
Abstract
We studied the toxic effects of glyphosate in vitro on HepG2 cells exposed for 4 and 24 h to low glyphosate concentrations likely to be encountered in occupational and residential exposures [the acceptable daily intake (ADI; 0.5 μg/mL), residential exposure level (REL; 2.91 μg/mL) and occupational exposure level (OEL; 3.5 μg/mL)]. The assessments were performed using biomarkers of oxidative stress, CCK-8 colorimetric assay for cell proliferation, alkaline comet assay and cytokinesis-block micronucleus (CBMN) cytome assay. The results obtained indicated effects on cell proliferation, both at 4 and 24 h. The levels of primary DNA damage after 4-h exposure were lower in treated vs. control samples, but were not significantly changed after 24 h. Using the CBMN assay, we found a significantly higher number of MN and nuclear buds at ADI and REL after 4 h and a lower number of MN after 24 h. The obtained results revealed significant oxidative damage. Four-hour exposure resulted in significant decrease at ADI [lipid peroxidation and glutathione peroxidase (GSH-Px)] and OEL [lipid peroxidation and level of total antioxidant capacity (TAC)], and 24-h exposure in significant decrease at OEL (TAC and GSH-Px). No significant effects were observed for the level of reactive oxygen species (ROS) and glutathione (GSH) for both treatment, and for 24 h for lipid peroxidation. Taken together, the elevated levels of cytogenetic damage found by the CBMN assay and the mechanisms of primary DNA damage should be further clarified, considering that the comet assay results indicate possible cross-linking or DNA adduct formation.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Glyphosate [N-(phosphonomethyl)glycine] is a non-selective broad spectrum systemic organophosphorus herbicide that effectively acts to control all plant types such as grasses, perennials, vines, corn, soy, shrubs and trees (Dill et al. 2010). Its main mechanism of action is inhibition of the 5-enolpyruvylshikimate-3-phosphate-synthase involved in shikimate pathway. It interferes with the production of the aromatic amino acids phenylalanine, tyrosine and tryptophan essential for plant growth.
Since shikimate pathway exists only in plants and microorganisms, glyphosate has long been considered safe for humans (de María et al. 1996). The widespread and constant intense use of glyphosate in the last decades has also raised questions about weed resistance, the need for higher doses due to adaptive/resistance response (Duke and Powless 2009) and increasing amounts of the herbicide in soil, underground water and rivers (IFEN 2006).
There are evidences that connect glyphosate with pregnancy outcomes, miscarriages, children developmental malformations (Benítez-Leite et al. 2009; Carrasco 2013), increasing number of chronic kidney disease in agricultural workers (Jayasumana et al. 2014, 2015), metabolic disorders (celiac disease and gluten intolerance) (Samsel and Seneff 2013a) and with increased incidence of rare tumour disease such as non-Hodgkin lymphoma, B cell lymphoma and one subtype of non-Hodgkin lymphoma (hairy cell leukaemia) in case-control studies (McDuffie et al. 2001; Hardell et al. 2002; De Roos et al. 2003, 2005; Eriksson et al. 2008; Schinasi and Leon 2014). These findings and the results of other studies dealing with glyphosate and estimation of risk for different cancer types were mentioned in the World Health Organization (WHO) and International Agency for Research on Cancer (IARC) report on glyphosate in 2015 (WHO 2015). They also raised the question of whether glyphosate itself is safe and if it has other mechanisms of action that were not detected when it gained approval for use, due to a possible inadequacy of animal and in vitro models and the time period for gaining results (Antoniou et al. 2012). As an endocrine disruptor, glyphosate can exhibit an inverted dose-response relationship, with more acute effects from very low doses than from higher doses (Gasnier et al. 2009). Recently, WHO and IARC changed its classification to probably carcinogenic (Group 2A) (WHO 2015). This change has raised the question about the safety of low-dose exposures.
The aim of this study was to examine the effects of acute exposure (4 and 24 h) to glyphosate active compound on the human hepatoma HepG2 cell model. Four-hour treatment is a common period for short-term in vitro treatments (3 to 6 h) and 24 h for long-term in vitro treatment (1.5–2 cell cycle) according to the OECD guidelines for chemical testing (for example, OECD Guideline No. 487). Glyphosate was applied at low concentrations as encountered in everyday life. To obtain more information on the possible mechanism(s) of action of glyphosate active compound on the level of primary DNA damage, cell proliferation and oxidative stress, a battery of biochemical and molecular biomarkers was selected and evaluated simultaneously.
Materials and methods
Chemicals
All of the chemicals were of analytical grade and purchased from Sigma-Aldrich Chemical Co. (St. Louis, MO, USA), unless otherwise specified.
Cell line
HepG2 cell line (ATCC® HB8065™) was purchased from the American Type Culture Collection (Rockville, MD, USA), cultivated in Eagle’s minimum essential medium (EMEM) supplemented with 10% foetal bovine serum (FBS) and 1% penicillin-streptomycin (10,000 U/mL) and maintained in a humidified atmosphere (95% relative humidity) with 5% CO2 at 37 °C. Experiment was performed when the duplication time has been established constant, and the passage step was more than 3 but less than 10 cell passages.
Treatment conditions
Glyphosate (CAS No. 1071-83-6) was purchased as analytical standard purity grade (≤100%) as Pestanal®, a registered trademark of Sigma-Aldrich Laborchemikalien GmbH (Germany). The stock solution was prepared in phosphate-buffered solution (PBS). The tested concentrations were derived from toxicological reference values obtained by regulatory calculations as those encountered in everyday life. Concentrations were chosen corresponding to the values of acceptable daily intake (ADI) (Annex I, EU Directive 91/414/EEC), residential exposure level (REL) (US EPA 2002, 2004) and occupational exposure level (OEL) (US EPA 2002, 2004). They corresponded to OEL 3.5 μg/mL, ADI 0.5 μg/mL and REL 2.91 μg/mL. The calculation of tested concentrations was based on average male human with 60 kg of body weight and total volume of 36 L of extracellular liquids, simulating submersion of cultured cells in the culture medium with the exact glyphosate concentration. Extrapolation was made according to the Guyton and Hall (1996).
HepG2 cells were grown until 80% confluence, trypsinized and transferred in cell culture flasks (filter-top flasks, 25 cm2, TPP) for the micronucleus and comet assay, and in 96 wells for studying cell proliferation and markers of oxidative stress. Prior to treatment, the whole cultivation medium was removed, cells were washed with PBS and fresh complete medium with different glyphosate concentration was added. Cells were treated for 4 or 24 h at 37 °C in a humidified atmosphere (95% relative humidity) with 5% CO2. Negative and positive controls were studied in parallel.
Cell proliferation assay
Proliferation of HepG2 cells after the 4 and 24-h treatment with glyphosate was studied by means of CCK-8 colorimetric assay, based on the use of Cell Counting Kit-8 (Sigma-Aldrich, 96992). This kit uses 2-(2-methoxy-4-nitrophenyl)-3-(4-nitrophenyl)-5-(2,4-disulfophenyl)-2H-tetrazolium, monosodium salt (WST-8) that is bioreduced by cellular dehydrogenases to an orange formazan product, soluble in tissue culture medium in the presence of an electron carrier, 1-methoxy PMS. The amount of formazan produced is directly proportional to the number of living cells.
Briefly, 100 μL of cell suspension (105 cells/mL) was added to each well in 96-well plates and seeded in EMEM medium, low concentration of glucose, supplemented with 10% FBS, 20 IU/mL penicillin and 20 mg/mL streptomycin in order to get 104 cells in each well. After overnight incubation at 37 °C in a CO2 incubator, cells were treated 4 and 24 h with different concentrations of glyphosate. Negative (EMEM medium plus 10 μL of the PBS since glyphosate was diluted in the PBS) cell samples were studied in parallel. At the end of the treatment, cells were washed with PBS and 10 μL of WST-8 solution was added to each well. After 4 and 24 h of incubation at 37 °C in a CO2 incubator, the optical density at 450 nm was determined for each well using a Victor3™ Multilabel Plate Reader (PerkinElmer, MA, USA). Data were expressed as a percentage absorbance compared to relevant negative controls. Positive control was 10% DMSO in the complete nutrient medium. The experiment was done twice independently with four repeated measurements in each.
