500 compounds are enlisted worldwide as pesticides and their use cannot be avoided as they improve diet, good health, and decrease cost (WHO1990). Increasing use of pesticides in food production has in recent years become a global challenge because of their perceived level of toxicity. Above a specified level, they are considered harmful to human health as they affect cholinesterase enzyme with persistence and bioaccumulation (Tankiewicz et al. 2010; Benelli et al. 2018; Benelli 2018a, b).
Organophosphorus pesticides (OPPs) are one of the largest classes of agricultural insecticides currently in use due to better pest control, low cost, and faster degradation (Fenik et al. 2011). OPP residual permissible maximum limit is between 0.1 and 0.5 ng mL−1 as set by the Environmental Protection Agency (EPA 2003). In general, OPP residue maximum levels set by the European Union from 0.1 to 0.3 mg kg−1 is considered safe (Picó and Kozmutza 2007). Widespread use of pesticides in vegetable production and presence of a wide variety of toxic pesticide contamination expose consumers to low levels of pesticides, especially when these commodities are freshly consumed (Bidari et al. 2011). Thus, monitoring and evaluation of pesticide residue in vegetables is necessary to warrant consumers’ safety (Mahmood and Malik 2014). Direct instrumental determination of OPPs is often limited due to their low concentration levels in sample solution and presence of matrix interferences. Therefore, analysis of OPPs requires a good sample preparation method to isolate the analytes from complex matrices and remove the interfering compounds before instrumental analysis to achieve a sufficient sensitivity and improve limit of detection (Rashidi Nodeh et al. 2017).
Several methods have been successfully demonstrated for extraction of OPPs in various matrices. These include liquid-liquid extraction (LLE) (Igarashi and Yotsuyanagi 1992), solid phase extraction (SPE) (Sanagi et al. 2013; Wu et al. 2011; Wan Ibrahim et al. 2012), solid phase micro-extraction (SPME) (Bidari et al. 2011; Wan Ibrahim et al. 2010), stir bar sorptive micro-extraction (SBSE) (Zhou and Fang 2015; Wan Ibrahim et al. 2011), and dispersive solid phase extraction (DSPE) (Zheng et al. 2010). Use of the LLE method for determination of OPPs is tedious, time consuming, and labor intensive and consumes large amount of toxic solvents which pose disposal and environmental problem, thereby, limiting its use. Though considered a bit expensive, the later three methods are environmental friendly. Of the various methods, SPE is the most popular method used in various applications due to its simplicity and high preconcentration factor achieved (Sanagi et al. 2013). In comparison to LLE, SPE handles small samples (50–100 mL), consumes less solvent (2–20 mL), and has reasonable cost and time with no emulsion formation. SPE limitations such as time consumption (as it encompasses multi-step operations including evaporation of elute), channeling problem, and cost led to search for a simple, low-cost, and environment-friendly method (Bianchi et al. 2012). Therefore, a new SPE method involving the introduction of magnetic nanoparticles known as magnetic solid phase extraction (MSPE) evolved and has gained prodigious attention.
Accordingly, this new method has been adjudged as a rapid, efficient, and excellent treatment method (Šafařı́ková and Šafařı́k 1999). In MSPE, the magnetic adsorbent can be readily isolated from the sample matrix with an external magnet without filtration or centrifugation. After adsorption, the analytes held in the solid adsorbent are eluted with a suitable solvent (Barakat 2011; Rashidi Nodeh et al. 2017). The properties and nature of the solid adsorbent in MSPE are paramount. In practice, solid adsorbent for MSPE should contain high surface area, high adsorption capacity, efficient dispersability in liquid samples, appropriate surface for interaction, and good stability and magnetic property for fast and quantitative sorption and elution (Ali et al. 2015; Xu et al. 2015). In this context, Fe3O4 magnetic nanoparticles (MNPs) as MSPE adsorbent are not reliable due to its shortcomings such as aggregation, less surface for interaction, and low stability in acidic medium and are easily oxidized which leads to their modification and development (Zheng et al. 2013). The modification will provide a more efficient method at the same time enhance the selectivity of the modified adsorbent toward the target analytes. Thus, an investigation into the synthesis of a novel and simple adsorbent with excellent adsorption performance is desirable (Xu et al. 2015).
Over the years, several developments on the MSPE adsorbent have been reported (Kamboh et al. 2016; Ali et al. 2015; Rashidi Nodeh et al. 2017; Yusuff et al. 2018; Sun et al. 2015; Mahpishanian et al. 2015), whereas, using graphene oxide (GO) (Zheng et al. 2013; Sitko et al. 2013; Sun et al. 2015) and sporopollenin (SP) (Gubbuk 2011; Sargin and Arslan 2015; Kamboh et al. 2016) is of great interest. Sporopollenin, a chemical co-polymer that makes up the outer layer (exine) of pollen grains with approximate composition C90H144O27, has remarkable physical and chemical stability. The hollow nature of its exines and thick walls (2 mm) perforated with a variety of channels and porous outer and inner surface are readily available for binding (Kamboh et al. 2016). Its profound attribute is its ability to act like a sponge around substances such as pesticides, toxins, and toxic metals by soaking them up and bring them together (Gubbuk 2011). It has high selectivity for metals but has less selectivity for polar OPPs (Sargin and Arslan 2015). To increase its selectivity toward polar OPP analytes, GO has been considered. GO has high surface area (3700 m2 g−1 theoretically) (Sitko et al. 2013), variety of benzene rings, functional groups, noticeable π-π electron, significant high adsorption capacity, wide range of application, and good selectivity toward polar OPPs (Sitko et al. 2013; Sun et al. 2015). Both GO and SP combine to form hydrophobic interaction, strong hydrogen bonding, and π-π interaction which is enough to keep the analytes adsorbed. Therefore, combination of magnetite (Fe3O4) with SP and GO is expected to promote high surface area for adsorption, excellent selectivity, good stability, and ease of separation. To the best of our knowledge, this is the first report of the MSPE method based on Fe3O4-SP/GO for polar OPPs.
