
Abstract
The competitive binding trends of Ni(II) and Eu(III) on montmorillonite in the absence/presence of Na-oxalate are explored by using batch sorption/desorption technique, speciation modeling, and X-ray diffraction (XRD) analysis. With a series of molar Ni:Eu ratios (i.e., 1:1, 5:1, 10:1, 1:5, and 1:10), the coexisting Ni(II) did not affect the sequestration behaviors and immobilization mechanisms of Eu(III). In contrast, the presence of Eu(III) obviously suppressed the sorption percentages of Ni(II) in the acidic pH range. Even though no obvious influence of Eu(III) on the macroscopic binding trends of Ni(II) was observed under alkaline conditions, the fraction of Ni(II) adsorbed by the inner-sphere complexation mechanism decreased and that of Ni(II) precipitation increased with rising molar Ni:Eu ratio. The coexisting Na-oxalate did not influence Eu(III) sorption, while inhibited the sorption of Ni(II). The XRD analysis indicated the potential formation of two Eu-oxalate precipitate phases (i.e., Eu2(C2O4)3·xH2O(s)-1 and Eu2(C2O4)3·xH2O(s)-2) at different pH values (4.0 and 6.5) and Na-oxalate concentrations (ranging from 0.5 to 5.0 mM). Interestingly, the Eu2(C2O4)3·xH2O(s)-2 phase would be transformed into the Eu2(C2O4)3·xH2O(s)-1 solid with the increase of Na-oxalate concentration. The research findings could provide essential data for evaluating the fate of coexistent Eu(III) and Ni(II) in the complicated aquatic environment.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
The contamination of environmental systems by radionuclides with high and long-term radioactivity has received worldwide concern. 63Ni(II) (T1/2 = 96 a) is an activation product of steam-generating heavy water reactors (Steeb et al. 2009). 152+154Eu(III) is usually used as a neutron absorber for the control rods of the nuclear reactors (Xu et al. 2016). Numerous studies have proven that the coexistence of two metal elements induces a synergistic toxicity effect on aquatic organisms (Svecevicius et al. 2012; Pujol et al. 2013). Therefore, it is significant to have a clear understanding on the sorption behaviors and environmental fate of Ni(II) and Eu(III) in each other’s presence.
A series of studies have been performed to investigate the competitive immobilization mechanisms of coexisting metal ions on natural minerals (Bradbury and Baeyens 2005; Antoniadis and Tsadilas 2007; Sheikhhosseini et al. 2013; Yang et al. 2015). For instance, Bradbury and Baeyens (2005) explored the sorption trends of Co(II), Ni(II), Zn(II), Eu(III), Nd(III), Am(III), Th(IV), and U(VI) on montmorillonite under various combinations and concentrations of these metals. Interestingly, the competitive sorption behaviors only occurred between the metal ions with similar chemical properties (i.e., valence state, hydrolysis behavior, etc.), while the metal ions with diverse properties did not compete with one another. However, another study regarding the competitive sorption of Cu(II) (8.0 × 10−5~1.2 × 10−3 mol/L) and Eu(III) (1.0 × 10−4~1.2 × 10−3 mol/L) on titanium dioxide mineral showed a relative binding affinity of Eu(III) > Cu(II) via the cation exchange mechanism (Konstantinou and Pashalidis 2008). Keeping in mind that no compatible conclusion can be obtained from the previous literatures, the competitive sequestration mechanisms of Ni(II) and Eu(III) at the mineral/water interfaces should be further explored at different concentrations and molar ratios.
Oxalate, a widespread organic substance in the soil and aquatic systems, can alter the surface properties of minerals and the physicochemical behaviors of radionuclides via the dissolution, complexation, and/or precipitation reactions. Previous studies showed that oxalate strongly decreased Ni(II) sorption on hydrous silica and montmorillonite (Pathak and Choppin 2006; Marcussen et al. 2009), while synergistically enhanced the sorption of Eu(III) on hydrous silica (Pathak and Choppin 2007). More specifically, Guo et al. (2015) proposed that the effects of oxalate on Eu(III) sorption by Na-bentonite were closely related with the molar ligand/Eu ratios. However, Verma et al. (2014) found that the sorption of Eu(III) on bentonite was not affected by the coexisting oxalate. In summary, the foregoing studies focused on the role of oxalate on the sorption behaviors of individual Ni(II) or Eu(III). However, considering the heterogeneity and complication of the real environment, more research work is needed to further explore the effects of oxalate on the migration and transformation trends of coexisting Ni(II) and Eu(III) in the binary-metal systems.
In the present study, the competitive sequestration behaviors of Ni(II) and Eu(III) on a montmorillonite mineral were explored under various molar Ni:Eu ratios (i.e., multiple combinations of Ni(II) and Eu(III) concentrations), solution pH values, Na-oxalate concentrations, and contact time. Based on the experimental findings, a detailed analysis was performed to deduce the main factors that determine the relative sorption affinity of Ni(II) and Eu(III) on montmorillonite.
Materials and methods
Chemicals and materials
Ni(NO3)2·6H2O and Eu(NO3)3·6H2O in analytical pure were purchased from ENERGY CHEMICAL Co., Ltd. The stock solutions of Ni(II) and Eu(III) were prepared by dissolving Ni(NO3)2·6H2O and Eu(NO3)3·6H2O into the Milli-Q water, respectively. Montmorillonite mineral, sodium oxalate (Na-oxalate), and disodium ethylenediamine tetraacetate (2Na-EDTA) were obtained from Sinopharm Chemical Reagent Co., Ltd.
Characterization
The FTIR spectrum within 4000 to 400 cm−1 was obtained with a Thermo Nicolet 6700 spectrometer via the KBr pellet method. The XRD pattern of the montmorillonite sample was collected on a Bruker D8 Advance diffractometer with Cu-Kα (λ = 1.54056 Å) radiation. The zeta potentials and particle size were measured by a Malvern Zetasizer (632.8 nm, He-Ne laser). The specific surface area was determined from the N2 adsorption/desorption isotherms measured on a Quantachrome Autosorb gas sorption analyzer IQ2 at 77 K.
Macroscopic sorption and desorption experiments
The batch experiments of Ni(II) and Eu(III) sorption in the single- and binary-solute systems with various molar Ni:Eu ratios and Na-oxalate concentrations were performed under ambient conditions (see the details in the Supporting Information (denoted as SI in the following text)). In brief, the montmorillonite suspension, NaNO3 electrolyte solution, Na-oxalate, and Ni(II) and/or Eu(II) stock solutions were added to a series of 10-mL Teflon centrifuge tubes. The pH values were carefully adjusted by adding tiny HNO3 and/or NaOH solutions. The preliminary kinetics experiment indicated that the sorption of Ni(II) and Eu(III) on montmorillonite could reach equilibrium within a short contact time of 30 min (see Figure S1 in the Supporting Information (denoted as SI in the following text)). In view of this, the suspensions were gently shaken for 24 h to ensure complete equilibrium. Afterwards, the suspensions were centrifuged at 9000 rpm for 10 min and the supernatants were filtrated with 0.45-μm polypropylene membrane. The Ni(II) and/or Eu(III) concentrations in the filtrates were measured by inductively coupled plasma-atomic emission spectrometry (ICP-AES) (Thermo, iCAP-7000). The sorption percentages of Ni(II) and Eu(III) (sorption% = (C0-Ce)/C0 × 100%) were calculated from their initial (C0, mol/L) and equilibrium (Ce, mol/L) concentrations.
Desorption experiments by using CH3COONH4 and 2Na-EDTA as the eluents were further carried out to identify the contribution of ion exchange, inner-sphere surface complexation, and precipitation to total Ni(II) and/or Eu(III) adsorption under various conditions. The proportions of Ni(II) and/or Eu(III) retained by different mechanisms were expressed relative to their adsorption percentages (more detailed information of the desorption experiments is described in the SI).