Alkaline comet assay
After 4 or 24 h of treatment, cells were washed twice with PBS, detached by trypsinization (trypsin 0.25% EDTA), centrifuged at 90×g for 8 min and resuspended in complete EMEM. To prepare agarose microgels, 7 μL of single cell suspension (104 cells/mL) was mixed with 100 μL of 0.5% low melting point (LMP) agarose and layered on slides precoated with 0.6% normal melting point (NMP) agarose. Gels were kept on 4 °C for 10 min until solidification. Afterwards, another 100 μL of 0.5% LMP agarose was layered and kept on 4 °C for 10 min until solidification. Duplicate slides were prepared for each time period and concentration, together with negative and positive controls. Hydrogen peroxide was used (cells embedded in agarose were treated with 50 μM H2O2 for 10 min at 4 °C) as positive control. All the cells were processed, and the slides were prepared within 1 h after the treatment has finished, including trypsinization, centrifugation, mixing with agarosis layers and solidification. Slides were not exposed to sunlight in any moment, only to artificial light. Following preparation of all microgels, they were immersed into fresh cold lysis solution (2.5 M NaCl, 100 mM Na2EDTA, 10 mM Tris-HCl, 1% Na lauroyl sarcosinate, 1% Triton X-100 and 10% dimethyl sulfoxide, pH 10) for 24 h at 4 °C. Afterwards, slides were washed with bidistilled water to remove lysis debris and denatured in freshly prepared cold denaturation/electrophoresis buffer (300 mM NaOH, 1 mM Na2EDTA, pH 13.0). After 20 min at 4 °C, cells were placed in the electrophoretic chamber and subjected to electrophoresis for 20 min at 0.9 V/cm. Slides were neutralized in three changes of buffer (0.4 mol/L Tris-HCl, pH 7.5) at 5-min intervals and dehydrated using 70 and 96% EtOH for 10 min each. After drying, the slides were kept in a dry and dark chamber until scoring. Before scoring, the slides were stained with ethidium bromide (20 μg/mL). The slides were analysed with fluorescent microscope Olympus BX51 (×200 magnification) using the Comet Assay IV software for image analysis (Perceptive Instruments, UK). A total of 100 nucleoids (50 per slide) were measured per each experimental point. The level of DNA damage was estimated based on tail intensity (TI), which points to the %DNA in the comet tail. The experiment was performed once with two replicates that were compared, and if no difference (statistically significant) was found, they were combined together. Throughout comet measurements, nucleoids doubled in size, and apoptotic and necrotic nucleoids were also seen, but were not scored.
Cytochalasine B-blocked micronucleus cytome assay
Cells were seeded at concentration of 104 cells/mL in complete EMEM medium. After 4 or 24 h of treatment with glyphosate, cells were washed twice with PBS and fresh complete medium was added. At the 44th hour, cytochalasin B (3 μg/mL) was added, and the cell cultures were harvested for 24 h thereafter. Positive control was incubated simultaneously (cyclophosphamide at a final concentration of 0.28 mg/mL). After harvesting, the medium was discarded, and cells were washed twice with PBS, detached by trypsinization (trypsin 0.25% EDTA), rinsed and resuspended in complete EMEM medium. Cell suspensions in complete EMEM were transferred to tubes and centrifuged at 45×g for 5 min. The pellet was resuspended in PBS and centrifuged, and Carnoy’s fixative (methanol/glacial acetic acid, 3:1) with two drops of formaldehyde was added to the pellet. After centrifugation, cells were washed two times with only Carnoy’s fixative. Slides were prepared by dropping and air drying. Afterwards, the slides were stained with 2% Giemsa stain (Merck) solution for 8 min and air-dried. The experiment had been performed once with two replicates.
Micronuclei (MNi), nucleoplasmic bridges (NPBs), nuclear buds (NBUDs) and apoptotic and necrotic cells were scored in binucleated (BN) cells according to the criteria of Fenech et al. (2003) and Fenech (2007). A total of 2000 BN cells per each experimental point were scored to determine parameters of CBMN Cyt assay. Furthermore, by scoring 1000 cells with one to four nuclei, nuclear division index (NDI) was also calculated, using the following formula: NDI = [M1 + (2 × M2) + (3 × M3) + (4 × M4)] / N, where M1, M2, M3 and M4 represent the number of cells with one, two, three or four nuclei, and N is the total number of cells scored (Eastmond and Tucker 1989).
Measurement of oxidative stress parameters
Lipid peroxidation
The end products of lipid peroxidation, e.g. malondialdehyde, were measured using thiobarbituric acid reactive substance (TBARS) assay with some modification (Ohkawa et al. 1979; Pareek et al. 2013; Mateos et al. 2004). Briefly, 0.5 mL of sample was added to 0.5 mL of thiobarbituric-trichloroacetic acid (TBA-TCA) reagent and heated at 90 °C for 30 min. Then, it was immediately cooled in an ice bath and centrifuged at 90×g for 8 min. Absorbances were measured with a microplate spectrophotometric system (Victor3™ Multilabel Plate Reader) at 530 nm. Two independent experiments were performed, and each sample was measured in duplicate. TBARS concentration in unknown samples was calculated using a standard curve constructed with 1,1,3,3-tetrametoxypropane (0.3–6.07 μM) and expressed as μmol/L.
Total antioxidant capacity
Total antioxidant capacity (TAC) was investigated using ferric-reducing ability of plasma (FRAP) assay, based upon reduction of Fe3+-TPTZ complex under acidic conditions (Benzie and Strain 1996). The FRAP assay used in this study was slightly modified on the basis of previous reports (Kozics et al. 2013). Briefly, 100 μL of cell suspension (104 cells in total) was added to 1.0 mL of FRAP reagent. Absorbance was measured after 4 min of incubation at 593 nm on spectrophotometer Cecil 9000 (Cecil Instruments Limited, Cambridge, UK) using FRAP working solution as blank. Two independent experiments were performed, and each sample was measured in duplicate. The results were calculated on a basis of a standard curve obtained using Fe2SO4·7H2O (0.05–4.0 mM).
ROS detection
The amount of intercellular reactive oxygen species (ROS) was measured using 2′,7′-dichlorodihydrofluorescein diacetate (DCFH-DA). DCFH-DA is deacetylated by cellular esterases in a non-fluorescent compound which is then in the presence of hydroxyl, peroxyl and other ROS oxidized to the fluorescent 2′,7′-dichlorodihydrofluorescein (DCF) (Griffiths et al. 2011).
All of the measurements were performed in quadruplicate in dark-sided 96-well microplates in which each well was added 100 μL of cell suspension meaning that every well comprised 104 cells. Cells were grown in the same medium and conditions as for cell proliferation assay and treated with the same concentrations of glyphosate for 4 and 24 h as for cell proliferation assay. After the treatment, the cells were washed with PBS and 100 μL of 50 μM DCFH-DA dye diluted in PBS was added and kept on cells for 30 min at 37 °C in a CO2 incubator. Control for dye autofluorescence was prepared without addition of dye. Negative (non-treated) cell controls (in EMEM) were included in each experiment. Positive control was 100 mM H2O2.
The results after the cell treatment and DCFH-DA dyeing were analysed using a Victor3™ Multilabel Plate Reader at an excitation wavelength of 485 nm and an emission wavelength of 535 nm. Data were expressed as fluorescence arbitrary units (AUs) and later transformed into percentages compared to control values.
Quantification of glutathione
Fluorogenic bimane probe, monochlorobimane (MBCl), reacts specifically with glutathione (GSH) and forms a fluorescent product equal to the amount of GSH. Analysis of GSH level was performed using an adduct that can be measured fluorometrically (Kamencic et al. 2000).
Briefly, cell cultures in 96-well plates were prepared such that each well of 100 μL of cell suspension comprised of 104 cells, and each well was treated with the same amount of glyphosate for 4 and 24 h. After washing with PBS, cells were incubated with 100 μL of 50 μM MBCl in PBS for 20 min at 37 °C in a CO2 incubator. The amount of GSH in the cell samples was analysed using a Victor3™ Multilabel Plate Reader at an excitation wavelength of 355 nm and an emission wavelength of 460 nm. Negative (non-treated) cell controls were included in each experiment. Data were expressed as fluorescence AUs and later transformed into percentages compared to control values. All of the measurements were performed in quadruplicate.