In this current work, MSPE based on Fe3O4-SP/GO as an active new adsorbent is presented. Selectivity and adsorption capacity of the Fe3O4 have been improved by SP and GO to adapt various targets. The nature of the magnetic material and method provides time saving and is a convenient, low toxic, inexpensive, environmental friendly, effective, and sensitive procedure for determination of selected polar OPPs in selected vegetables. The OPPs and the vegetable samples were selected due to ground-level survey and literature review which revealed that dimethoate, phenthoate, and phosphamidon were frequently used to control pest in Malaysian vegetables such as bean species, peppers, tomatoes, cucumbers, and cabbages. Therefore, Fe3O4-SP/GO MSPE is proposed for simultaneous extraction of three selected polar OPPs in four selected vegetables, namely cucumber, green bell pepper, green long beans, and reddish tomato.
Materials and methods
Materials
Dimethoate, phosphamidon, and phenthoate standard pesticides were purchased from Sigma-Aldrich (St. Louis, MO, USA). Hydrochloric acid (37%), sulfuric acid (98%), Ar grade acetone, ethanol, potassium permanganate, hydrogen peroxide, and ammonia solution were purchased from Mark (Darmstadt, Germany). Ferric and ferrous chloride were obtained from Fluke (Seelze, Hanover, Germany). The graphite and sporopollenin powder were purchased from Sigma-Aldrich (St. Louis, MO, USA). The vegetable samples were obtained from Sri-Pulai market, Skudai, Johor, Malaysia.
Preparation of the Fe3O4-sporopollenin
The Fe3O4 was prepared as described by Li et al. (2015). Briefly, 1.36 g of FeCl2·4H2O and 1.7 g of FeCl3·6H2O (1:2) were carefully mixed in deionized water (DW), after which the solution was sonicated and degassed for 30 min and stirred for 1 h at 50 °C in the presence of N2 gas. A volume of 10 mL of 32% ammonia solution was added dropwise with continuous stirring until a black precipitate solution was observed at pH 9. The mixture was stirred for 2 h and the heat was then turned off, but continued stirring for about 5 h. The obtained product was divided into two portions. One portion was washed with DW and then oven dried at 100 °C. To the other potion, an amount 0.03 g SP was dispersed gently onto the prepared Fe3O4 with continued stirring and 2 mL methanol was added to enhance solubility. Then, the solution was stirred for 2 h at 50 °C. The mixture was continuously stirred for 4 h without heat. The product (Fe3O4-SP) obtained was washed with DW until a neutral pH value and oven dried at 100 °C overnight.
Synthesis of graphene oxide
GO was prepared from graphite powder using the modified Hummers method (Hummers and Offeman 1958). An amount of 1.0 g graphite powder was weighted into a 250-mL conical flask and 40 mL DW was added. The mixture was then stirred for 20 min at 50 °C and then put in an ice bath. Then, 100 mL H2SO4 was gently added into the mixture with vigorous magnetic stirring for 1 h. Then, 5.0 g KMnO4 was gradually added to the mixture with continued stirring at 50 °C for about 2 h, after which the resulting mixture was cooled to room temperature and poured into ice (300 mg), then added 10 mL of 30% H2O2 to produce a yellowish color solution. The solution was then diluted with DW (600 mL) and left overnight for precipitation. Finally, the supernatant was decanted and the product was diluted and washed with DW until pH 7. The product obtained was filtered (suction pump) and oven dried at 100 °C overnight.
Preparation of Fe3O4-SP/GO
The freshly prepared graphene oxide was dispersed wisely into the magnetic Fe3O4-SP. The mixture was then stirred at 50 °C for 3 h. Thereafter, the heat was removed, and the mixture was allowed to continue stirring for the next 8 h. Finally, the product was washed until pH 7 and was oven dried at 100 °C overnight.
Characterization Fe3O4-SP/GO adsorbent
The prepared materials were characterized using the Fourier transformed infrared (FTIR) spectroscopy, SEM, thermogravimetric-differential thermogravimetric analysis (TGA-DTA), and vibrating sample magnetometry (VSM). The FTIR spectra of the materials were recorded with a PerkinElmer FTIR spectrometer (MA, USA) in the range of 4000–400 cm−1 using the KBr pellet method. Surface morphology and size of the synthesized materials were observed using SEM JEOL JSM-6390LV system (Tokyo, Japan). The SEM shows the morphology of the material. In this study, the prepared sample for SEM was placed in a sample holder and coated with gold sputtering using a double-sided tape as adhesive and analyzed. The resultant signal was obtained due to the electron beam contact with the material in the column and was recorded at a magnification of × 10,000 (10 K). The TGA-DTA demonstrates the thermal stability of the adsorbent. Stability and weight loss of the magnetic material were determined using PerkinElmer STA 800 TGA-DTA (MA, USA). The TGA-DTA was performed by placing 14.647 mg of the sample in a crucible pans ceramic material. Subsequently, the sample was inserted into the instrument furnace and heated over a temperature range of 30 to 1000 °C at 10 °C min−1 in a stream of nitrogen gas (N2). The graph of the result was plotted as mass change (mg) against the change in temperature and was displayed using a “PYRIS ONE SOFTWARE” of the PerkinElmer instrument. Magnetic properties were observed using Lake Shore 7404 (USA) VSM at room temperature. The VSM as a function of magnetic field, temperature, and time shows its magnetic properties for easy separation from sample matrices. In this study, each VSM sample was prepared using a transparent tape. It was placed within a suitable placed sensing coil at room temperature (25 °C) and allow to undergo mechanical vibration over an amplitude of 1.5 mm. The superparamagnetic parameter of the sample was calculated by extrapolating high-field M(H) data to M axis at H = 0 value.
Real sample preparation
The samples were prepared as described by Mahpishanian et al. (2015) and Li et al. (2012). For brevity and clarity, vegetable samples obtained from Sri-Pulai market, Skudai, Johor, Malaysia, were cut into small pieces and homogenized with a laboratory stainless steel blender. Then, 20 g of the sample was accurately weighed into a 50-mL centrifugal tube, and 10 mL DW and 2 mL methanol (MeOH) were added. The mixture was vortex mixed for about 2 min and then centrifuged for 5 min at 4000 rpm. The resulting supernatant was then filtered through a 0.45-mm filter paper. Finally, the mixture was transferred into a 50-mL volumetric flask and diluted to the mark with distilled water and kept at 4 °C for further analysis. For spiked samples, a specific volume (1 ng mL−1) of standard solutions of pesticides was added to the blank vegetable samples and then prepared as before.