XRD analyses
The XRD characterization would help verify the phase transformation of minerals and the potential formation of new metal-bearing precipitates. Herein, the Ni- and Eu(III)-loaded samples were prepared by using the same procedures as the above sorption experiments (“Macroscopic sorption and desorption experiments” section). Particularly, the reaction volumes were expanded to 250 mL so as to confirm the collection of enough samples. The wet pastes collected after centrifugation were dried in a vacuum oven and then grinded to powder. Afterwards, the powder samples were collected and analyzed by XRD.
Results and discussion
Characterization
Figure 1 shows the functional groups, mineralogy, surface potentials, and particle size of the purchased montmorillonite sample. Specifically for the FTIR spectrum (Fig. 1a), the peak at 3623 cm−1 is attributed to the stretching vibration of hydroxyl (–OH) groups that coordinated to the octahedral cations. The band at 1635 cm−1 originates from the bending vibration of –OH groups in H2O molecules (Tyagi et al. 2006). The peaks at 1010 cm−1 and 523 cm−1 represent the stretching and bending vibrations of Si–O bond, respectively (Tyagi et al. 2006; Paluszkiewicz et al. 2011). The weak absorption bands at 916 and 839 cm−1 are assigned to the bending vibrations of AlAlOH and AlMgOH, respectively (Madejová et al. 1999; Jóna et al. 2007; Paluszkiewicz et al. 2008; Paluszkiewicz et al. 2011). The peak at 467 cm−1 is due to the deformation vibration of Si–O–Si bond (Jóna et al. 2007). The XRD pattern (Fig. 1b) is consistent with the Ca-montmorillonite phase (marked by M, JCPDS card no. 13-0135) (Chen et al. 2017). The appearance of a characteristic (001) diffraction plane at two theta degrees of 5.87° corresponds to a basal spacing of 15.04 Å (Hu et al. 2018). The other diffraction peaks correspond to the quartz impurity (marked by Q) in the montmorillonite mineral. As shown in Fig. 1c, the montmorillonite surfaces would be negatively charged (i.e., negative zeta potential) at pH > 2.0. The particle size of montmorillonite distributes in the range of 200–400 nm (Fig. 1d). The specific surface area was measured to be 70.8 m2/g by using the N2-BET method.

FTIR spectrum (a), XRD pattern (b), zeta potential (c), and particle size distribution (d) of the montmorillonite sample
Macroscopic sorption data in the absence of Na-oxalate
Effect of solution pH value and initial metal concentration
Figure 2 shows the pH-dependent sorption trends of Eu(III) and Ni(II) in the single- and binary-solute systems with a molar Ni:Eu ratio of 1:1. For the single-solute system with an initial Eu(III) concentration of 5.0 × 10−4 mol/L, the sorption percentage increases from ~ 50 to ~ 100% as the pH value rises from 2.0 to 7.5 (see Fig. 2a). Afterwards, the sorption percent maintains at ~ 100% at alkaline pH level, which can be potentially attributed to the formation of hydroxide and/or carbonate precipitates as well as surface complexes (Rabung et al. 2005; Fan et al. 2009; Tan et al. 2009; Yang et al. 2014; Cai et al. 2015). Specifically, the sorption percentage of Eu(III) decreases with the increase of its initial concentration within the low pH range (Fig. 2a–c). This result can be interpreted by considering the initial concentration of Eu(III) and the active sites of montmorillonite. At lower Eu(III) concentration, the active sites of montmorillonite are abundant for its binding, and therefore the sorption percentage is high. With the increase of initial Eu(III) concentration, the aqueous Eu(III) species would compete for binding on the finite montmorillonite sites, which would result in the decrease of total Eu(III) sorption percentage. As shown in Fig. 2a–c, the sorption curves of Eu(III) in the binary-solute systems overlap well with those in the single-solute systems. This phenomenon suggests the weak competition of coexisting Ni(II) on Eu(III) sorption.

pH-dependent sorption trends of Eu(III) and Ni(II) in the single- and binary-solute systems at molar Ni/Eu ratio of 1:1 (CEu(III)initial = CNi(II)initial = 5.0 × 10−4 mol/L for a and d, CEu(III)initial = CNi(II)initial = 5.0 × 10−5 mol/L for b and e, CEu(III)initial = CNi(II)initial = 5.0 × 10−6 mol/L for c and f). T = 298 K, m/V = 0.5 g/L, I = 0.01 mol/L NaNO3
As shown in Fig. 2d, the sorption percentage of Ni(II) in the single-solute system with an initial concentration of 5.0 × 10−4 mol/L gradually rises from ~ 20 to ~40% with pH value increasing from 2.0 to 7.5, then sharply increases to ~ 100% as the pH value reaches 9.0. Previous surface complexation modeling, XRD, and extended X-ray absorption fine structure (EXAFS) analyses indicated that Ni(II) was retained by montmorillonite via the cation exchange reaction at low pH values, resulting in the formation of outer-sphere surface complexes. At neutral and alkalescent pH values, Ni(II) tended to form inner-sphere complexes by directly binding on the surface ≡AlOH and ≡SiOH sites. In contrast, Ni(II) would form the Ni-Al-layered double hydroxides (abbreviated to LDH), the Ni phyllosilicate, and/or the Ni(OH)2(s) precipitates at high alkaline pH values (Livi et al. 2009; Peltier et al. 2010; Yang et al. 2011, 2015). While in the binary-solute system (CNi(II)initial = CEu(III)initial = 5.0 × 10−4 mol/L, molar Ni:Eu = 1:1), the sorption of Ni(II) is significantly reduced by the coexisting Eu(III) at pH < 7.5 (Fig. 2d). Similarly, the sorption of Ni(II) for the other two concentrations (herein, 5.0 × 10−5 and 5.0 × 10−6 mol/L) is also inhibited in the presence of equimolar Eu(III) (Fig. 2e, f). The results herein suggest that the sorption affinity of montmorillonite for Ni(II) is lower than that for Eu(III). This conclusion is further supported by the experimental phenomenon that the sorption curves of Ni(II) are much lower than those of Eu(III) under the same concentrations (Fig. 2d vs. a, e vs. b, and f vs. c).
Effect of molar Ni:Eu ratios
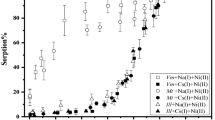
To further verify the sorption affinities of Eu(III) and Ni(II) on montmorillonite, additional experiments for the binary-solute systems were performed at different molar Ni:Eu ratios and the results are shown in Figs. 3 and 4. Meanwhile, the sorption curves of Eu(III) or Ni(II) in the corresponding single-solute systems are also illustrated for comparison. One can see from Fig. 3a that the coexisting Ni(II) with an initial concentration of 5.0 × 10−4 mol/L has no distinct influence on the sorption of Eu(III) at equal (CEu(III)initial = 5.0 × 10−4 mol/L, molar Ni:Eu ratio = 1:1) or even lower (CEu(III)initial = 1.0 × 10−4 mol/L, molar Ni:Eu ratio = 5:1 and CEu(III)initial = 5.0 × 10−5 mol/L, molar Ni:Eu ratio = 10:1) concentrations. In contrast, the presence of Eu(III) obviously reduces the sorption of Ni(II) at pH < 7.5 (Fig. 3b). More specifically, the competitive effect of Eu(III) gradually degrades with the increase of molar Ni:Eu ratio from 1:1 to 10:1 (i.e., the decrease of initial Eu(III) concentration from 5.0 × 10−4 to 5.0 × 10−5 mol/L). Similar results can be observed for the competitive sorption trends of Eu(III) and Ni(II) with molar Ni:Eu ratios of 1:1, 1:5, and 1:10 (Fig. 4). Interestingly, an enhanced inhibitory effect of Eu(III) on Ni(II) sorption (CNi(II)initial = 5.0 × 10−5 mol/L) is observed with the decrease of molar Ni:Eu ratio from 1:1 (CEu(III)initial = 5.0 × 10−5 mol/L) to 1:5 (CEu(III)initial = 2.5 × 10−4 mol/L), while the coexisting Eu(III) shows no further competitive effect as the molar Ni:Eu ratio reaches to 1:10 (CEu(III)initial = 5.0 × 10−4 mol/L) (Fig. 4b). This phenomenon indicates that the competitive sorption trends of Eu(III) and Ni(II) are greatly dependent on their initial concentration (or the molar Ni:Eu ratios) in solution. According to the experimental data herein, one can infer that Eu(III) and Ni(II) tend to bind on the same type of montmorillonite sites with an affinity order of Eu(III) > Ni(II). In the binary-solute systems, the active sites of montmorillonite would be preferentially occupied by Eu(III). As a result, fewer sites are available for binding Ni(II). The specific factors that account for this competitive sorption sequence would be analyzed and summarized in the later part of the text.