Glutathione peroxidase activity
The glutathione peroxidase (GSH-Px) activity in HepG2 cells was measured by the European standardized method (Belsten and Wright 1995). In order to increase assay sensitivity for the measurement of GSH-Px, samples were prediluted 60 times instead of a recommended 110-fold dilution used for blood samples.The amount of glutathione oxidized by t-butyl hydroperoxide was determined by following the decrease in the β-NADPH concentration, and the decrease in absorbance at 340 nm was measured by spectrophotometry (Cary 50 UV-Vis, Varian Inc., CA, USA). Two independent experiments were performed, and each sample was measured in duplicates. One unit of GSH-Px was expressed as the amount of enzyme that oxidizes 1 μmol β-NADPH/min at 37 °C. Activity of GSH-Px was expressed per gramme of total protein (U/g) and was determined by the Bradford method (1976).
Protein determination
Protein concentration of cell lysates was measured by Bradford method (1976) using Total Protein Kit, Micro (Sigma-Aldrich). Samples, standards and blanks were done in triplicate, and absorbance was measured in 96-well plates at 570 nm (1420 Multilabel Counter Victor3™, PerkinElmer).
Statistical analysis
Statistical analysis was performed using StatSoft Dell (StatSoft Dell; Tulsa, USA) Statistica 13 package program. Descriptive statistics was used to determine the basic statistical parameters (mean, standard error and deviation, median and minimum and maximum values). Data gathered using Comet Assay IV software were logarithmically transformed prior to statistical evaluation, with the aim of normalizing distribution and equalizing variances. The intragroup and intergroup comparisons between samples were performed using one-way analysis of variance (ANOVA) with post hoc Scheffé’s test. Comparisons between values obtained for CBMN Cyt assay values and HepG2 proliferation kinetics measured with CBMN were made by Pearson’s χ 2 test for two-by-two contingency tables. For the biochemical assays and cell proliferation measured as CCK-8 assay data analysis, we used descriptive statistics and t test for comparisons between independent samples. The level of statistical significance was set at p < 0.05.
Results
Cell proliferation measured by CCK-8 assay
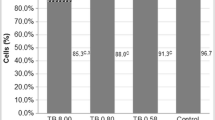
In glyphosate-treated HepG2 cells, we observed a slight increase of cell proliferation after 4-h treatment; it was about 9% higher as compared to control at ADI, and almost 8% higher at REL (Fig. 1), but without statistical significance. The values recorded for OEL-treated cells did not differ from control.

Cell proliferation in HepG2 cells after 4 and 24-h treatments with glyphosate. Results are expressed as means ± SD; p < 0.05. *Significantly different vs. negative control. C control (EMEM plus 10 μL PBS), ADI acceptable daily intake, REL residential exposure level, OEL occupational exposure limit, PC positive control (10% DMSO in the complete EMEM)
After 24 h of treatment, glyphosate-treated HepG2 cells showed higher, but not statistically significant, cell proliferation. At REL, it was 3%, and at OEL 1% higher than in the control. Proliferation of ADI-treated cells did not differ from control (Fig. 1).
Primary DNA damage measured by the alkaline comet assay
The results of the alkaline comet assay are reported in Table 1.
After 4 h of exposure, the values of TI at all of the tested concentrations were lower compared to controls, and these differences were statistically significant.
After 24 h of exposure, TI values for ADI, OEL and REL did not significantly differ from the control values.
Throughout comet measurements, apoptotic and necrotic nucleoids were also counted, but as there were no significant deviations in their number from the control, these data are not shown.
Cytogenetic damage measured using CBMN cytome assay
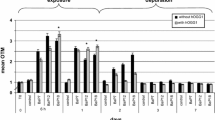
As demonstrated in Table 2, after 4 h of exposure, HepG2 cells treated with glyphosate at all three concentrations had significantly more MN than control, with the increase of the concentration of glyphosate. In HepG2 cells treated with glyphosate at ADI and REL, we observed a significantly increased NBUD frequency, as compared to control (for ADI, χ 2 was 10.42, df = 1, p = 0.0012; for REL, χ 2was 7.34, df = 1, p = 0.0067). Furthermore, ADI had a significantly higher NBUD number than OEL (86 vs. 60; χ 2 = 4.71, df = 1, p = 0.03). As for NPB, their incidence was again the highest in cells treated with glyphosate at ADI (6 per 2000 BN cells). Considering apoptotic and necrotic cells scored in the CBMN cytome assay, only 2 apoptotic cells per 2000 cells were found in the sample treated with glyphosate at OEL.
The values of NDI in glyphosate-treated cells differed from the control sample. In the OEL-treated sample, there were marked changes in the distribution of M1–M4 cells as compared to control (χ 2 = 8.377, df = 1, p = 0.0388), and OEL treatment resulted with more cells in the M2 phase. The same was observed when ADI-treated and OEL-treated samples were compared (χ 2 = 9.491, df = 1, p = 0.0234), as well as when REL-treated and OEL-treated samples were compared (χ 2 = 8.781, df = 1, p = 0.0324) (see Supplementary Table 1).
As shown in Table 2, after 24 h of exposure, a lower number of micronucleated BN cells was found at all three of the tested glyphosate concentrations when compared to control. Statistically significant differences were the following: REL-treated sample vs. control (χ 2 = 4.33, df = 1, p = 0.0374), OEL-treated sample vs. control (χ 2 = 14.69, df = 1, p = 0.0001) and OEL-treated vs. ADI-treated samples (χ 2 = 9.83, df = 1, p = 0.0017).
NBUD frequency was significantly lower in all treated samples: ADI (there was in total 51 nuclear buds; χ 2 = 14.07, df = 1, p = 0.0002), OEL (χ 2 = 64.25, df = 1, p < 0.0001) and REL (χ 2 = 70.92, df = 1, p < 0.0001). Also, the NBUD frequency at ADI was significantly higher than at REL (χ 2 = 27.84, df = 1, p < 0.0001) and at OEL (χ 2 = 22.79, df = 1, p < 0.0001).
Control and glyphosate-treated cells did not significantly differ in the number of NPBs. When considering apoptotic and necrotic cells, at ADI we found 7 and at REL 5 apoptotic cells per 2000 BN cells.
NDI values did not differ between glyphosate-treated samples and control. We found only slightly changed incidences of M2 and M3 cells in treated samples as compared to control (see Supplementary Table 1).
Lipid peroxidation
After 4 h of exposure, TBARS concentrations in samples treated with glyphosate at ADI and OEL were significantly lower than control values (Fig. 2). Twenty-four-hour treatment of HepG2 cells with glyphosate resulted in lower TBARS concentrations at REL, but the difference was not significant as compared to control. Different values were observed for samples treated with glyphosate at OEL and ADI at 4 and 24-h treatment for which a similar TBARS concentration was measured after 4 or 24 h (Fig. 2).

Effects of 4 and 24-h treatment with glyphosate on the extent of lipid peroxidation in HepG2 cells. Lipid peroxidation was determined by level of thiobarbituric reactive substances (TBARS). Results are expressed as means ± SD of two independent measurements; p < 0.05. *Significantly different vs. negative control. C control (EMEM), ADI acceptable daily intake, REL residential exposure level, OEL occupational exposure limit
Total antioxidant capacity
After the shorter period of treatment (4 h), the TAC value in samples treated with OEL was significantly lower when compared to controls (Fig. 3). ADI had similar values to OEL, but due to slightly higher standard deviation, no significant difference from control values was found. Twenty-four-hour treatment of HepG2 cells with glyphosate generally resulted in lower TAC values, with OEL treatment significantly different than control values. Interestingly, after 24 h of exposure, FRAP values for all of the treated samples diminished by 2-fold.

Effects of 4 and 24-h treatment with glyphosate on the total antioxidant capacity in HepG2 cells. Results are expressed as means ± SD of two independent measurements; p < 0.05. *Significantly different vs. negative control. C control (EMEM), ADI acceptable daily intake, REL residential exposure level, OEL occupational exposure limit
ROS level
The level of ROS did not change significantly after 4 and 24-h treatments, whereas only values measured in the OEL-treated sample at 24 h were lower than control values (Fig. 4), but none of the values observed was statistically significantly different than control values.