Fe3O4-SP/GO magnetic solid phase extraction
The process of MSPE parameters was optimized using one-variable-at-a-time (OVAT). Initially, 1 ng mL−1 (1 ppb) of the OPP standards with internal standard (IS) prepared was measured into a 50-mL centrifuge tube containing 40-mL sample volume. Then, 10 mg of the adsorbent was added into the mixture and shaken for 5 min with the aid of an orbital shaker at a maximum speed of 250 rpm. The adsorbed OPPs were then separated from solution with the aid of an external magnet. The liquid phase was decanted and the adsorbed analytes were eluted using 2 mL of different polar solvents (methanol, ethanol, acetone, and acetonitrile) aided by shaking. The adsorbent was separated from the extract using an external magnet and transferred into a 2-mL centrifuge tube. A mild stream of N2 was passed through the eluate to dryness and the resultant residue was reconstituted with 1 mL methanol. Finally, 1 μL of the extracted sample was injected into the gas chromatography analysis with electron capture detection (GC-μECD) instrument for analysis. Based on the highest peak area ratio observed (peak area of OPPs/peak area IS), acetone was chosen as the best desorption solvent. Next, the other parameters were optimized as well using the OVAT procedure such as desorption solvent volume (0.5–5 mL), sample volume (10–90 mL), extraction time (1–50 min), desorption shaken time (1–10 min) (desorption shaking time is the time needed to assist desorption of adsorbed analytes from adsorbent during desorption process), mass of adsorbent (5–40 mg), and sample solution pH level (3–11).
GC conditions
Agilent 7820A (Santa Clara, CA, USA) GC-μECD was used for the analysis and identification of the selected polar OPPs (dimethoate, phenthoate, and phosphamidon) with dicrotophos as internal standard (IS) in vegetable samples. The column used was a HP-5 (5% phenyl methyl siloxane 325 °C, 30 m × 250 μm × 0.25 μm). The optimum GC-μECD conditions include helium was used as the back-inlet carrier gas at flow rate of 2.5 mL min−1 and nitrogen as the back detector make up gas at flow rate 5 mL min−1. The temperature program was at 70–280 °C, starting at 70 °C (held for 1 min). Ramp 1 is at 50 °C min−1 to 190 °C (held for 1 min); ramp 2 is at 30 °C to 280 °C (held for 2 min). The detector temperature was set at 300 °C and back-inlet port at 280 °C (pressure 25 psi).
Results and discussion
Characterization of Fe3O4-SP/GO magnetic adsorbent
FTIR analysis
The FTIR spectrum (see Fig. 1) provides abundant information about the presence of functional groups in the synthesized Fe3O4, GO, SP, Fe3O4-SP, and Fe3O4-SP/GO materials. The broad peak at 3404 cm−1 corresponds to –OH stretching vibration in all the synthesized materials. Peaks appearing at 1725 cm−1, 1712 cm−1, and 1713 cm−1 are attributed to the C=O stretching vibration of COOH group of Fe3O4-SP/GO, SP, and GO, respectively. Peaks at 2852 cm−1, 2820 cm−1, and 570 cm−1 correspond to CH2, C–H, and Fe–O vibrations, respectively, present in SP and Fe3O4-SP which shows that Fe3O4 was successfully modified to Fe3O4-SP. The peaks at 1573 cm−1 and 1626 cm−1 correspond to C=C stretching vibrations present in Fe3O4-SP/GO and Fe3O4-SP, respectively. Peaks around 1216 cm−1 and 1148 cm−1 correspond to C–O (carboxyl) stretching vibration present in Fe3O4-SP/GO and GO. The C–O (epoxy) stretching vibration peak around 1440 cm−1 and 1451 cm−1 belongs to Fe3O4-SP and SP. –O (alkoxy) stretching peak is around 868 cm−1 for Fe3O4-SP/GO. This indicated the presence of abundant functional groups (–OH, C=O, C–O, –O, CH2) on the surface of GO and SP. The peak at 1633 cm−1 is attributed to –OH bending vibration for Fe3O4. The adsorption bands around 570 cm−1 and 556 cm−1 correspond to Fe–O stretching mode in Fe3O4, Fe3O4-SP, and Fe3O4-SP/GO which shows successful modification of Fe3O4, Fe3O4-SP, and Fe3O4-SP/GO. Figure 1(c, d) shows a gradual disappearance of the CH2 and CH3 of the SP and in Fe3O4-SP. This noticeable change from Fig. 1(c, d) indicated a successful modification of SP unto Fe3O4 and GO unto Fe3O4-SP. In summary, Fe3O4 has no epoxy, C=C, and CH2 functional groups, but when modified with SP gives epoxy, C=C, and CH2 peaks at 1440, 1626, and 2852 cm−1, respectively, indicating successful modification of Fe3O4 to Fe3O4-SP. When further modified with GO, Fe3O4-SP gives additional functional groups (C=O, C–O, and –O at 1725, 1216, and 868 cm−1, respectively) and slight disappearance of CH2 and C–H groups at 2852 and 2820 cm−1 respectively.

FTIR spectra of (a) Fe3O4, (b) GO, (c) SP, (d) Fe3O4-SP, and (e) Fe3O4-SP/GO with KBr pellet successful modification of Fe3O4-SP to Fe3O4-SP/GO
SEM analysis
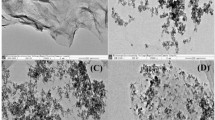
The morphology of the Fe3O4-SP/GO was investigated using SEM. In Fig. 2(a), the bare Fe3O4 nanoparticles were spherical and partially aggregated due to the existence of magnetic attraction. In Fig. 2(b), the sheet of the GO is noticeable. The morphology of SP in Fig. 2(c) shows the presence of benzene ring structure and cavities (hole). After dispersing with Fe3O4, Fig. 2(d) showed clearly the distribution of the Fe3O4 in-between the cavities of the SP, indicating that the surface was successfully modified (incorporation of Fe3O4). From Fig. 2(e), a significant change was observed when the Fe3O4-SP (depicted in Fig. 2(d)) was further modified with GO. The GO sheets were noticeable, where it was incorporated on the surface of the SP-modified magnetite. The morphology showed that the analytes can access both the SP and GO functional groups for better adsorption, interaction, and analyte retention.