pH-dependent sorption trends of Eu(III) (a) and Ni(II) (b) (CNi(II)initial = 5.0 × 10−4 mol/L) in the single- and binary-solute systems at molar Ni:Eu ratios of 1:1 (CEu(III)initial = 5.0 × 10−4 mol/L), 5:1 (CEu(III)initial = 1.0 × 10−4 mol/L), and 10:1 (CEu(III)initial = 5.0 × 10−5 mol/L). T = 298 K, m/V = 0.5 g/L, I = 0.01 mol/L NaNO3

pH-dependent sorption trends of Eu(III) (a) and Ni(II) (b) (CNi(II)initial = 5.0 × 10−5 mol/L) in the single- and binary-solute systems at molar Ni:Eu ratios of 1:1 (CEu(III)initial = 5.0 × 10−5 mol/L), 1:5 (CEu(III)initial = 2.5 × 10−4 mol/L), and 1:10 (CEu(III)initial = 5.0 × 10−4 mol/L). T = 298 K, m/V = 0.5 g/L, I = 0.01 mol/L NaNO3
Desorption results
Figure 5 shows the relative contribution of ion exchange, surface complexation, and precipitation to the immobilization of Ni(II) and Eu(III) by montmorillonite. These data are summarized from the desorption experiments. For all the single- and binary-solute systems at an acidic pH of 4.0, Eu(III) is predominately adsorbed via inner-sphere surface complexation with an extremely low contribution of ion exchange/outer-sphere complexation (Fig. 5a). At an alkaline pH value of 8.0, surface complexation remains the primary retention mechanism for most of the cases with lower Eu(III) concentrations (Fig. 5b). However, precipitation becomes an important driving force for Eu(III) immobilization at higher concentrations of 2.5 × 10−4 (binary-solute system with molar Ni:Eu = 1:5) and 5.0 × 10−4 mol/L (single-solute and binary-solute system with molar Ni:Eu = 1:10). More than 50% of Eu(III) is retained by forming insoluble precipitates. As shown in Fig. 5b, the relative contribution of inner-sphere surface complexation and precipitation to total Eu(III) adsorption in the binary-solute system with a molar Ni:Eu ratio of 1:10 (CNi(II)initial = 5.0 × 10−5 mol/L, CEu(III)initial = 5.0 × 10−4 mol/L) is comparable to that in the corresponding single-solute system with the same Eu(III) concentration. Similar phenomena can be also observed for the other systems with various Ni:Eu ratios.

Desorption of Eu(III) and Ni(II) in the single- and binary-solute systems at pH 4.0 (a, c) and 8.0 (b, d). T = 298 K, m/V = 0.5 g/L, I = 0.01 mol/L NaNO3
As shown in Fig. 5c, Ni(II) is adsorbed via both ion exchange and inner-sphere surface complexation at an acidic pH value of 4.0. In the binary-solute systems with various Ni:Eu ratios, the coexisting Eu(III) reduces the proportion of Ni(II) adsorbed by surface complexation, while scarcely influences the fraction of Ni(II) retained by ion exchange. Herein, the montmorillonite mineral used in this study shows a higher affinity towards Eu(III) than Ni(II). Under such circumstances, Eu(III) preferentially occupies the active sites on montmorillonite surfaces and correspondingly inhibits the binding of Ni(II). When comparing Fig. 5d with Fig. 5c, one can clearly find that the proportion of Ni(II) inner-sphere surface complexation increases as the solution pH value rises from 4.0 to 8.0. In addition, a portion of Ni(II) is also immobilized via the formation of insoluble precipitates at an alkaline pH value of 8.0, the amounts of which depend on the initial Ni(II) concentration and molar Ni:Eu ratio. Specifically for an initial Ni(II) concentration of 5.0 × 10−4 mol/L, the relative proportion of precipitation shows an apparently decreasing trend with the increase of molar Ni:Eu ratio from 1:1 to 5:1 and 10:1 (i.e., CEu(III)initial decreases from 5.0 × 10−4 mol/L to 1.0 × 10−4 mol/L and 5.0 × 10−5 mol/L) (Fig. 5d). Accordingly, the fraction of inner-sphere complexation increases with increasing Ni:Eu ratio. As shown in Fig. 3b, the coexisting Eu(III) with different concentrations has no obvious effect on the macroscopic adsorption behaviors of Ni(II) (CNi(II)initial = 5.0 × 10−4 mol/L) at pH 8.0. However, the desorption results herein verify the alteration of the Ni(II) immobilization mechanism in the presence of Eu(III). For an initial Ni(II) concentration of 5.0 × 10−4 mol/L, inner-sphere surface complexation is the predominant immobilization mechanism of Ni(II) and its fraction is similar for different Ni:Eu ratios of 1:1 (CEu(III)initial = 5.0 × 10−5 mol/L), 1:5 (CEu(III)initial = 2.5 × 10−5 mol/L), and 1:10 (CEu(III)initial = 5.0 × 10−4 mol/L). By combining the experimental results illustrated in Figs. 2, 3, 4, and 5, one can make a conclusion that the coexisting Ni(II) has no influence on both the macroscopic adsorption behaviors and microscopic sequestration mechanisms of Eu(III) by montmorillonite over a wide range of pH value and Ni:Eu ratio. In contrast, the presence of Eu(III) obviously changes the immobilization mechanisms of Ni(II) under some specific conditions.
Macroscopic sorption data in the presence of Na-oxalate
Figure 6 shows the sorption percentages of Eu(III) and Ni(II) in the absence/presence of Na-oxalate. Specifically, the coexisting Na-oxalate has no apparent effect on Eu(III) sorption, while obviously reduces the sorption of Ni(II). In general, the coexisting ligands can enhance the immobilization of metal ions on solid surfaces by creating a favorable electrostatic environment, by the generation of ternary surface complexes and by the occurrence of surface co(precipitation). In contrast, the complexing agents can also inhibit metal ion sequestration by competing for the same sites, by producing a barrier that prevents metal ions from binding on the available sites and by forming soluble complexes with metal ions (Collins et al. 1999; Buerge-Weirich et al. 2002; Yang et al. 2011, 2013; Guo et al. 2015). Herein, the distinct effects of Na-oxalate can be interpreted from their intrinsic properties and complexation/precipitation behaviors with Eu(III) and Ni(II). To help interpret the macroscopic sorption data, the pH-dependent speciation of Eu(III) and Ni(II) in the absence/presence of Na-oxalate is simulated by using Visual Minteq 3.1 (Gustafsson n.d.) and the results are illustrated in Figures S2-S4. Since the coexisting Na-oxalate exhibits a similar impact on the sorption behaviors of Eu(III) and Ni(II) at pH values of 4.0 and 6.5 (Fig. 6), the discussion herein is only based on the speciation of metal ions at pH 4.0.