Percentage of ROS after exposure to 4 and 24-h treatment with glyphosate in HepG2 cells. Results are expressed as means ± SD. C control (EMEM), ADI acceptable daily intake, REL residential exposure level, OEL occupational exposure limit
GSH level
The level of GSH did not significantly differ from negative control for all concentrations and exposure time. After 4-h treatment, the ADI-treated sample had a 4% higher level of GSH than control, and REL-treated and OEL-treated cells had a value of GSH similar to negative control values. The 24-h ADI-treated sample had the lowest GSH concentration, which showed for 5% lower values than control samples, and again, REL-treated and OEL-treated cells had a value of GSH similar to negative control values (Fig. 5).

Percentage of glutathione (GSH) after exposure to 4 and 24-h treatments with glyphosate in HepG2 cells. Results are expressed as means ± SD. C control (EMEM), ADI acceptable daily intake, REL residential exposure level, OEL occupational exposure limit
GSH-Px activity
The 4-h exposure to glyphosate at its ADI concentration significantly decreased GSH-Px activity in HepG2 cells compared to control. After 24 h of treatment, a statistically significant decrease in the GSH-Px activity was observed in the sample treated with the OEL concentration of glyphosate when compared to the control sample (Fig. 6).

Effects of 4 and 24-h treatment with glyphosate on the activity of glutathione peroxidase (GSH-Px) in HepG2 cells. Results are expressed as means ± SD; p < 0.05. *Significantly different vs. negative control. C negative control (EMEM), ADI acceptable daily intake, REL residential exposure level, OEL occupational exposure limit
Discussion
New evidence on glyphosate carcinogenicity and a lack of knowledge regarding the mechanisms of its toxicity at cell level prompted us to study the effects of exposure to low concentrations of the compound, likely to be encountered in everyday life. We decided to focus on “realistic” concentrations, such as ADI (0.5 μg/mL), OEL (3.5 μg/mL) and REL (2.91 μg/mL), calculated according to the Annex I, EU Directive 91/414/EEC and US EPA level from 2002 and 2004, having in mind that many environmental hazards and adverse effects due to high concentrations of glyphosate are already known or have been documented in a plethora of scientific papers, as well as in evaluation reports and/or official papers issued by international regulatory or government bodies. On the other hand, the effects of such low concentrations are poorly or not even described at all in the existing literature. To the best of our knowledge, until now, no study of glyphosate toxicity at cell level has employed the same comprehensive array of biomarkers. Thus, the present study evaluated how low-dose exposure for 4 and 24 h affected cytotoxic, genotoxic and biochemical parameters in the human hepatoma cell line HepG2. We have selected this cell type as a suitable model considering that most xenobiotics are commonly metabolized in the liver, and these cells retain many of the genotypic and phenotypic features of liver cells, including an intrinsic metabolism (Knasmüller et al. 1998).
To prove whether after exposure to low concentrations of glyphosate there are any signs of increased cell proliferation, which is a prerequisite for carcinogenicity (Columbano et al. 1981, 1987; Wood et al. 2015), our first goal was to establish whether there are changes in cell proliferation using the CCK-8 cell proliferation assay. The results obtained indicate that, at the tested concentrations, glyphosate stimulated HepG2 cell proliferation at both exposure times. However, the effect was more pronounced after 4 h of exposure (8–9% as compared to control), than after 24-h exposure (3% as compared to control). Although this was not a statistically significant increase, our results are quite comparable with findings by Mesnage et al. (2014), who discovered similar values at the same concentration range. Thus, our results are a confirmation of their study on the HepG2 cell line. It seems that stimulation of cell proliferation by glyphosate is not an exclusive phenomenon for only one cell type, since the same authors have also shown the same effect on other human cell lines: embryonic (HEK293) and placental (JEG3) cells. Considering that in our study, glyphosate was tested at low concentrations, and its concentrations in a previous study by Mesnage et al. (2014) were even lower (up to the solubility limits), it is important to stress that the active ingredient alone caused the same amount of cell proliferation in all three cell types. The same authors also demonstrated that after 24 h of exposure, cell proliferation starts to decrease only after 1 ppm (1 mg/L) of the treatment with an active compound and that can explain why studies using higher concentrations did not demonstrate proliferative effect. Our results are also in agreement with other investigators who have already shown that glyphosate can induce cell proliferation in non-tumorigenic HaCaT cell line in concentrations 0.01–0.1 mM after exposure during 24, 48 and 72 h, where only after 72-h exposure, proliferation at 0.1 mM was significantly different than control values (George and Shukla 2013). It is worth mentioning here that our concentrations were 0.002957 mM (ADI), 0.0172 mM (REL) and 0.0207 mM (OEL). Two of them, REL and OEL, were within a range similar as used in the HaCaT cell line experiment. Interestingly, at ADI, which was lower than the concentrations tested in the above mentioned study, we observed the highest increase of HepG2 cell proliferation (9%). It seems that really low concentrations effectively stimulate cell proliferation, especially in already unstable and cell proliferation keen cells such as tumour cells. A similar finding was shown in a study by Thongprakaisang et al. (2013), where glyphosate increased proliferation of human breast cancer cell T47D in vitro for 15–30% when presented even in parts per trillion (10−6–1 μM). It is worth mentioning that T47D cells are hormone-dependent and the influence was seen when endogenous oestrogen in cell culture medium was diminished. Glyphosate did not change the proliferation rate in hormone-independent breast cancer cell line MDA-MB231 (Thongprakaisang et al. 2013).
In our experiment, during comet assay analysis, it was consistently observed that HepG2 cells differed in the amount of DNA contained in their nuclei, which also indicate the influence of glyphosate on cell proliferation. We found a clear distinction between cells with normal ploidy (as compared to control samples) and those with doubled nucleoid size. Such a finding indicates that glyphosate-treated cells were not synchronous as in the negative control sample, but a fraction of them prepared for division. Keeping in mind that cells differ in the ploidy, scoring of comets was performed only by selecting normal-sized nucleoids.
The comet assay results we obtained shed new light on the primary damage levels in glyphosate-treated HepG2 cells. Existing literature reports contradictory results regarding glyphosate genotoxicity, both in vivo and in vitro. Dermal or inhalative exposure to glyphosate formulations led to increased DNA damage in peripheral lymphocytes of exposed workers (Paz-y-Miño et al. 2007). From environmental studies conducted so far, it could be concluded that various organisms are differently sensitive and that several mechanisms are responsible for DNA damage. For instance, in the fish species Anguilla anguilla, short-term exposure to the glyphosate commercial formulation Roundup at 58 and 116 μg/L resulted in oxidative DNA damage as measured by comet assay in hepatic cells. DNA damage levels normalized on the first day after cessation of exposure (Marques et al. 2014). Exposure of the same fish species to glyphosate main metabolite aminomethylphosphonic acid (AMPA) in environmentally realistic concentrations of 11.8 and 23.6 μg/L for 1 and 3 days also resulted in genotoxicity (Guilherme et al. 2014).
The genotoxic effects of glyphosate at cell level were reported in several comet assay studies: on TR146 buccal cell line (20 mg/L or higher, 20 min; Koller et al. 2012), HT1080 fibrosarcoma cell line (0.9–3 mM; Monroy et al. 2005), GM38 fibroblast cells (5.2–8.5 mM; Monroy et al. 2005), human lymphocytes (20 h, 0.0007–0.7 mM; Alvarez-Moya et al. 2014) (3.5–580 μg/mL 4 h with S9 metabolic activation; Mladinic et al. 2009a) and HepG2 cell line (3–7 mM; Mañas et al. 2009a). The genotoxicity of its main metabolite AMPA was found in HepG2 after 4-h treatment in concentrations of 2.5–7.5 mM (Mañas et al. 2009b), while the genotoxicity of its formulation was observed in the TR146 buccal cell line (Koller et al. 2012). In spite of these previous observations, the results of our study did not confirm DNA-damaging effects of glyphosate when tested as a pure active compound. We found that the values for comet parameter measured in ADI-treated, REL-treated and OEL-treated HepG2 cells were significantly lower than control values. Our previous study on glyphosate-treated human lymphocytes (Mladinic et al. 2009a) showed genotoxic effects only at 580 μg/mL without metabolic activation, and in the range of 3.5, 92.8 and 580 μg/mL at activation with S9. Using the hOGG1 comet assay, a significant increase in tail intensity was observed at 2.91 μg/mL with S9 and 580 μg/mL without S9. Koller et al. (2012) also demonstrated that buccal cell line TR146 treated with glyphosate and its formulation Roundup for 20 min showed higher levels for comet assay only at doses of 20 mg/L and above. Using the same HepG2 model, Gasnier et al. (2010) demonstrated significant DNA damage only after exposure to 5 ppm (5 mg/L) of glyphosate, which was a concentration far above ours. Monroy et al. (2005) showed genotoxic effects in GM38 cells only at concentrations of 4–6.5 mM glyphosate and in HT1080 cells at concentrations of 4.75–5.75 mM.