SEM image of (a) Fe3O4, (b) GO, (c) SP, (d) Fe3O4-SP, and (e) Fe3O4-SP/GO
TGA/DTA
The TGA and DTA of the Fe3O4-SP/GO were performed to demonstrate its thermal stability and illustrated in Fig. 3. The TGA (see Fig. 3(a)) and DTA (see Fig. 3(b)) were applied over a temperature range from 30 to 1000 °C in N2 flow at a thermal rate of 10 °C min−1. The thermogram shows three degradation steps of Fe3O4-SP/GO from 30 to 900 °C. The first degradation step, 4.6% weight loss from 36 to 100 °C, is attributed to water loss and volatile component loss adsorbed on the material structure in Fig. 3(a, b). At 200 °C, oxygenated functional group loss was observed for SP and GO and at 700 °C. The loss was attributed to GO skeletal with a weight loss of about 2.9% in Fig. 3(a) and 10.9% in Fig. 3(b) respectively. The DTA curve showed a significant stability between 200 and 900 °C which shows the material is highly stable before a collapse at 900 °C was observed.

(a) TGA and (b) DTA of Fe3O4-SP/GO over a range temperature of 30 °C to 900 °C heated at 10 °C min−1 in N2 flow
VSM analysis
The magnetic properties of Fe3O4, Fe3O4-SP, and Fe3O4-SP/GO adsorbents were analyzed using VSM at room temperature. The result obtained was then plotted (magnetization (emu/g) vs applied magnetic field (Oe)) at 300°K and magnetic parameter was calculated (see Fig. 4). The magnetization hysteresis loop curves illustrate that all the materials (Fe3O4, Fe3O4-SP, and Fe3O4-SP/GO) are superparamagnetic with magnetization saturations of 65.6 emu g−1, 51.2 emu g−1, and 27.8 emu g−1 respectively. A noticeable decrease in magnetic strength illustrated in Fig. 4(a–c) indicates successful modification. The result showed that sufficient magnetic separation can be achieved using an external magnet with 27.8 emu g−1 superparamagnetic saturation. A value of 16.3 emu g−1 is reported to be sufficient for magnetic separation (Ma et al. 2005).

Hysteresis loops of (a) Fe3O4, (b) Fe3O4-SP, and (c) Fe3O4-SP/GO at room temperature. Sample weight, 6.6 mg Fe3O4, 6.5 mg Fe3O4-SP, and 6.8 mg Fe3O4-SP/GO
Selection of prepared adsorbents
The adsorption ability of Fe3O4, Fe3O4-SP, and Fe3O4-SP/GO adsorbent was compared. The magnetite shows poor adsorption efficiency toward the polar OPPs. This could be due to less interaction between Fe3O4 and the selected OPPs. When Fe3O4 was modified with sporopollenin (SP), the adsorption performance increases significantly. This is attributed to the increase in surface area, interaction, and change in charge density. Further modification of Fe3O4-SP with graphene oxide (GO) to produce Fe3O4-SP/GO gives better performance. The extraordinary properties of GO such as high surface area, π-π bonds, and functional groups (–OH, C=O) promote great interaction with the selected OPPs. The combination of Fe3O4, SP, and GO is considered a great adsorbent as it combines the properties of all the compounds. That is, Fe3O4 promotes fast separation of analyte from sample solution, and SP provides more functional groups and stability. Therefore, Fe3O4-SP/GO was chosen to obtain good extraction efficiency for the selected OPPs in selected vegetables.
Optimization of MSPE procedure
MSPE optimization parameters on the extraction of dimethoate, phenthoate, and phosphamidon performance under experimental conditions were investigated to obtain the best result from the real sample analysis and assess the ability of the Fe3O4-SP/GO MSPE adsorbent. Seven parameters were optimized, namely type of desorption solvent, volume of desorption solvent, sample volume, extraction time, shaking time in desorption process, adsorbent mass, and sample pH value. Each sample was spiked with 10 ng mL−1. All experiments were performed in triplicate and the result was plotted. The highest peak area ratio response of the selected OPPs and IS after analysis was considered as the optimum condition for each parameter.
Effect of desorption solvent
Desorption solvent plays a vital role in MSPE process to ensure high recovery and sensitivity on method performance. Thus, it is critical to select an appropriate desorption solvent. For the desorption of the selected polar OPPs from the magnetic adsorbent, four polar solvents with decreasing polarity order, namely methanol, ethanol, acetonitrile, and acetone with relative polarity of 0.76, 0.65, 0.46, and 0.36, respectively, were used to elute the adsorbed analyte from the magnetic Fe3O4-SP/GO adsorbent. The highest response and best extraction performance measured by ratio of OPPs peak area/peak area of IS were achieved when acetone was used as the desorption solvent (Fig. 5). All the solvents are polar, so polarity did not play a significant role in the desorption process. Acetone is the least viscous solvent compared to the other three solvents. The response falls with an increasing viscosity (0.32, 0.37, 0.55, and 1.10 centipoise at 20 °C for acetone, acetonitrile, methanol, and ethanol, respectively) of the desorption solvent. The lower the viscosity of the polar solvent, the more likely the analyte will desorb into the solvent from the adsorbent. Thus, acetone was selected as the desorption solvent for further analysis.