Sorption of Eu(III) and Ni(II) in the binary-solute systems at pH 4.0 and 6.5 with different concentrations of Na-oxalate. T = 298 K, m/V = 0.5 g/L, CEu(III)initial = CNi(II)initial = 5.0 × 10−5 mol/L, I = 0.01 mol/L NaNO3
In the absence of oxalate, Eu(III) in solution is present as ~ 90% of Eu3+ ions and ~ 10% of EuNO32+ species at pH 4.0 (Figure S2A). The presence of Na-oxalate at 0.5 mM and 5.0 mM alters the species of Eu(III) from Eu3+ ions (Figure S2A) to Eu-(oxalate)2− and Eu-oxalate+ (Figures S3A and S4A). It is worth noting that the expectative Eu-oxalate precipitate (Eu2(C2O4)3(s)) is absent due to the lack of thermodynamic data for this phase in the database of Visual Minteq. Oxalate is extensively used as a precipitant for heavy metal ions in solution (Sun and Qiu 2012). Herein, the aqueous medium conditions applied in our experiments (i.e., CEu(III)initial = 5.0 × 10−5 mol/L, CNa-oxalate = 0.5 mM or 5.0 mM) are supersaturated with respect to Eu2(C2O4)3(s) with a solubility product constant of 10−26 at 25 °C (Tsukahara et al. 2004). Hence, the potential formation of Eu2(C2O4)3(s) phase may be predominantly responsible for the high sorption percentage of Eu(III) with 0.5 mM or 5.0 mM Na-oxalate (Fig. 6). However, Alliot et al. (2006) found that the increase of oxalate concentration from 0.1 mM to 0.1 M resulted in a monotonic decrease on the sorption amount of Eu(III) by α-alumina. In contrast, the coexisting oxalate with a concentration of 1.0 mM significantly enhanced the sorption of Eu(III) (1.0 × 10−4 mol/L) on suspended silica (Pathak and Choppin 2007). More interestingly, Guo et al. (2015) reported that the presence of oxalate enhanced Eu(III) sorption at pH 4.0 when their concentrations were 2.0 × 10−4 mol/L. In contrast, the coexisting oxalate reduced Eu(III) sorption when the initial concentration of Eu(III) was changed to 2.0 × 10−7 mol/L. In summary, the different experimental findings as described above suggest that the specific effect of oxalate on Eu(III) sorption is dependent on their relative concentrations and the physicochemical properties of different minerals. In the case of Ni(II), the speciation modeling (Figures S3B and S4B) does not reflect the presence of Ni-oxalate precipitate (NiC2O4·2H2O(s)) as reported in previous studies (Allen 1953; Magyarosy et al. 2002). Herein, the solution conditions (i.e., CNi(II)initial = 5.0 × 10−5 mol/L, CNa-oxalate = 0.5 mM or 5.0 mM) are supersaturated with respect to NiC2O4·2H2O(s) with a solubility product constant of 10−10 to 10−8 (Gavris et al. 2008). If this is the case, the sorption of Ni(II) would be enhanced. However, a contrary phenomenon is observed, i.e., the presence of Na-oxalate greatly suppresses the sorption of Ni(II) (Fig. 6). It seems that the precipitation of Eu(III) with Na-oxalate may inhibit the potential precipitation of Ni(II) with this ligand. Alternatively, Ni(II) tends to interact with Na-oxalate in solution and forms soluble Ni-oxalate(aq) and Ni-(oxalate)22− species (see Figures S3B and S4B), which competitively diminish the binding of Ni(II) on montmorillonite.
XRD analysis of metal-loading samples
To help identify the underlying adsorption mechanisms of Eu(III) and Ni(II), the XRD patterns of metal ion-loaded montmorillonite samples in the absence/presence of Na-oxalate were collected and analyzed in detail. As shown in Fig. 7, no obvious changes on the XRD pattern of montmorillonite can be observed after the sorption of Eu(III) and Ni(II). According to the desorption experimental results (Fig. 5 and “Effect of molar Ni:Eu ratios” section ), nearly all the Eu(III) is sequestrated by the inner-sphere surface complexation mechanism at pH 4.0, while Ni(II) is immobilized via the combination of ion exchange (~ 10%) and inner-sphere surface complexation (~ 20%). Anyway, the total amount of adsorbed Ni(II) is too low to alter the crystalline structure of montmorillonite. Besides, the absence of new diffraction peaks after Eu(III) and Ni(II) sorption (Fig. 7) rules out the potential precipitation of metal hydroxides and carbonate phases.

XRD patterns of metal-loaded samples at pH 4.0 and 6.5 with 5.0 mM Na-oxalate. T = 298 K, m/V = 0.5 g/L, CEu(III)initial = CNi(II)initial = 5.0 × 10−5 mol/L, I = 0.01 mol/L NaNO3
In the presence of 5.0 mM Na-oxalate, the first diffraction peak of montmorillonite sample shows an apparent shift to a higher 2θ value after Eu(III) and Ni(II) loading (Fig. 7). In other words, the basal spacing of montmorillonite decreases after its interaction with Eu(III), Ni(II), and Na-oxalate. It is possible that the interlaminar Ca2+ ions with a hydrated ionic radius (RH) of 4.12 Å are captured by the coexisting Na-oxalate via the precipitation reaction (Slngh et al. 1991; Christensen et al. 2003; Ihli et al. 2015). Instead, the Na+ (originating from the addition of electrolyte NaNO3) and H+ (originating from the acidity of the solution) ions with smaller hydrated RH values would enter the interlayer to maintain the charge balance of the montmorillonite structure (Fourest et al. 1984; Volkov et al. 1997; Yang et al. 2015). In addition, the appearance of new diffraction peaks at 12.2°, 14.3°, 14.8°, and 24.4° (marked as asterisks in Fig. 7) clearly implies the formation of a new solid phase. Specifically, this solid is expected to be the metal-oxalate precipitates due to their low solubility products (Tsukahara et al. 2004; Gavris et al. 2008).
To verify the origin and chemical phase of the formed precipitates, the single Eu(III)-loaded montmorillonite sample in the presence of 5.0 mM Na-oxalate was also prepared and the XRD pattern was collected for comparison. As shown in Fig. 8, some new diffraction peaks appear at 12.2°, 14.3°, 14.8°, and 24.4°, all of which are present in the XRD pattern of Eu(III)- and Ni(II)-loaded sample with coexisting Na-oxalate (Fig. 7). In contrast, the XRD pattern of the Eu(III)- and Ni(II)-loaded sample is apparently different from that of NiC2O4·2H2O(s). These two phenomena suggest that the precipitate formed in the binary-solute system is the Eu-oxalate solid rather than the Ni-oxalate phase. As shown in Fig. 8, the Eu-oxalate solid formed in our experiments is not the europium oxalate decahydrate (Eu2(C2O4)3·10H2O(s)) (Hong et al. 2014). Theoretically, there are a series of Eu-oxalate phases containing different H2O molecules in their crystal structures (Balboul et al. 2002; Kustaryono et al. 2010; Zhan et al. 2012; Hong et al. 2014). Owing to the lack of the crystallographic information files for the other Eu2(C2O4)3·xH2O(s) solids, it is impossible to clearly identify the specific chemical phase of the formed precipitate herein. Nevertheless, the precipitation of Eu-oxalate phase is undeniably an important driving force for the immobilization of Eu(III). In addition, Eu(III) and Ni(II) can be also adsorbed via the formation of binary (e.g., montmorillonite-Eu(III)/Ni(II)) and/or ternary complexes (e.g., montmorillonite-oxalate-Eu(III)/Ni(II) or montmorillonite-Eu(III)/Ni(II)-oxalate) as proposed in a series of sorption systems (Montavon et al. 2002, 2004; Strathmann and Myneni 2005; Yang et al. 2011, 2013, 2015). The broadening of the first diffraction peak of montmorillonite after metal sorption may be a circumstantial evidence for the occurrence of this binding mode. Additional desorption/dissolution experiments and advanced spectral techniques (e.g., time-resolved laser fluorescence spectroscopy and EXAFS) are needed in the following studies to further identify the surface complexes.