It is possible that the two exposure times selected for our study were too short for a reliable estimation of glyphosate genotoxicity. Benachour et al. (2007) observed a time-amplifying effect: the differential toxicity between the active ingredient glyphosate and its formulation Roundup increased by five times over 72 h of exposure. Mesnage et al. (2014) made the hypothesis that using short exposures, the direct toxicity of the tested compounds could be significantly underestimated in comparison with long-term exposures. Furthermore, it is also important to know whether the tests were performed using complete or serum-free media, considering that the serum in media can mask the damage that would occur in serum-free media, with a delay of 1 or 2 days (Richard et al. 2005).
In our study, cell proliferation was also measured with CBMN cytome assay, and the deviations from control were seen only for the 4-h treatment, where OEL-treated cells had more M2-phase cells than other concentrations and control. However, the overall values of NDI obtained for these samples did not significantly differ.
The other possible explanation of outstanding low genotoxicity as observed by the comet assay in our study is related to mechanisms of DNA damage infliction. As known, some compounds generate covalent adducts in DNA leading to interstrand cross-linking. This formation can cause a DNA structure distortion that includes unwinding and bending. The formation of DNA adducts causes distortion and later obstruction in DNA replication, enhanced cell death in the form of apoptosis and cell cycle arrest in G2 phase. Significantly lower DNA damage can be a sign of possible adduct formation. There is evidence about adduct formation of glyphosate in mouse liver following in vivo exposure (Bolognesi et al. 1997) and dose-dependent adduct formation in kidney and liver of mice with glyphosate formulation Roundup (Peluso et al. 1998).
Glyphosate genotoxicity was also investigated using the micronucleus assay both in vivo and in vitro. Bolognesi et al. (2009) found increased micronucleus frequency in lymphocytes of occupationally exposed workers on days 5 and 15 following the spraying in three out of five examined Columbian territories. Two previous studies evaluated glyphosate toxicity using in vitro CBMN assay. Koller et al. (2012) found elevated levels of micronuclei and nuclear buds in human buccal epithelial cell line TR146 after 20 min of exposure to glyphosate and its formulation (10–20 mg/L). Mladinic et al. (2009a) found that micronuclei, nuclear buds and nucleoplasmic bridge frequency in human lymphocytes treated without S9 increased at 3.5 μg/mL and higher and that S9 significantly elevated the frequency of nuclear instabilities only at 580 μg/mL.
The results of this study on HepG2 cells have demonstrated that 4-h exposure to glyphosate led to slightly increased cytogenetic damage in terms of MN, which was statistically significant only at OEL. Despite the non-significant increase in MN frequencies at ADI and REL, we found significant increases in nuclear bud frequency. Since nuclear budding represents a mechanism of MN formation, our results indicate even a low dose of glyphosate as ADI influences the level of DNA damage and cell stability. At 24 h of exposure, all of the treated cells had lower values than control samples considering the frequency of all parameters as micronuclei, nuclear buds and nucleoplasmic bridges. The results we obtained speak in favour of previous observations (Richard et al. 2005; Benachour and Seralini 2009) that glyphosate exerts the highest DNA-damaging potential after 48 or even 72 h since the beginning of treatment. Considering that in our study, shorter exposure lasted only 4 h, while total cell culture lasted for 72 h before harvesting, we had an opportunity to better examine the effects of low-dose exposure on DNA damage. On the contrary, in the case of longer exposure—24 h—it seems that this effect only started to be slightly visible (based on lower concentrations such as ADI that had significantly higher DNA damage). Richard et al. (2005) demonstrated under in vitro conditions that long exposure times allowed low concentrations to present toxic effects, and since the time of exposure especially to low doses of pollutants in vivo conditions may be longer, observing the effects 48 or 72 h from exposure could finally show what is the exact effect of exposure to low doses.
If glyphosate really induced cross-linking, as indicated by comet assay results, the repair of such complex DNA damage needs more time than available to cells following 24-h treatment. It also has to be taken into account that the presence of serum can also delay (buffer) the toxic effect as already demonstrated by George and Shukla (2013) and Richard et al. (2005). Further evidence which supports such conclusions are findings from our previous study on lymphocytes where no concentration-related increase of centromere (C+) and DAPI signals (DAPI+) for glyphosate treatment was found (Mladinic et al. 2009b).
Glyphosate induces cell death through autophagy pathways in addition to activating apoptotic pathways (Gui et al. 2012). It does not influence the level of apoptosis through membrane disruption by the release of intracellular adenylate kinase in HepG2, HEK293 and JEG3 (Mesnage et al. 2014). The same authors demonstrated that all three cell lines did not differ from control values in percentage of apoptotic cells measured by the level of caspase-3/7. Koller et al. (2012) found that Roundup induced acute cytotoxic effects in the buccal epithelial cell line (TR146) at concentrations >40 mg/L, which were due to membrane damage and impairment of mitochondrial functions, while glyphosate induced increase in extracellular lactate dehydrogenase at doses >80 mg/L. Gasnier et al. (2010) demonstrated that after exposure of the HepG2 cell line to glyphosate, the cytotoxic effect starts from 10 ppm (10 mg/L) as measured with the alamarBlue Assay (the most sensitive), although they also used other assays such as MTT, which is similar to ours. Monroy et al. (2005) demonstrated that GM38 cells showed cytotoxic effects after exposure to 4.0–7.0 mM glyphosate and HT1080 cells to 4.5–5.8 mM glyphosate. Apoptosis and necrosis effect were not seen in this experiment.
According to Johansson et al. (2006), exposure to pesticides at levels as low as those found in nature usually does not cause mortality. In the present study, we evaluated frequencies of apoptotic and necrotic cells both by comet assay and CBMN cytome assay. Considering that apoptotic and cytotoxic influence was not observed, it seems that glyphosate applied at such low doses affects cells with some other mechanisms.
Since most of the pesticides produce oxidative damage that can influence the level of DNA damage, the logical step was to examine the most usual parameters measured in cells for oxidative damage estimation. The results we obtained were again interesting. There was no increase in ROS levels after 4 and 24-h exposure. Probably, the amount of ROS created during 4 and 24-h exposure was efficiently removed by an antioxidant system that has been also confirmed by lower values of TAC-FRAP (and their double lower values after 24 h) and slightly lower GSH levels. Lower levels of GSH can be explained by earlier reports of Gasnier et al. (2010) who found that treatment of human hepatic cell line with glyphosate and its four formulations caused the lowering of glutathione transferase levels and at the same time an increase in levels of CYP3A4 and CYP1A2. Kwiatkowska et al. (2014) obtained similar results in erythrocytes exposed to glyphosate and its metabolites at concentrations of 0.01–5 mM for 1, 4 and 24 h. They found a very small percentage of haemolysis and haemoglobin oxidation even after 24 h, with induced reactive oxygen compounds only after 0.25 mM concentrations.
The significantly lower values of GSH-Px activity observed only after 4-h treatment with ADI concentration also show that possible oxidative damage does not include peroxydes and again does not include lipids. After 24 h of exposure, significantly lower values of GSH-Px activity were observed only at OEL concentration compared to control, although a decrease in GSH-Px was also observed at ADI and REL concentrations. With regard to time of treatment, the results for 24-h treatment were almost twice as high as for the 4-h treatment for C, ADI and REL. Elevated GSH-Px enzyme activities after exposure to glyphosate have been associated with tolerance of non-target species to deleterious effects caused by different insecticides and herbicides. Slaninova et al. (2009) demonstrated that tissue GSH levels are often depleted after a short period of oxidative stress, but elevated after long-term oxidant exposures, and that GSH levels after exposure to glyphosate could be dependent on the administration dose as well as on the extent of the exposure to herbicide. Lioi et al. (1998) demonstrated altered cell metabolism in bovine lymphocyte cultures exposed to glyphosate (17–170 μM) through generating reactive oxygen intermediates, and with observed depletion of intracellular-reduced GSH level, there was subsequent activation of glucose-6-phosphate dehydrogenase in these cells.