Effect of different types of desorption solvents on the desorption efficiency of selected OPPs from vegetable samples using Fe3O4-SP/GO MSPE. Spiked OPP level, 10 ng mL−1. MSPE conditions: desorption solvent volume, 2 mL; sample volume, 40 mL; extraction time, 3 min; desorption shaking time, 5 min; adsorbent mass, 10 mg; and sample solution, pH 6
Effect of desorption solvent volume
The effect of desorption solvent volume on MSPE performance is also important to achieve good result as it determines the maximum preconcentration factor for target analytes to achieve good sensitivity. The aim is to determine the volume of desorption solvent which gives the highest signal. Thus, the effect of different acetone volumes was investigated from 0.5 to 5 mL. It was found that the peak area ratio increased when the solvent volume was increased from 0.5 to 2 mL probably due to the highest analyte preconcentration and subsequently decreased after 2 mL (see Fig. 6) which could be due to lower analyte preconcentration and re-adsorption of the analyte from the adsorbent. The highest peak ratio of all the selected OPPs was observed when 2 mL acetone volume was used. Hence, 2 mL acetone volume was selected for further analysis.

Effect of desorption volumes on the extraction efficiencies of the selected OPPs from vegetable samples using Fe3O4-SP/GO MSPE. Spiked OPP level, 10 ng mL−1. MSPE conditions: type of desorption solvent, acetone; sample volume, 40 mL; extraction time, 3 min; desorption shaking time, 5 min; adsorbent mass, 10 mg; and sample solution pH, 6
Effect of sample volume
Choice of a suitable optimum sample volume for MSPE performance is important to obtain high enrichment factor and recovery. Sample volume can significantly affect the recovery values and the sensitivity of the methodology. To evaluate this effect, different sample volumes were investigated ranging from 10 to 90 mL. The peak area ratio of the OPPs extracted increased when the sample volume was increased from 10 to 50 mL probably due to high enrichment factor achieved with increasing sample volume but decreased significantly when the sample volume was further increased to 70 mL and 90 mL (see Fig. 7). The decrease could be due to decrease in analyte concentration from analyte breakthrough volume of sample (breakthrough volume exceeded). Thus, 50-mL sample volume was selected as the optimum volume for further analysis as it produced the highest peak area ratio response for all the OPPs. The highest response was observed for phenthoate which means that the Fe3O4-SP/GO has the highest selectivity for phenthoate which could be due to the presence of benzene rings, thus, more π systems to interact with Fe3O4-SP/GO.

Effects of sample volume on the extraction efficiency of the selected OPPs from vegetable samples using Fe3O4-SP/GO MSPE. Spiked OPP level, 10 ng mL−1. MSPE conditions: desorption solvent, acetone; desorption solvent volume, 2 mL; extraction time, 3 min; desorption shaking time, 5 min; adsorbent mass, 10 mg; and sample solution pH, 6
Effect of extraction time
The aim of extraction time is to find an optimum time required to achieve adsorption equilibrium. The study of the effect of extraction time on the extraction efficiency of OPPs using Fe3O4-SP/GO MSPE was performed to achieve the highest sensitivity and recovery. Extraction was performed in a 50-mL centrifuge tube with the aid of an orbital shaker at maximum speed of 250 rpm. The extraction efficiency depends on the mass transfer between the Fe3O4-SP/GO and analytes in the sample solution. Because mass transfer is a time-dependent process, the effect of extraction time was varied from 1- to 50-min range while other parameters were kept constant. The sample solution was continuously agitated at 25 °C on an orbital shaker (250-rpm rate) to facilitate the mass transfer. The highest peak area ratio was observed at the 5-min extraction time as depicted in Fig. 8. At 10-min extraction time, the response decreased and remained almost constant at 30- and 50-min extraction time probably due to desorption of the analyte from the adsorbent. Thus, 5 min was taken as the optimum extraction time for further analysis.

Effect of extraction time on extraction efficiency of the selected OPPs from vegetable samples using Fe3O4-SP/GO MSPE. Spiked OPP level, 10 ng mL−1. MSPE conditions: desorption solvent, acetone; desorption solvent volume, 2 mL; sample volume, 50 mL; desorption shaking time, 5 min; adsorbent mass, 10 mg; and sample solution pH, 6
Effect of desorption shaking time
Shaking time during desorption process is another important parameter affecting MSPE efficiency. The study of the effect of shaking time during desorption process was performed with the aid of an orbital shaker at 250 rpm (maximum) to enhance the mass transfer rate of analytes. Five different desorption shaking times from 1 to 10 min were evaluated. It was observed that the peak area ratio increased significantly with an increase in desorption shaking time from 1 to 3 min (see Fig. 9), but decreased significantly when increased at 5 and 10 min. The increase in response of analyte from 1- to 3-min shaking time is because of the desorption of analyte from sorbent into solution. However, the decrease in the response of analyte at 5 min is probably due to re-adsorption of analyte onto the sorbent and re-adsorption remains constant at 10 min. Thus, desorption shaking time of 3 min was found to be optimum.

Effect of shaking time during desorption on extraction efficiency of the selected OPPs from vegetable samples using Fe3O4-SP/GO MSPE. Spiked OPP level, 10 ng mL−1. MSPE conditions: desorption solvent, acetone; desorption volume, 2 mL; sample volume, 50 mL; extraction time, 5 min; adsorbent mass, 10 mg; and sample solution pH, 6
Effect of adsorbent dosage
The MSPE optimum adsorbent (Fe3O4-SP/GO) mass is one of the critical conditions that can improve the recovery of the selected OPPs from solution. The effect of the mass of the adsorbent on the extraction of the OPPs was studied using five different quantities ranging from 5 to 40 mg. The peak area ratio of the extracted OPPs increased with an increased in dosage amount up to 20 mg before stabilizing at 30 to 40 mg as illustrated in Fig. 10. The decrease in response could be due to saturation of active sites on the adsorbent that has been exceeded. Thus, 20-mg adsorbent mass was selected for further analysis.