XRD patterns of standard Eu(III)- or Ni(II)-oxalate solids and metal-loaded samples at pH 4.0 with 5.0 mM Na-oxalate. T = 298 K, m/V = 0.5 g/L, CEu(III)initial = CNi(II)initial = 5.0 × 10−5 mol/L, I = 0.01 mol/L NaNO3
According to the macroscopic data as shown in Fig. 6, the variation of pH values from 4.0 to 6.5 and the increase of Na-oxalate from 0.5 to 5.0 mM have no detectable impact on the sorption percentage of Eu(III). In view of this, the XRD patterns of the sorption samples are analyzed herein to determine whether the same solid phase is formed in these systems. In the presence of 5.0 mM Na-oxalate, the XRD pattern of the sample prepared at pH 4.0 is identical to that prepared at pH 6.5 (Fig. 9), pointing to the formation of the same Eu-oxalate precipitate phase (denoted as Eu2(C2O4)3·xH2O(s)-1). In contrast, the XRD pattern of the sample prepared at pH 4.0 in the presence of 0.5 mM Na-oxalate exhibits some other diffraction peaks at 12.7°, 13.2°, 18.0°, and 18.6° (marked as plus signs in Fig. 9), which suggests the generation of a different Eu-oxalate solid (denoted as Eu2(C2O4)3·xH2O(s)-2). More interestingly, the XRD pattern of the sample prepared at pH 6.5 with 0.5 mM Na-oxalate shows the diffraction peaks of Eu2(C2O4)3·xH2O(s)-1 (marked as asterisks) and Eu2(C2O4)3·xH2O(s)-2 (marked as plus signs), implying the simultaneous presence of these two solid phases under the applied solution conditions. Based on these observations, one can deduce that Eu(III) and the coexisting oxalate tend to form disparate kinds of precipitates at different pH values and Na-oxalate concentrations, even though the sorption percentage is identical under these conditions (Fig. 6).

XRD patterns of metal-loaded samples at pH 4.0 and 6.5 with 0.5 mM and 5.0 mM Na-oxalate. T = 298 K, m/V = 0.5 g/L, CEu(III)initial = CNi(II)initial = 5.0 × 10−5 mol/L, I = 0.01 mol/L NaNO3
It is worth noting that there is a ten-fold difference on the Na-oxalate concentrations (i.e., 5.0 mM vs. 0.5 mM) used in the foregoing experiments, under which conditions we observe the generation of two different Eu-oxalate solids. In view of this, the XRD patterns of the metal-loaded samples with Na-oxalate concentration ranging from 0.5 to 5.0 mM are analyzed to carefully determine the effects of this carboxylate. At pH 4.0 with 0.5 mM and 2.5 mM Na-oxalate, the XRD patterns indicate the formation of a single Eu2(C2O4)3·xH2O(s)-2 phase (with the specific diffraction peaks marked as plus signs in Fig. 10a). While at a higher Na-oxalate concentration of 3.3 mM, it is interesting to find that the Eu2(C2O4)3·xH2O(s)-1 precipitate (with the characteristic diffraction peaks marked as asterisks in Fig. 10a) begins to form along with the Eu2(C2O4)3·xH2O(s)-2 phase. However, the Eu2(C2O4)3·xH2O(s)-2 phase disappears as the concentration of Na-oxalate increases up to 4.0 mM and 5.0 mM, while the Eu2(C2O4)3·xH2O(s)-1 phase remains with good crystallinity. The XRD patterns at pH 6.5 show a different trend with the change of Na-oxalate concentration (Fig. 10b). None of the XRD patterns indicates the presence of a pure Eu2(C2O4)3·xH2O(s)-1 solid. Instead, the simultaneous existence of Eu2(C2O4)3·xH2O(s)-1 (with the characteristic diffraction peaks marked as asterisks) and Eu2(C2O4)3·xH2O(s)-2 (with the specific diffraction peaks marked as plus signs) phases is observed even at a low Na-oxalate concentration of 0.5 mM. As the concentration of Na-oxalate rises to 2.5 mM or higher, the XRD patterns show a single Eu2(C2O4)3·xH2O(s)-1 phase. These observations mean that the Eu2(C2O4)3·xH2O(s)-2 phase formed at lower Na-oxalate concentration would gradually transforms into the Eu2(C2O4)3·xH2O(s)-1 solid with the increase of ligand concentration. Specifically, the critical concentration of Na-oxalate for this phase transformation process is identified to be in the range of 2.5–4.0 mM at pH 4.0, while in the range of 0–2.5 mM at pH 6.5. The concentration of natural organic matters in the aquatic environment is usually variable along with the water flowing and biological activities (Jones 1998). As a result, the metal ions therein would form diverse precipitates with disparate thermodynamic stability, which would correspondingly influence their immobilization and environmental fate.

XRD patterns of metal-loaded samples at pH 4.0 (a) and 6.5 (b) with different Na-oxalate concentrations. T = 298 K, m/V = 0.5 g/L, CEu(III)initial = CNi(II)initial = 5.0 × 10−5 mol/L, I = 0.01 mol/L NaNO3
Aging time is an important factor that influences the formation of metal precipitates (Reiller and Casanova 2005; Singh et al. 2010; Yang et al. 2012, 2013). In view of this, the time-dependent XRD patterns of the metal-loaded samples with 5.0 mM and 0.5 mM Na-oxalate were analyzed to identify the kinetics growing process of the Eu2(C2O4)3·xH2O(s) phase. In the presence of 5.0 mM Na-oxalate, the appearance of a rough diffraction peak at 24.4° (marked as asterisk in Fig. 11a) after a short reaction time of 10 min suggests that the formation of a new phase begins. As the aging time prolonged to 60 min, the XRD pattern shows additional broad and rough diffraction peaks at 12.2°, 14.3°,and 14.8° (marked as asterisks), pointing to the gradual crystal growth of the Eu2(C2O4)3·xH2O(s)-1 phase. Afterwards, these diffraction peaks become more smooth and sharp at an aging time of 300 min. This phenomenon can be ascribed to the increased amount and the enhanced crystallinity of the Eu2(C2O4)3·xH2O(s)-1 phase. The XRD patterns of the metal-loaded samples at 600 min and 1440 min are almost the same as those at 300 min. This phenomenon indicates that an aging time of 300 min is necessary for the precipitation of Eu2(C2O4)3·xH2O(s)-1 phase. Similarly, the variation trend for the time-dependent XRD patterns with the presence of 0.5 mM Na-oxalate indicates the formation of Eu2(C2O4)3·xH2O(s)-2 phase after an aging time of 300 min (Fig. 11b). By comparing Fig. 11a with Fig. 11b, one can see that the increase of aging time only improves the amount and crystallinity of a specific Eu-oxalate solid, while does not alter its chemical form during the period of 10 min to 1440 min. The migration of heavy metal ions in the real water systems may undergo over a relatively long period from days to months even years (Reiller and Casanova 2005; Singh et al. 2010; Yang et al. 2012, 2013). In view of this, more studies, e.g., desorption/dissolution experiments, XRD, and advanced spectral analysis for metal-loaded samples at prolonged aging time (i.e., 24 h–1 year), are needed to clearly predict the migration and transformation behaviors of Eu(III) and Ni(II) with coexisting Na-oxalate.