Lipid peroxidation is probably the best predictor of systemic level of damage induced by ROS and is therefore through this mechanism also involved in the toxicity of pesticides. Previous studies focussed on exposure to glyphosate, and its formulations indicate significant increase in lipid peroxidation in amphibians exposed to concentrations below legal limits (Dornelles and Oliveira 2016). Other authors have reported a dose-dependent increase in TBARS after 1-h exposure of erythrocytes to glyphosate and its formulation Roundup (Pieniazek et al. 2004) and also a significant increase in pregnant rat livers and foetus livers after 21-day glyphosate oral administration (Beuret et al. 2005).
Lipid peroxidation measured as TBARS is a measure of oxidative damage on lipids and not necessarily of oxidative stress in cells per se. In our previous study (Mladinic et al. 2009a), elevated TBARS and FRAP values were found only at 580 μg/mL. Mañas et al. (2009a) demonstrated that at higher concentrations of glyphosate, such as 400 mg/kg in mice, there were elevated levels of TBARS, superoxide dismutase (SOD) and catalase (CAT) activities in mouse organs. Slaninova et al. (2009) also did not find marked modifications in lipid peroxidation levels in rats exposed to glyphosate through drinking water, while the production of TBARS tended to be lower. Since there was no cell disruption or cell killing, that could be one explanation, the other could be that glyphosate created special groups of oxidative radicals that did not attack lipids. An interesting study from Pieniazek et al. (2004) on human erythrocytes treated with glyphosate and its formulation for 1, 4 and 24 h showed that 1-h exposure to glyphosate increased levels of lipid peroxidation only at 1000 ppm (1000 mg/L), with haemolysis at the highest dose of 1500 ppm (1500 mg/L) and exposure time of 24 h. In that study, glyphosate formulation and glyphosate did not statistically change GSH levels, but have increased CAT activity. Larsen et al. (2012) in Wistar rats treated for 30 or 90 days with dietary water with the highest concentration of glyphosate allowed in water for human consumption (US EPA 1993, 2011; 0.7 mg/L) and a 10-fold higher concentration (7 mg/L) showed that TBARS production did not change or was even lower compared to controls not exposed to glyphosate. It seems that increased levels of reduced GSH and enhanced GSH-Px activities can act as a protective mechanism that acts as an adaptive response during moderate oxidative stress (Slaninova et al. 2009; Larsen et al. 2012) and during deleterious effects caused by different insecticides and herbicides.
Therefore, the results of our experiments still do not answer the question: what does actually happen when glyphosate enters the cell? There is evidence that it affects proliferation in small doses. There is also evidence that it influences cell membrane channels and sulphate levels (George and Shukla 2013; Samsel and Seneff 2013b). Although glyphosate, if we consider its structure, should not pass through a cell membrane easily, after dissociation in solution, it becomes anionic and then it can pass the lipid barrier and enter into a cell (Kwiatkowska et al. 2013). On the other hand, due to its amino acid-like structure (glycine analogue, Samsel and Seneff 2016) glyphosate can be also actively taken up by mammalian cells (nasal and cells of gastrointestinal epithelium Caco-2 cell line) along L-type amino acid transporters such as LAT1/LAT2 (Xu et al. 2016), with ATP and Na+-independent uptake (Xu et al. 2016). Although glyphosate is a glycine analogue, transporters for the glycine uptake pathway are Na+-dependent and therefore not responsible for the glyphosate uptake (Xu et al. 2016). Vasiluk et al. (2005) demonstrated on the human Caco-2 cell line that glyphosate in concentrations of 10 mg/mL has led to a significant increase in paracellular permeability.
It has been shown that glyphosate induces imbalance and oxidative stress, increasing the level of ROS involved in the elevation of calcium ions which in turn are a crucial element in the progression of glyphosate-induced cell proliferation. There are studies that demonstrated that glyphosate promotes proliferation via modulation of Ca2+ levels. The lowering of calcium with glyphosate treatment suppressed the activation of caspases and Bax/Bcl-2 ratio and thereby possibly prevented apoptosis (George and Shukla 2013). The imbalance between Ca2+ homeostasis and cellular oxidative stress is mainly responsible for glyphosate-induced hyperproliferation of HaCaT cells (George and Shukla 2013). Gniadecki and Gajkowska (2003) showed that emptying of intracellular Ca2+ stores in keratinocytes facilitated basal cell carcinomas or squamous cell carcinoma development. Cattani et al. (2014) demonstrated that glyphosate can activate N-methyl-D-aspartate (NMDA) receptors in rat hippocampal cells and through this activation influence the voltage-dependent Ca2+ channels and calcium levels inside the cell. Since HepG2 have also NMDA receptors, a similar example can be also transferred into this model. Changes in the calcium uptake can induce mitogen-activated protein kinases/extracellular-regulated kinase [mitogen-activated protein kinase (MAPK)/extracellular signal-regulated kinases (ERKs)] signalling expression, which, in turn, induces cell proliferation as demonstrated by Zhang and Liu (2002) and White and Sacks (2010). L-Type calcium channels and MAPK/ERK signal are essential for mast cell activation (Suzuki et al. 2010) and L929 fibroblast proliferation (Sazonova et al. 2007), and calcium ions are the best regulator of the voltage-dependent calcium channels and on their intracellular stores (White and Sacks 2010). Changes in calcium are associated in the progression of the cell cycle and in the activation of ERK and AKT signalling pathways, and Zhang et al. (2013) demonstrated that those steps are crucial in the proliferation, transformation and collagen synthesis in fibroblast and then in the development and progression of pulmonary fibrosis. Cattani et al. (2014) demonstrated that 30-min acute exposure to glyphosate formulation Roundup® can change the calcium ion influx by activating NMDA receptors, ERK and voltage-dependent calcium channels, leading to oxidative stress and neural cell death.
Furthermore, Wright et al. (2008) demonstrated that activated MAPK has a downstream target Nf2, and Nf2 after transfer into the nucleus upregulates antioxidant proteins, and therefore can reduce the oxidative damage to lipids, as observed in our experiments. Also, as a glycine analogue, glyphosate can be a substitute for glycine and therefore disrupt protein function during protein synthesis. Kelch-like ECH-associated protein 1 (KEAP1) is a protein that regulates Nrf2 activity. Nrf2 is a transcription factor that regulates important antioxidant and phase II detoxification genes, and it depends on a conserved glycine to prevent Nrf2 migration into the nucleus to activate multiple genes. Glyphosate substitution for glycine in KEAP1 would interfere with KEAP1’s ability to suppress the overexpression of Nrf2 (Samsel and Seneff 2016). This would result in a hyperphosphorylated state, which could explain both the increased proliferation (MAPK/ERK phosphorylation signalling cascade) and the reduced oxidative damage (through hyperphosphorylation of Nrf2) observed in our experiments. Samsel and Seneff (2016) have also proposed that the combination of phosphatase inhibition and enhanced kinase activity can cause excessive phosphorylation systemically, and systemic phosphorylation can have an impact on elevated risk to both Alzheimer’s disease and cancer. Furthermore, glycine substitution can enhance the level of DNA damage, as measured in our experiment with comet assay. Although there is a possibility that exposure to white light (from the sun) can cause a higher level of DNA damage in alkaline comet assay, it should not be the case in our study, since the samples were all prepared within 1 h from sampling and were exposed only to the light of the lamp while preparing the slides. Samsel and Seneff (2016) in their review mentioned that glyphosate exposure for 12 or 24 h can cause achromatic lesions (gaps) in chromatids, in both in vitro and in vivo experiments. Those gaps are caused by the endonuclease activity which is a part of the DNA repair pathway (Harvey et al. 1997). If those gaps are unrepaired, they are a measure of unrejoined DNA double-strand breaks and lesions that usually include pyrimidine bases and oxidative lesions in DNA (Collins 2004). As we already mentioned, serum in the cell culture can mask the development of DNA damage observed in micronucleus assay results; this can be the explanation why the standard time period used in this experiment did not demonstrate a similar effect as in the case of comet assay where primary DNA damage demonstrated higher damage levels. It would be also interesting to see whether the prolonged cell culture would also demonstrate similar effect if we count in this prolonged period the time necessary for discovering damage that was masked due to the serum level in the cell medium for cell cultivation.