Effects of adsorbent mass on the extraction efficiency of the selected OPPs from vegetable samples using Fe3O4-SP/GO MSPE. Spiked OPP level, 10 ng mL−1. MSPE conditions: desorption solvent, acetone; desorption solvent volume, 2 mL; sample volume, 50 mL; extraction time, 5 min; desorption shaking time, 3 min; and sample solution pH, 6
Effect of sample pH level
The sample solution pH level plays a key part during adsorption process by affecting the existing forms of the analytes, regulating the charge species and density on the adsorbent surface. The influence of sample pH level on the extraction efficiency of the OPP peak area was studied within the range of 3 to 11 (see Fig. 11). The result shows that there is a significant increment in signal (peak area ratio) as the pH level rises from 3 to 7. This could be due to increase in stability of the OPPs because OPPs are highly stable at pH levels from 4.5 to 7. However, subsequent increase in the sample pH level decreases the peak area ratio significantly. This is because on approaching pH 9, OPPs start to be hydrolyzed and the higher the pH level, the faster the hydrolysis, hence the decrease in OPP response from pH value of 9 to 11 (Jiang et al. 2016). At pH 7, the sample solution was highly stabilized; hence, the OPPs are highly stable and the adsorbent could not undergo protonation or deprotonation which allowed the OPPs to hydrogen bond with the adsorbent and to undergo π-π interaction and hydrophobic interaction as well (Kamboh et al. 2016). Extraction at a slightly acidic pH level could be due to surface complexation through association between acidic hydroxyl (+OH2) groups of Fe3O4-SP/GO nanomaterial and negative charge sites (Cl (in phosphamidon), S (in phenthoate and dimethoate), and O (in all selected OPPs)). Excessive H+ at high acidic pH levels could lead to protonation of hydroxyl groups in GO and SP, lowering the electron-donating ability of N (in dimethoate and phosphamidon) and O atoms (in all selected OPPs) (Rashidi Nodeh et al. 2017). Thus, pH 7 was selected as the optimum pH value for subsequent analysis as it showed the highest signal.

Effect of sample solution pH level on extraction efficiency of the selected OPPs in vegetable sample using Fe3O4-SP/GO MSPE. Spiked OPP level, 10 ng mL−1. MSPE conditions: desorption solvent, acetone; desorption solvent volume, 2 mL; sample volume, 50 mL; extraction time, 5 min; shaking time, 3 min; and adsorbent mass, 20
Method validation
The proposed Fe3O4-SP/GO MSPE method was investigated and validated using the following optimum MSPE conditions: acetone as desorption solvent, 2 mL acetone as desorption solvent volume, 50 mL sample volume, 5 min as extraction time, 3 min as desorption shaking time to aid desorption process and mass transfer of analyte, 20 mg as mass of adsorbent and sample solution pH 7. The parameters involved are linearity, limit of detection (LOD), limit of quantification (LOQ), precision, and recovery (%R). The method validation was chosen according to SANTE/11945/2015 guidelines of the European Commission. Linearity values of the coefficient of determination (R2) were calculated using the calibration graph to determine the linear relationship between the signal (peak area ratio of OPPs/internal standard) and the concentration of the mixed standards. The LOD and LOQ were calculated based on the three times signal-to-noise ratio (S/N = 3) and ten times signal-to-noise ratio (S/N = 10) respectively. The precisions were expressed based on the %RSD for both intra-day (n = 3) and inter-day (n = 9) and the percentage recovery (%R) was calculated based on Eq. (1):
Matrix match calibration
Matrix match calibration was applied due to the expected complex nature of the vegetable samples and the presence of matrix interference since these samples contain pigments, fatty acids, carotenoids, and sugars. Matrix match calibration plot using vegetable samples was constructed for each OPP for a concentration range from 0.1 to 500 ng mL−1 for phosphamidon, dimethoate, and phenthoate. Good linearity was obtained for all the three selected OPPs using the magnetic Fe3O4-SP/GO MSPE method with good coefficient of determination (R2) of 0.9994, 0.9996, and 0.9997 for dimethoate, phosphamidon, and phenthoate, respectively. The LODs were calculated as 0.05, 0.04, and 0.02 ng mL−1 (3 × S/N, n = 3) whereas the LOQs calculated were 0.17, 0.13, and 0.10 ng mL−1 based on 10 × S/N ratios (n = 10) for phosphamidon, dimethoate, and phenthoate, respectively (see Table 1).
Real sample analysis
The applicability of the proposed Fe3O4-SP/GO MSPE method was used for the determination of selected polar OPPs in several vegetable sample matrices obtained from Sri-Pulai Perdana market Skudai, Johor Bahru, Malaysia. Prior to real sample analysis, the accuracy of the method was investigated by spiking OPP standards at level 1 and 50 ng mL−1 into the vegetable samples, namely 4 bell peppers, 3 cucumbers, 6 green long beans, and 5 reddish tomatoes. Each sample was analyzed three times using the GC-μECD (see Fig. 12). Good recoveries of the method were observed in the range of 81.0 to 120% (n = 3) with repeatability, %RSD (n = 3) of 1.4 to 7.8% and reproducibility, %RSD (n = 9) ranging from 3.7 to 8.1%. Poorer recoveries were obtained for tomato samples probably due to matrix interference such as carotenoid (lycopene) as is the major component of about 80% in tomato. From this study, the amount of OPPs in all vegetables was not detected by the instrument. Quantitative recoveries of OPPs were obtained with good repeatability and reproducibility which shows the method is applicable and reliable to real sample analysis. Table 2 summarized the precision recoveries achieved using the developed Fe3O4-SP/GO MSPE method.