Time-dependent XRD patterns of the metal-loaded samples at pH 4.0 in the presence of 5.0 mM and 0.5 mM Na-oxalate. T = 298 K, m/V = 0.5 g/L, CEu(III)initial = CNi(II)initial = 5.0 × 10−5 mol/L, I = 0.01 mol/L NaNO3
Selective order and competitive binding mechanisms
According to the above macroscopic and XRD observations, Eu(III) is preferentially sequestrated on montmorillonite with no distinct interference from the coexisting Ni(II) in the whole pH range. In contrast, the sorption behavior of Ni(II) is obviously impacted by the presence of Eu(III). It seems that the same active sites of montmorillonite are involved in the immobilization of Eu(III) and Ni(II). Specifically, Eu(III) exhibits a higher affinity for binding on these sites and its sorption percentage is higher than that of Ni(II) over a wide pH range. In addition, the tendency of Eu(III) to form precipitates with Na-oxalate is higher than that of Ni(II). The potential sequestration mechanisms of Eu(III) and Ni(II) by montmorillonite in the absence and presence of Na-oxalate are schematically illustrated in Fig. 12. An in-depth discussion is required to verify the influencing factors for the selective order of Eu(III) > Ni(II) derived herein.

Potential sequestration mechanisms of Eu(III) and Ni(II) by montmorillonite in the absence and presence of Na-oxalate
The different interaction affinities of metal ions with minerals and organic ligands may be induced by their differences in the hydrated ionic radius, electrovalence, acid-base property, first hydrolysis constant, and Misono softness parameter (Shaheen et al. 2013; Sheikhhosseini et al. 2013; Yang et al. 2015). In general, the strength of electrostatic interaction between metal ions and clay minerals is in inverse proportion to their hydrated ionic radius (RH). Specifically, in the case of Eu(III) and Ni(II), the RH value of Eu3+ ions (4.58 Å) is greater than that of Ni2+ ions (Fourest et al. 1984; Volkov et al. 1997; Trivedi et al. 2001; Yang et al. 2015). Hence, Eu3+ ions are expected to be weakly retained by montmorillonite. However, a contrary result is obtained in our experiments. Hence, the hydrated ionic radius and electrostatic interaction are not responsible for the selective order of Eu(III) > Ni(II). Alternatively, one can consider the electrovalence of metal ions and the corresponding cation exchange mechanism. The Eu3+ ions with a higher electrovalence would have a stronger competitiveness for binding on montmorillonite (Flogeac et al. 2007; Valisko and Boda 2007). According to the hard and soft acids and bases theory, Eu3+ is a hard Lewis acid, while Ni2+ is a borderline Lewis acid (Remko et al. 2008; Suneesh et al. 2012; Kaur et al. 2013). Hence, Eu(III) has a greater affinity for complexing with the hydroxyl sites on montmorillonite surfaces or the carboxylic groups of the coexisting Na-oxalate, which are identified as hard Lewis bases. The relative capacity of metal ions to form inner-sphere complexes on the mineral surfaces (denoted as specific sorption) is closely related to their first hydrolysis products (i.e., the pK value of the first hydrolysis reaction: M2+ + H2O = MOH+ + H+). Generally, the metal ion with a lower first hydrolysis constant is easier to be specifically adsorbed (Saha et al. 2002). The first hydrolysis constant for Eu(III) is 7.3 (Caceci and Choppin 1983), whereas that for Ni(II) is 9.6 (McKenzie 1980). In view of this, Eu(III) is more preferentially retained on the montmorillonite surfaces than Ni(II). The Misono softness parameter is an index of the tendency of a metal ion to form covalent bonds with the active sites on minerals (Flogeac et al. 2007). Herein, the preferential retention of Eu(III) on montmorillonite is partly attributed to its higher Misono softness value of (~ 0.36) than that of Ni(II) (~ 0.25) (Misono et al. 1967; Thompson et al. 2013).
Conclusions
The competitive binding of coexisting toxic components at the natural mineral/water interfaces exhibits a significant impact on their physicochemical behaviors. Herein, the macroscopic data and the XRD analysis results are helpful to better understand the speciation, migration, transformation, and potential risk of the coexistent Ni(II) and Eu(III) in the aquatic systems. Specifically, Ni(II) has no obvious effect on the adsorption behaviors and sequestration mechanisms of Eu(III) by montmorillonite at a series of solution pH values and molar Ni:Eu ratios. In contrast, the coexisting Eu(III) reduces the fraction of Ni(II) inner-sphere complexation at both the acidic and alkaline pH values. It seems that the active sites of montmorillonite are preferentially occupied by Eu(III). As a result, a large proportion of Ni(II) would remain and continuously migrate in the aqueous solution, inducing serious threat to the health of the aquatic organisms. The low molecular weight Na-oxalate, being widely present in the soil and water systems, reduces the sorption of Ni(II) on montmorillonite. It is interesting to find that the presence of Na-oxalate does not influence the macroscopic behavior of Eu(III), while induces the generation of different Eu2(C2O4)3·xH2O(s) solids at different pH values and ligand concentrations. The formed precipitates would gradually sink into the sediments and correspondingly reduce the concentration and chemical mobility of Eu(III). Considering the complexity of actual aquatic systems and the limitations of our experimental conditions, further studies are ongoing in our lab to supplement the competitive sequestration mechanisms of Ni(II) and Eu(III) before extending to the real multicomponent environment.
Change history
17 February 2020
In the original publication Fig. 10b was erroneously plotted due to the authors��� carelessness and unintentional misoperation.
References
Allen JA (1953) The precipitation of nickel oxalate. J Phys Chem 57:715–716. https://doi.org/10.1021/j150508a027
Alliot C, Bion L, Mercier F, Toulhoat P (2006) Effect of aqueous acetic, oxalic, and carbonic acids on the adsorption of europium(III) onto α-alumina. J Colloid Interface Sci 298:573–581. https://doi.org/10.1016/j.jcis.2006.01.004
Antoniadis V, Tsadilas CD (2007) Sorption of cadmium, nickel, and zinc in mono- and multimetal systems. Appl Geochem 22:2375–2380. https://doi.org/10.1016/j.apgeochem.2007.06.001
Balboul BAA, El-Roudi AM, Samir E, Othman AG (2002) Non-isothermal studies of the decomposition course of lanthanum oxalate decahydrate. Thermochim Acta 387:109–114. https://doi.org/10.1016/S0040-6031(01)00834-6
Bradbury MH, Baeyens B (2005) Experimental measurements and modeling of sorption competition on montmorillonite. Geochim Cosmochim Acta 69:4187–4197. https://doi.org/10.1016/j.gca.2005.04.014
Buerge-Weirich D, Hari R, Xue HB, Behra P, Sigg L (2002) Adsorption of Cu, Cd, and Ni on goethite in the presence of natural groundwater ligands. Environ Sci Technol 36:328–336. https://doi.org/10.1021/es010892i
Caceci MS, Choppin GR (1983) The determination of the first hydrolysis constant of Eu(III) and am(III). Radiochim Acta 33:101–104. https://doi.org/10.1524/ract.1983.33.23.101