Conclusion
Taken together, the results of all cytogenetic, molecular and biochemical methods used in this study indicate that glyphosate applied at low concentrations, likely to be encountered in everyday life, possess toxic potential towards HepG2 cells, which has to be further explained. It seems that the theory of different effects of low-dose vs. high-dose exposure, and more deleterious effects at low doses, is true.
Although at ADI, REL and OEL, we did not measure drastically different levels of oxidative damage, the elevated level of permanent DNA damage found with micronucleus assay call for concern, especially if it led to adduct formation, as shown by comet assay results. Based on the obtained results, we cannot say without doubt whether glyphosate acts as an aneugen or a clastogen (Li et al. 1983), but there are indications from previous studies that the aneugenic effect plays an important role in the formation of micronuclei. This study did not evaluate the effects on cytoskeleton and proteins, but this would be a direction for future evaluations of glyphosate toxicity, together with the clarification of its effects on cell membrane level, especially in different phases of the cell cycle.
References
Alvarez-Moya C, Silva MR, Ramírez CV, Gallardo DG, Sánchez RL, Aguirre AC, Velasco AF (2014) Comparison of the in vivo and in vitro genotoxicity of glyphosate isopropylamine salt in three different organisms. Genet Mol Biol 37(1):105–110
Antoniou M, Habib ME, Howard CV, Jennings RC, Leifert C, Nodari RO, Robinson CJ, Fagan J (2012) Teratogenic effects of glyphosate-based herbicides: divergence of regulatory decisions from scientific evidence. J Environ Anal Toxicol S4 006:1–13
Belsten JL, Wright AJ (1995) European community—FLAIR common assay for whole-blood glutathione peroxidase (GSH-Px); results of an inter-laboratory trial. Eur J Clin Nutr 49(12):921–927
Benachour N, Seralini G (2009) Glyphosate formulations induce apoptosis and necrosis in human umbilical, embryonic, and placental cells. Chem Res Toxicol 22(1):97–105
Benachour N, Sipahutar H, Moslemi S, Gasnier C, Travert C, Seralini GE (2007) Time- and dose-dependent effects of roundup on human embryonic and placental cells. Arch Environ Contam Toxicol 53(1):126–133
Benítez-Leite S, Macchi ML, Acosta M (2009) Malformaciones congenítas asociadas a agrotóxicos. Arch Pediatr Urug 80:237–247
Benzie IF, Strain JJ (1996) The ferric reducing ability of plasma (FRAP) as a measure of “antioxidant power”: the FRAP assay. Anal Biochem 239:70–76
Beuret CJ, Zirulnik H, Gimenez MS (2005) Effect of the herbicide glyphosate on liver lipoperoxidation in pregnant rats and their foetuses. Reprod Toxicol 19:501–504
Bolognesi C, Bonatti S, Degan P, Gallerani E, Peluso M, Rabboni R, Roggieri P, Abbondandolo A (1997) Genotoxic activity of glyphosate and its technical formulation roundup. J Agric Food Chem 45(5):1957–1962
Bolognesi C, Carrasquilla G, Volpi S, Solomon KR, Marshall EJ (2009) Biomonitoring of genotoxic risk in agricultural workers from five Colombian regions: association to occupational exposure to glyphosate. J Toxicol Environ Health A 72(15–16):986–997
Bradford M (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72:248–254
Carrasco A (2013) Teratogenesis by glyphosate based herbicides and other pesticides: relationship with the retinoic acid pathway. In: Breckling B, Verhoeven R (eds) GM-Crop Cultivation – Ecological Effects on a Landscape Scale.Theorie in der Ökologie 17. Peter Lang, Frankfurt, pp 113–117
Cattani D, de Liz Oliveira Cavalli VL, Heinz Rieg CE, Domingues JT, Dal-Cim T, Tasca CI, Mena Barreto Silva FR, Zamoner A (2014) Mechanisms underlying the neurotoxicity induced by glyphosate-based herbicide in immature rat hippocampus: involvement of glutamate excitotoxicity. Toxicology 320:34–45
Collins AR (2004) The comet assay for DNA damage and repair: principles, applications, and limitations. Mol Biotechnol 26(3):249–261 Review
Columbano A, Rajalakshmi S, Sarma DSR (1981) Requirement of cell proliferation for the initiation of liver carcinogenesis as assayed by three different procedures. Cancer Res 41:2079–2083
Columbano A, Ledda-Columbano GM, Coni P, Sarma DSR, Pani P (1987) Initiation of chemical hepatocarcinogenesis: compensatory cell proliferation verus mitogen induced hyperplasia. In: Roberfroid MB, Préat V (eds) Experimental Hepatocarcinogenesis. Proceedings of European meeting on Experimental Hepatocarcinogenesis. Plenum Press, New York, pp 133–144
de María N, Becerril JM, Garca-Plazaola JI, Hernandez AH, de Felipe MR, Fernández-Pascual M (1996) New insights on glyphosate mode of action in nodular metabolism: role of shikimate accumulation. J Agric Food Chem 54:2621–2628
De Roos AJ, Zahm SH, Cantor KP, Weisenburger DD, Holmes FF, Burmeister LF, Blair A (2003) Integrative assessment of multiple pesticides as risk factors for non-Hodgkin’s lymphoma among men. Occup Environ Med 60(9):E11 1-9
De Roos AJ, Blair A, Rusiecki JA, Hoppin JA, Svec M, Dosemeci M, Sandler DP, Alavanja MC (2005) Cancer incidence among glyphosate-exposed pesticide applicators in the Agricultural Health Study. Environ Health Perspect 113(1):49–54
Dill GM, Sammons RD, Feng PCC, Kohn F, Kretzmer K, Mehrsheikh A, Bleeke M, Honegger JL, Farmer D, Wright D, Haupfear EA (2010) Glyphosate: discovery, development, applications, and properties. In: Nandula VK (ed) Glyphosate Resistance in Crops and Weeds: History, Development, and Management. Wiley, Hoboken, pp 1–34
Dornelles MF, Oliveira GT (2016) Toxicity of atrazine, glyphosate and quinclorac in bullfrog tadpoles exposed to concentrations below legal limits. Environ Sci Pollut Res Int 23(2):1610–1620
Duke S, Powless SB (2009) Glyphosate-resistant crops and weeds: now and in the future. AgBioforum 12(3,4):346–357
Eastmond DA, Tucker JD (1989) Identification of aneuploidy–inducing agents using cytokinesis–blocked human lymphocytes and an antikinetochore antibody. Environ Mol Mutagen 13:34–43
Eriksson M, Hardell L, Carlberg M, Akerman M (2008) Pesticide exposure as risk factor for non-Hodgkin lymphoma including histopathological subgroup analysis. Int J Cancer 123(7):1657–1663
Fenech M (2007) Cytokinesis-block micronucleus cytome assay. Nat Protoc 2(5):1084–1104
Fenech M, Chang WP, Kirsch-Volders M, Holland N, Bonassi S, Zeiger E, Human MicronNucleus project (2003) HUMN project: detailed description of the scoring criteria for the cytokinesis-block micronucleus assay using isolated human lymphocyte cultures. Mutat Res 534(1–2):65–75
Gasnier C, Dumont C, Benachour N, Clair E, Chagnon MC, Séralini GE (2009) Glyphosate-based herbicides are toxic and endocrine disruptors in human cell lines. Toxicology 262:184–191
Gasnier C, Benachour N, Clair E, Travert C, Langlois F, Laurant C, Decroix-Laporte C, Séralini GE (2010) Dig1 protects against cell death provoked by glyphosate-based herbicides in human liver cell lines. J Occup Med Toxicol 27(5):29 1-13
George J, Shukla Y (2013) Emptying of intracellular calcium pool and oxidative stress imbalance are associated with the glyphosate-induced proliferation in human skin keratinocytes HaCaT cells. ISRN Dermatol 825180:1–12
Gniadecki R, Gajkowska B (2003) Intracellular calcium pool emptying induces DNA synthesis in HaCaT keratinocytes. Exp Dermatol 12(4):453–459