Chromatogram of (a) blank tomato sample, (b) unspiked tomato sample, and (c) spiked OPP tomato sample (10 ng mL−1) extracted using Fe3O4-SP/GO MSPE. MSPE conditions: acetone as desorption solvent, 2 mL as desorption solvent volume, 50 mL sample volume, 5 min extraction time, 3 min desorption shaking time, 20 mg adsorbent mass, and sample solution pH 7. GC conditions: flow rate (2.5 mL min−1 helium and 5 mL min−1 N2), pressure (25 psi). Temperature program: start 70 °C held for 1 min, ramp 1 (50 °C min−1 to 190 °C held for 1 min), ramp 2 (30 °C min−1 to 280 °C, held for 2 min), and μECD at 300 °C. Peak identification: 1. Phosphamidon (4.35 min), 2. Dimethoate (5.68 min), 3. Phenthoate (6.01 min) and IS (internal standard)
Comparison of Fe3O4-SP/GO MSPE with other methods
The results obtained were compared to those of some reported works (Table 3). The result shows that the MSPE method has considerably low LODs than SPME (Wan Ibrahim et al. 2010; Jabbari et al. 2016), DSPE (Zheng et al. 2013), and SPE (Sanagi et al. 2013; Wan Ibrahim et al. 2012). In this work, result showed that the Fe3O4-SP/GO MSPE developed for OPP analysis using vegetable samples is suitable when compared with recent works in the literature (Mahpishanian et al. 2015; Rashidi Nodeh et al. 2017; Wan Ibrahim et al. 2012; Wu et al. 2011). The new adsorbent showed high affinity toward OPPs probably due to good interaction with the OPPs. The LOD is well below OPP permissible limit (0.1 to 0.5 ng mL−1) established (EPA 2003). Thus, analysis can be done based on the EPA method. The performance of the developed Fe3O4-SP/GO MSPE method in terms of the recoveries in vegetable samples is satisfactory when compared with most works in the literature (Mahpishanian and Sereshti 2016; Mahpishanian et al. 2015; Wu et al. 2011; Guan et al. 2008; Zheng et al. 2013).
Conclusion
Investigation in this work proved successful application of Fe3O4-SP/GO MSPE for determination of selected polar OPPs in vegetable samples. The prepared adsorbent showed high potential as MSPE sorbent for the selected polar OPPs analysis in selected vegetables (green bell pepper, cucumber, green long beans, and orange tomato). Good accuracy and precision of OPPs analysis from vegetables were obtained using the new Fe3O4-SP/GO MSPE GC-μECD method developed. The LOD and LOQ obtained allowed the method to be used for real sample analysis based on the Environmental Protection Agency (EPA) method. Thus, this work offers several benefits for analytical analysis as it provides a simple, cheap, efficient, sensitive, fast, easy, and environmental friendly method for extraction of polar OPPs from vegetable complex sample matrix.
References
Ali AA, Sara K, Homeira E, Nafiseh S, Niloofar J (2015) Polypyrrole/magnetic nanoparticles composite as an efficient sorbent for dispersive micro solid phase extraction of antidepressant drugs from biological fluids. Int J Pharm 494:102–112
Barakat MA (2011) New trends in removing heavy metals from industrial wastewater. Arab J Chem 4:361–367
Benelli G (2018a) Mode of action of nanoparticles against insects. Environ Sci Pollut Res 25(13):12329–12341
Benelli G (2018b) Plant-borne compounds and nanoparticles: challenges for medicine, parasitology and entomology. Environ Sci Pollut Res 25(11):10149–10150
Benelli G, Pavela R, Petrelli R, Cappellacci L, Santini G, Fiorini D, Sut S, Dall’Acqua S, Canale A, Maggi F (2018) The essential oil from industrial hemp (Cannabis sativa L.) by-products as an effective tool for insect pest management in organic crops. Ind Crop Prod 122:308–315
Bianchi F, Chiesi V, Casoli F, Luches P, Nasi L, Careri M, Mangia A (2012) Magnetic solid phase extraction based on diphenyl functionalization of Fe3O4 magnetic nanoparticles for the determination of polycyclic aromatic hydrocarbons in urine samples. J Chromatogr A 1231:8–15
Bidari A, Ganjali MR, Norouzi P, Hosseini MRM, Assadi Y (2011) Sample preparation method for the analysis of some organophosphorus pesticides residues in tomato by ultrasound-assisted solvent extraction followed by dispersive liquid–liquid microextraction. Food Chem 126:1840–1844
EPA (2003) Guidelines establishing test procedures for the analysis of pollutants; analytical methods for biological pollutants in ambient water; final rule. US EPA federal method registry 40 CFR part 136
Fenik J, Tankiewicz M, Biziuk M (2011) Properties and determination of pesticides in fruits and vegetables. TrAC Trends Anal Chem 30:814–826
Guan W, Wang Y, Xu F, Guan Y (2008) Poly(phthalazine ether sulfoneketone) as novel stationary phase for stir bar sorptive extraction of organochlorine compounds and organophosphorus pesticides. J Chromatogr A 1177(1):28–35
Guan H, Brewer EW, Garris ST, Morgan SL (2010) Disposable pipette extraction for the analysis of pesticides in fruits and vegetables using gas chromatography/mass spectrometry. J Chromatogr A 1217(12):1867–1874
Gubbuk IH (2011) Isotherms and thermodynamics for the sorption of heavy metal ions onto functionalized sporopollenin. J Hazard Mater 186:416–422
Hummers WS, Offeman RE (1958) Preparation of graphitic oxide. J Am Chem Soc 80(6):1339–1339
Igarashi, Yotsuyanagi (1992) Homogeneous liquid-liquid extraction by pH dependent phase separation with a fluorocarbon ionic surfactant and its application to the preconcentration of porphyrin compounds. Microchim Acta 106(1–2):37–44
Jabbari M, Razmi H, Farrokhzadeh S (2016) Application of magnetic graphene nanoparticles for determination of organophosphorus pesticides using solid phase microextraction. Chromatographia 79(15–16):985–993
Jiang L, Huang T, Feng S, Wang J (2016) Zirconium(IV) functionalized magnetic nanocomposites for extraction of organophosphorus pesticides from environmental water samples. J Chromatogr A 1456:49–57
Kamboh MA, Wan Ibrahim WA, Rashidi Nodeh H, Sanagi MM, Sherazi STH (2016) The removal of organophosphorus pesticides from water using a new amino-substituted calixarene based magnetic sporopollenin. New J Chem 40(4):3130–3138