Cai YW, Ren XM, Lang Y, Liu ZY, Zong PF, Wang XK, Yang ST (2015) Sequestration and speciation of Eu(III) on gamma alumina: role of temperature and contact order. Environ Sci Processes Impacts 17:1904–1914. https://doi.org/10.1039/c5em00412h
Chen TX, Zhao YL, Li HL, Song SX (2017) Effects of colloidal montmorillonite particles on froth flotation of graphite, galena and fluorite. Physicochem Probl Mi 53:699–713. https://doi.org/10.5277/ppmp170202
Christensen T, Gooden DM, Kung JE, Toone EJ (2003) Additivity and the physical basis of multivalency effects: a thermodynamic investigation of the calcium EDTA interaction. J Am Chem Soc 125:7357–7366. https://doi.org/10.1021/ja021240c
Collins CR, Ragnarsdottir KV, Sherman DM (1999) Effect of inorganic and organic ligands on the mechanism of cadmium sorption to goethite. Geochim Cosmochim Acta 63:2989–3002. https://doi.org/10.1016/S0016-7037(99)00226-4
Fan QH, Tan XL, Li JX, Wang XK, Wu WS, Montavon G (2009) Sorption of Eu(III) on attapulgite studied by batch, XPS, and EXAFS techniques. Environ Sci Technol 43:5776–5782. https://doi.org/10.1021/es901241f
Flogeac K, Guillon E, Aplincourt M (2007) Competitive sorption of metal ions onto a north-eastern France soil. Isotherms and XAFS studies. Geoderma 139:180–189. https://doi.org/10.1016/j.geoderma.2007.01.016
Fourest B, Duplessis J, David F (1984) Comparison of diffusion coefficients and hydrated radii for some trivalent lanthanide and actinide ions in aqueous solution. Radiochim Acta 36:191–195. https://doi.org/10.1524/ract.1984.36.4.191
Gavris G, Stanãsel O, Pode R, Stoia M, Chitac V (2008) Study upon the recuperative purging of nickel and cobalt ions from residual solutions by means of chemical precipitation. Rev Chim 59:61–64
Guo N, Yang JW, Zhang R, Ye YL, Wu WS, Guo ZJ (2015) Effects of organic acids on Eu(III) sorption on Na-bentonite. J Radioanal Nucl Ch 303:2185–2192. https://doi.org/10.1007/s10967-014-3768-8
Gustafsson JP (n.d.) Visual MINTEQ ver. 3.1. Department of Land and Water Resources Engineering, KTH (Royal Institute of Technology), SE-100 44, Stockholm, Sweden, Available at http://vminteq.lwr.kth.se/download/
Hong F, Zhou LQ, Li L, Xia Q, Ye ML (2014) Hydrothermal synthesis and properties of a red-emitting phosphor of fully concentrated Eu3+ based oxalate Eu2(C2O4)3·10H2O. Mater Res Bull 60:252–257. https://doi.org/10.1016/j.materresbull.2014.08.036
Hu W, Lu S, Song W, Chen T, Hayat T, Alsaedi NS, Chen C, Liu H (2018) Competitive adsorption of U(VI) and Co(II) on montmorillonite: a batch and spectroscopic approach. Appl Clay Sci 157:121–129. https://doi.org/10.1016/j.clay.2018.02.030
Ihli J, Wang YW, Cantaert B, Kim YY, Green DC, Bomans PHH, Sommerdijk NAJM, Meldrum FC (2015) Precipitation of amorphous calcium oxalate in aqueous solution. Chem Mater 27:5347–5367. https://doi.org/10.1021/acs.chemmater.5b01642
Jóna E, Rudinská G, Sapietová M, Pavlík V, Drábik M, Mojumdar SC (2007) Interactions of different heterocyclic compounds with monoionic forms of montmorillonite: thermal, IR-spectral and X-ray studies of Ni(II)-montmorillonite with 3-R-and 2-R pyridines (R=CH3, Cl, NH2). J Therm Anal Calorim 90:687–691. https://doi.org/10.1007/s10973-007-8523-8
Jones DL (1998) Organic acids in the rhizosphere - a critical review. Plant Soil 205:25–44. https://doi.org/10.1023/A:1004356007312
Kaur M, Zhang HJ, Martin L, Todd T, Qiang Y (2013) Conjugates of magnetic nanoparticle-actinide specific chelator for radioactive waste separation. Environ Sci Technol 47:11942–11959. https://doi.org/10.1021/es402205q
Konstantinou M, Pashalidis L (2008) Competitive sorption of Cu(II), Eu(III) and U(VI) ions on TiO2 in aqueous solutions-a potentiometric study. Colloid Surf A 324:217–221. https://doi.org/10.1016/j.colsurfa.2008.04.020
Kustaryono D, Kerbellec N, Calvez G, Freslon S, Daiguebonne C, Guillou O (2010) New family of porous lanthanide-containing coordination polymers: [Ln2(C2O4)3(H2O)6·12H2O](infinity) with Ln=La-Yb or Y. Cryst Growth Des 10:775–781. https://doi.org/10.1021/cg9011668
Livi KJT, Senesi GS, Scheinost AC, Sparks DL (2009) Microscopic examination of nanosized mixed Ni-Al hydroxide surface precipitates on pyrophyllite. Environ Sci Technol 43:1299–1304. https://doi.org/10.1021/es8015606
Madejová J, Arvaiová B, Komadel P (1999) FTIR spectroscopic characterization of thermally treated Cu2+, Cd2+, and Li+ montmorillonites. Spectrochim Acta A 55:2467–2476. https://doi.org/10.1016/S1386-1425(99)00039-6
Magyarosy A, Laidlaw RD, Kilaas R, Echer C, Clark DS, Keasling JD (2002) Nickel accumulation and nickel oxalate precipitation by Aspergillus niger. Appl Microbiol Biotechnol 59:382–388. https://doi.org/10.1007/s00253-002-1020-x
Marcussen H, Holm PE, Strobel BW, Hansen HCB (2009) Nickel sorption to goethite and montmorillonite in presence of citrate. Environ Sci Technol 43:1122–1127. https://doi.org/10.1021/es801970z
McKenzie RM (1980) The adsorption of lead and other heavy metals on oxides of manganese and iron. Aust J Soil Res 18:61–73. https://doi.org/10.1071/SR9800061
Misono M, Ochiai EI, Saito Y, Yoneda Y (1967) A new dual parameter scale for the strength of Lewis acids and bases with the evaluation of their softness. J Inorg Nucl Chem 29:2685–2691. https://doi.org/10.1016/0022-1902(67)80006-X
Montavon G, Markai S, Andres Y, Grambow B (2002) Complexation studies of Eu(III) with alumina-bound polymaleic acid: effect of organic polymer loading and metal ion concentration. Environ Sci Technol 36:3303–3309. https://doi.org/10.1021/es010312h
Montavon G, Rabung T, Geckeis H, Grambow B (2004) Interaction of Eu(III)/Cm(III) with alumina-bound poly (acrylic acid): sorption, desorption, and spectroscopic studies. Environ Sci Technol 38:4312–4318. https://doi.org/10.1021/es0301626
Paluszkiewicz C, Holtzer M, Bobrowski A (2008) FTIR analysis of bentonite in moulding sands. J Mol Struct 880:109–114. https://doi.org/10.1016/j.molstruc.2008.01.028
Paluszkiewicz C, Stodolak E, Hasik M, Blazewicz M (2011) FT-IR study of montmorillonite–chitosan nanocomposite materials. Spectrochim Acta A 79:784–788. https://doi.org/10.1016/j.saa.2010.08.053
Pathak PN, Choppin GR (2006) Nickel(II) sorption on hydrous silica: a kinetic and thermodynamic study. J Radioanal Nucl Ch 268:467–473. https://doi.org/10.1007/s10967-006-0192-8
Pathak PN, Choppin GR (2007) Effect of complexing anions on europium sorption on suspended silica: a TRLFS study for ternary complex formation. Radiochim Acta 95:267–273. https://doi.org/10.1524/ract.2007.95.5.267
Peltier E, Lelie DVD, Sparks DL (2010) Formation and stability of Ni-Al hydroxide phases in soils. Environ Sci Technol 40:302–308. https://doi.org/10.1021/es902332b
Pujol D, Bartrolí M, Fiol N, Torre FDL, Villaescusa I, Poch J (2013) Modelling synergistic sorption of Cr(VI), Cu(II) and Ni(II) onto exhausted coffee wastes from binary mixtures Cr(VI)-Cu(II) and Cr(VI)-Ni(II). Chem Eng J 230:396–405. https://doi.org/10.1016/j.cej.2013.06.033