Li Z, Bai S, Hou M, Wang C, Wang Z (2012) Magnetic graphene nanoparticles for the preconcentration of chloroacetanilide herbicides from water samples prior to determination by GC-ECD. Anal Lett 46:1012–1024
Li X, Liu Xu L, Wen Y, Ma J, Wu Z (2015) Highly dispersed Pd/PdO/Fe2O3 nanoparticles in SBA-15: for fenton-like process: confinement and synergistic effects. Appl Catal B Environ 165:79–86
Lucchi A, Benelli G (2018) Towards pesticide-free farming? Sharing needs and knowledge promotes integrated pest management. Environ Sci Pollut Res 25(14):13439–13445
Ma Z, Guan Y, Liu H, Liu H (2005) Preparation and surface modification of non-porous micro-sized magnetic poly (methyl acrylate) micros-pheres. J Polym Sci A Polym Chem 43:3433–3439
Mahmood A, Malik RM (2014) Human health risk assessment of heavy metals via consumption of contaminated vegetables collected from different irrigation sources in Lahore, Pakistan. Arab J Chem 7:91–99
Mahpishanian S, Sereshti H (2016) Three-dimensional graphene aerogel-supported iron oxide nanoparticles as an efficient adsorbent for magnetic solid phase extraction of organophosphorus pesticide residues in fruit juices followed by gas chromatographic determination. J Chromatogr A 1443:43–53
Mahpishanian S, Sereshti H, Baghdadi M (2015) Superparamagnetic core–shells anchored onto graphene oxide grafted with phenylethyl amine as a nano-adsorbent for extraction and enrichment of organophosphorus pesticides from fruit, vegetable and water samples. J Chromatogr A 1406:48–58
Picó Y, Kozmutza C (2007) Maximum residue levels of pesticides in or on food. Anal Bioanal Chem 389:1805–1814
Rashidi Nodeh H, Wan Ibrahim WA, Kamboh MA, Sanagi MM (2017) New magnetic graphene-based inorganic–organic sol-gel hybrid nanocomposite for simultaneous analysis of polar and non-polar organophosphorus pesticides from water samples using solid-phase extraction. Chemosphere 166:21–30
Šafařı́ková M, Šafařı́k I (1999) Magnetic solid phase extraction. J Magn Magn Mater 194(1):108–112
Sanagi MM, Salleh S, Wan Ibrahim WA, Naim AA, Hermawan D, Miskam M, Aboul-Enein HY (2013) Molecularly imprinted polymer solid phase extraction for the analysis of organophosphorus pesticides in fruit samples. J Food Compos Anal 32(2):155–116
Sargin I, Arslan G (2015) Chitosan/sporopollenin microcapsules: preparation, characterization and application in heavy metals removal. Int J Biol Macromol 75:230–238
Sitko R, Zawisza B, Malicka E (2013) Graphene as a new sorbent in analytical chemistry. TrAC Trends Anal Chem 51(0):33–43
Sun J, Liang Q, Han Q, Zhang X, Dingin M (2015) One-step synthesis of magnetic graphene oxide nanocomposite and its application in magnetic solid phase extraction of heavy metal ions from biological samples. Talanta 132:557–563
Tankiewicz M, Fenik J, Biziuk M (2010) Determination of organophosphorus and organonitrogen pesticides in water samples. TrAC Trends Anal Chem 29:1050–1063
Wan Ibrahim WA, Farhani H, Sanagi MM, Aboul-Enein HY (2010) Solid phase microextraction using new sol–gel hybrid polydimethylsiloxane-2-hydroxymethyl-18-crown-6 coated fiber for determination of organophosphorous pesticides. J Chromatogr A 1217:4890–4897
Wan Ibrahim WA, Ismail WNW, Keyon ASA, Sanagi MM (2011) Preparation and characterization of a new sol-gel hybrid based on tetraetho-xysilane-polydimethylsiloxane as a stir bar extraction sorbent materials. J Sol-Gel Sci Technol 58(3):602–611
Wan Ibrahim WA, Veloo KV, Sanagi MM (2012) Novel sol-gel hybrid methyltrimethoxysilane-tetraethoxysilane as solid phase extraction sorbent for organophosphorus pesticides. J Chromatogr A 1229:55–62
World Health Organisation (1990) Public health impact of pesticides uses in agriculture. 924 1561,94, 21–32. http://www.who.int/iris/handle/10665/39772
Wu Q, Zhao G, Feng C, Wang C, Wang Z (2011) Preparation of a graphene-based magnetic nanocomposite for the extraction of carbamate pesticides from environmental water samples. J Chromatogr A 1218(44):7936–7942
Xu B, Wang Y, Jin R, Li X, Song D, Zhang H, Sun Y (2015) Magnetic solid phase extraction based on Fe3O4@ polyaniline particles followed by ultrafast liquid chromatography for determination of Sudan dyes in environmental water samples. Anal Methods 7(4):1606–1614
Yusuff AS, Gbadamosi AO, Lala MA, Ngochindo JM (2018) Synthesis and characterization of anthill-eggshell composite adsorbent for removal of hexavlent chromium from aqueous solution. Environ Sci Pollut Res 1–12. https://doi.org/10.1007/s11356-018-2075-2
Zheng Z, Li H, Liu T, Cao R (2010) Monodisperse noble metal nanoparticles stabilized in SBA-15: synthesis, characterisation, and application in microwave-assisted Suzuku-Miyaura coupling reation. J Catal 270(2):268–274
Zheng HB, Zhao Q, Mo JZ, Huang YQ, Luo YB, Yu QW, Feng YQ (2013) Quick, easy, cheap, effective, rugged and safe method with magnetic graphitized carbon black and primary secondary amine as adsorbent and its application in pesticide residue analysis. J Chromatogr A 1300(0):127–133
Zhou Q, Fang Z (2015) Graphene-modified TiO2 nanotube arrays as an adsorbent in micro solid phase extraction for determination of carbamate pestici-des in water samples. Anal Chim Acta 869:43–49
Funding
One of the authors, Ayuba Markus, would like to convey a profound gratitude and sincere appreciation to Kaduna State Scholarship Board (KSSB), Nigeria, for financial support.
Author information
Authors and Affiliations
Corresponding author
Additional information
Responsible editor: Tito Roberto Cadaval Jr
Rights and permissions
About this article

Cite this article
Markus, A., Gbadamosi, A.O., Yusuff, A.S. et al. Magnetite-sporopollenin/graphene oxide as new preconcentration adsorbent for removal of polar organophosphorus pesticides in vegetables. Environ Sci Pollut Res 25, 35130–35142 (2018). https://doi.org/10.1007/s11356-018-3402-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11356-018-3402-3