Rabung T, Pierret MC, Bauer A, Geckeis H (2005) Sorption of Eu(III)/Cm(III) on Ca-montmorillonite and Na-illite. Part 1: batch sorption and time-resolved laser fluorescence spectroscopy experiments. Geochim Cosmochim Acta 69:5393–5402. https://doi.org/10.1016/j.gca.2005.06.030
Reiller P, Casanova F (2005) Influence of addition order and contact time on thorium(IV) retention by hematite in the presence of humic acids. Environ Sci Technol 39:1641–1648. https://doi.org/10.1021/es048856h
Remko M, Fitz D, Rode BM (2008) Effect of metal ions (Li+, Na+, K+, Mg2+, Ca2+, Ni2+, Cu2+, and Zn2+) and water coordination on the structure and properties of L-arginine and zwitterionic L-arginine. J Phys Chem A 112:7652–7661. https://doi.org/10.1021/jp801418h
Saha UK, Taniguchi S, Sakurai K (2002) Simultaneous adsorption of cadmium, zinc, and lead on hydroxyaluminum- and hydroxyaluminosilicate-montmorillonite complexes. Soil Sci Soc Am J 66:117–128. https://doi.org/10.2136/sssaj2002.0117
Shaheen SM, Tsadilas CD, Rinklebe J (2013) A review of the distribution coefficients of trace elements in soils: influence of sorption system, element characteristics, and soil colloidal properties. Adv Colloid Interf Sci 201-202:43–56. https://doi.org/10.1016/j.cis.2013.10.005
Sheikhhosseini A, Shirvani M, Shariatmadari H (2013) Competitive sorption of nickel, cadmium, zinc and copper on palygorskite and sepiolite silicate clay minerals. Geoderma 192:249–253. https://doi.org/10.1016/j.geoderma.2012.07.013
Singh A, Ulrich KU, Giammar DE (2010) Impact of phosphate on U(VI) immobilization in the presence of goethite. Geochim Cosmochim Acta 74:6324–6343. https://doi.org/10.1016/j.gca.2010.08.031
Slngh RP, Yeboah YD, Pambld ER, Debayle P (1991) Stability constant of the calcium-citrate(3-) ion pair complex. J Chem Eng Data 36:52–54. https://doi.org/10.1021/je00001a015
Steeb J, Josowicz M, Janata J (2009) Nickel-63 microirradiators. Anal Chem 81:1976–1981. https://doi.org/10.1021/ac802493m
Strathmann TJ, Myneni SCB (2005) Effect of soil fulvic acid on nickel(II) sorption and bonding at the aqueous-boehmite (γ-AlOOH) interface. Environ Sci Technol 39:4027–4034. https://doi.org/10.1021/es0481629
Sun L, Qiu K (2012) Organic oxalate as leachant and precipitant for the recovery of valuable metals from spent lithium-ion batteries. Waste Manag 32:1575–1582. https://doi.org/10.1016/j.wasman.2012.03.027
Suneesh AS, Venkatesan KA, Syamala KV, Antony MP, Rao PRV (2012) Mutual separation of americium(III) and europium(III) using glycolamic acid and thioglycolamic acid. Radiochim Acta 100:425–430. https://doi.org/10.1524/ract.2012.1941
Svecevicius G, Kazlauskiene N, Taujanskis E (2012) The acute and behavioral effects of a copper-nickel mixture on roach Rutilus rutilus. Bull Environ Contam Toxicol 89:147–151. https://doi.org/10.1007/s00128-012-0636-5
Tan XL, Fan QH, Wang XK, Grambow B (2009) Eu(III) sorption to TiO2 (anatase and rutile): batch, XPS, and EXAFS studies. Environ Sci Technol 43:3115–3121. https://doi.org/10.1021/es803431c
Thompson A, Amistadi MK, Chadwick OA, Chorover J (2013) Fractionation of yttrium and holmium during basaltic soil weathering. Geochim Cosmochim Acta 119:18–30. https://doi.org/10.1016/j.gca.2013.06.003
Trivedi P, Axe L, Dyer J (2001) Adsorption of metal ions onto goethite: single-adsorbate and competitive systems. Colloid Surf A 191:107–121. https://doi.org/10.1016/S0927-7757(01)00768-3
Tsukahara S, Takata A, Watarai H (2004) Magnetic field enhanced microextraction rate of europium(III) with 2-thenoyltrifluoroacetone and oxalate at dodecane-water interface. Anal Sci 20:1515–1521. https://doi.org/10.2116/analsci.20.1515
Tyagi B, Chudasama CD, Jasra RV (2006) Determination of structural modification in acid activated montmorillonite clay by FT-IR spectroscopy. Spectrochim Acta A 64:273–278. https://doi.org/10.1016/j.saa.2005.07.018
Valisko M, Boda D (2007) Selective adsorption of ions with different diameter and valence at highly charged interfaces. J Phys Chem C 111:15575–15585. https://doi.org/10.1021/jp073703c
Verma PK, Pathak P, Mohapatra PK (2014) Sorption of metal cations on suspended bentonite: effects of pH, ionic strength and complexing anions. Radiochim Acta 102:401–409. https://doi.org/10.1515/ract-2014-2160
Volkov AG, Paula S, Deamer DW (1997) Two mechanisms of permeation of small neutral molecules and hydrated ions across phospholipid bilayers. Bioelectrochem Bioenerg 42:153–160. https://doi.org/10.1016/S0302-4598(96)05097-0
Xu L, Zheng T, Yang ST, Zhang LJ, Wang JQ, Liu W, Chen LH, Diwu J, Chai ZF, Wang SA (2016) Uptake mechanisms of Eu(III) on hydroxyapatite: a potential permeable reactive barrier backfill material for trapping trivalent minor actinides. Environ Sci Technol 50:3852–3859. https://doi.org/10.1021/acs.est.5b05932
Yang ST, Ren XM, Zhao GX, Shi WQ, Montavon G, Grambow B, Wang XK (2015) Competitive sorption and selective sequence of Cu(II) and Ni(II) on montmorillonite: batch, modeling, EPR and XAS studies. Geochim Cosmochim Acta 166:129–145. https://doi.org/10.1016/j.gca.2015.06.020
Yang ST, Sheng GD, Guo ZQ, Tan XL, Xu JZ, Wang XK (2012) Investigation of radionuclide 63Ni(II) sequestration mechanisms on mordenite by batch and EXAFS spectroscopy study. Sci China Chem 55:632–642. https://doi.org/10.1007/s11426-011-4482-9
Yang ST, Sheng GD, Montavon G, Guo ZQ, Tan XL, Grambow B, Wang XK (2013) Investigation of Eu(III) immobilization on γ-Al2O3 surfaces by combining batch technique and EXAFS analyses: role of contact time and humic acid. Geochim Cosmochim Acta 121:84–104. https://doi.org/10.1016/j.gca.2013.07.013
Yang ST, Sheng GD, Tan XL, Jun H, Du JZ, Montavon G, Wang XK (2011) Determination of Ni(II) uptake mechanisms on mordenite surfaces: a combined macroscopic and microscopic approach. Geochim Cosmochim Acta 75:6520–6534. https://doi.org/10.1016/j.gca.2011.08.024
Yang ST, Zong PF, Sheng GD, Ren XM, Huang YY, Wang XK (2014) New insight into Eu(III) sorption mechanism at alumina/water interface by batch technique and EXAFS analysis. Radiochim Acta 102:143–153. https://doi.org/10.1515/ract-2014-2112
Zhan G, Yu JX, Xu ZG, Zhou F, Chi RA (2012) Kinetics of thermal decomposition of lanthanum oxalate hydrate. T Nonferr Metal Soc 22:925–934. https://doi.org/10.1016/S1003-6326(11)61266-1
Funding
Financial supports from the National Natural Science Foundation of China (21790370, 21790374), “Young Thousand Talented Program” in China, and the Priority Academic Program Development (PAPD) of Jiangsu Higher Education Institutions are acknowledged.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Responsible editor: Angeles Blanco
Electronic supplementary material
ESM 1
(DOCX 434 kb)
Rights and permissions
About this article

Cite this article
Xu, L., Liu, W., Cai, Y. et al. Competitive sequestration of Ni(II) and Eu(III) on montmorillonite: role of molar Ni:Eu ratios and coexisting oxalate. Environ Sci Pollut Res 25, 32617–32630 (2018). https://doi.org/10.1007/s11356-018-3252-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11356-018-3252-z