Griffiths HR, Dunston CR, Bennett SJ, Grant MM, Phillips DC, Kitas GD (2011) Free radicals and redox signalling in T-cells during chronic inflammation and ageing. Biochem Soc Trans 39(5):1273–1278
Gui YX, Fan XN, Wang HM, Wang G, Chen SD (2012) Glyphosate induced cell death through apoptotic and autophagic mechanisms. Neurotoxicol Teratol 34(3):344–349
Guilherme S, Santos MA, Gaivão I, Pacheco M (2014) DNA and chromosomal damage induced fish (Anguila agula L.) by aminomethylphosphonic acid (AMPA)—the major environmental breakdown product of glyphosate. Environ Sci Pollut Res Int 21(14):8730–8739
Guyton AC, Hall JE (1996) Textbook of medical physiology, 4th edn. W. B. Saunders Company, Philadelphia
Hardell L, Eriksson M, Nordstrom M (2002) Exposure to pesticides as risk factor for non-Hodgkin’s lymphoma and hairy cell leukemia: pooled analysis of two Swedish case-control studies. Leuk Lymphoma 43(5):1043–1049
Harvey AN, Costa ND, Savage JR, Thacker J (1997) Chromosomal aberrations induced by defined DNA double-strand breaks: the origin of achromatic lesions. Somatic. Cell Molec. Genet 23:211–219
IFEN (2006) Report on pesticides in waters: Data 2003–2004. In Dossier. Volume 5. Institut Français de l’Environnement, Orléans, France, pp 15–20
Jayasumana C, Gunatilake S, Senanayake P (2014) Glyphosate, hard water and nephrotoxic metals: are they the culprits behind the epidemic of chronic kidney disease of unknown etiology in Sri Lanka? Int J Environ Res Public Health 11:2125–2147
Jayasumana C, Paranagama P, Agampodi S, Wijewardane C, Gunatilake S, Siribaddana S (2015) Drinking well water and occupational exposure to herbicides is associated with chronic kidney disease, in Padavi-Sripura, Sri Lanka. Environ Health 14(6):1–10
Johansson M, Piha H, Kylin H, Merilä J (2006) Toxicity of six pesticides to common frog (Rana temporaria) tadpoles. Environ Toxicol Chem 25:3164–3169
Kamencic H, Lyon A, Paterson PG, Juurlink BH (2000) Monochlorobimane fluorometric method to measure tissue glutathione. Anal Biochem 286(1):35–37
Knasmüller S, Parzefall W, Sanyal R, Ecker S, Schwab C, Uhl M, MershSundermann V, Williamson G, Hietsch G, Langer T, Darroudi F, Natarajan AT (1998) Use of metabolically competent human hepatoma cells for the detection of mutagens and antimutagens. Mutat Res 402:185–202
Koller VJ, Fürhacker M, Nersesyan A, Mišík M, Eisenbauer M, Knasmueller S (2012) Cytotoxic and DNA-damaging properties of glyphosate and Roundup in human-derived buccal epithelial cells. Arch Toxicol 86(5):805–813
Kozics K, Klusová V, Srančíková A, Mučaji P, Slamenová D, Hunáková L, Kusznierewicz B, Horváthová E (2013) Effects of Salvia officinalis and Thymus vulgaris on oxidant-induced DNA damage and antioxidant status in HepG2 cells. Food Chem 141:2198–2206
Kwiatkowska M, Paweł J, Bukowska B (2013) Glyphosate and its formulations—toxicity, occupational and environmental exposure. Med Pr 64(5):717–729
Kwiatkowska M, Nowacka-Krukowska H, Bukowska B (2014) The effect of glyphosate, its metabolites and impurities on erythrocyte acetylcholinesterase activity. Environ Toxicol Pharmacol 37(3):1101–1108
Larsen K, Najle R, Lifschitz A, Virkel G (2012) Effect of sub-lethal exposure of rats to the herbicide glyphosate in drinking water: glutathione transferase enzyme activities, levels of reduced glutathione and lipid peroxidation in liver, kidneys and small intestine. Environ Toxicol Pharmacol 34(3):811–818
Li AP, Folk RM Monsanto Company Environmental Health Laboratory St Louis, Mo (1983) In vivo bone marrow cytogenetics study of glyphosate in Sprague Dawley rats. (unpublished study dated 20 October 1983, Study #ML-83-236, submitted by Monsanto Co. Washington DC to EFSA)
Lioi MB, Scarfi MR, Santoro A, Barbieri R, Zeni O, Di Berardino D, Ursini MV (1998) Genotoxicity and oxidative stress induced by pesticide exposure in bovine lymphocyte cultures in vitro. Mutat Res 403:13–20
Mañas F, Peralta L, Raviolo J, García OH, Weyers A, Ugnia L, Gonzalez CM, Larripa I, Gorla N (2009a) Genotoxicity of glyphosate assessed by the comet assay and cytogenetic test. Environ Toxicol Pharmacol 28:37–41
Mañas F, Peralta L, Raviolo J, García Ovando H, Weyers A, Ugnia L, Gonzalez Cid M, Larripa I, Gorla N (2009b) Genotoxicity of AMPA, the environmental metabolite of glyphosate, assessed by the comet assay and cytogenetic tests. Ecotoxicol Environ Saf 72(3):834–837
Marques A, Guilherme S, Gaivão I, Santos MA, Pacheco M (2014) Progression of DNA damage induced by a glyphosate-based herbicide in fish (Anguilla anguilla) upon exposure and post-exposure periods—insights into the mechanisms of genotoxicity and DNA repair. Comp Biochem Physiol C Toxicol Pharmacol 166:126–133
Mateos R, Goya L, Bravo L (2004) Determination of malondialdehyde by liquid chromatography as the 2,4-dinitrophenylhydrazone derivative: a marker for oxidative stress in cell cultures of human hepatoma HepG2. J Chromatogr B 805:33–39
McDuffie HH, Pahwa P, McLaughlin JR, Spinelli JJ, Fincham S, Dosman JA, Robson D, Skinnider LF, Choi NW (2001) Non-Hodgkin’s lymphoma and specific pesticide exposures in men: cross-Canada study of pesticides and health. Cancer Epidemiol Biomark Prev 10(11):1155–1163
Mesnage R, Defarge N, de Vendômois JS, Séralini GE (2014) Major pesticides are more toxic to human cells than their declared active principles. Biomed Res Int 179691:1–8
Mladinic M, Berend S, Vrdoljak AL, Kopjar N, Radic B, Zeljezic D (2009a) Evaluation of genome damage and its relation to oxidative stress induced by glyphosate in human lymphocytes in vitro. Environ Mol Mutagen 50(9):800–807
Mladinic M, Perkovic P, Zeljezic D (2009b) Characterization of chromatin instabilities induced by glyphosate, terbuthylazine and carbofuran using cytome FISH assay. Toxicol Lett 189(2):130–137
Monroy CM, Cortés AC, Sicard DM, de Restrepo HG (2005) Cytotoxicity and genotoxicity of human cells exposed in vitro to glyphosate. Biomedica 25(3):335–345
Ohkawa H, Ohishi N, Yagi K (1979) Assay for lipid peroxides in animal tissues by thiobarbituric acid reaction. Anal Biochem 95:351–358
Pareek A, Godavarthi A, Issarani R, Nagori BP (2013) Antioxidant and hepatoprotective activity of Fagonia schweinfurthii (Hadidi) Hadidi extract in carbon tetrachloride induced hepatotoxicity in HepG2 cell line and rats. J Ethnopharmacol 150:973–981
Paz-y-Miño C, Sánchez ME, Arévalo M, Muñoz MJ, Witte T, De-la-Carrera GO, Leone PE (2007) Evaluation of DNA damage in an Ecuadorian population exposed to glyphosate. Genet Mol Biol 30(2):456–460
Peluso M, Munnia A, Bolognesi C, Parodi S (1998) 32P-postlabeling detection of DNA adducts in mice treated with the herbicide Roundup. Environ Mol Mutagen 31(1):55–59
Pieniazek D, Bukowska B, Duda W (2004) Comparison of the effect of Roundup Ultra 360 SL pesticide and its active compound glyphosate on human erythrocytes. Pestic Biochem Physiol 79:58–63
Richard S, Moslemi S, Sipahtur H, Benachour N, Seralini GE (2005) Differential effects of glyphosate and roundup on human placental cells and aromatase. Environ Health Perspect 113(6):716–720
Samsel A, Seneff S (2013a) Glyphosate, pathways to modern diseases II: celiac sprue and gluten intolerance. Interdiscip Toxicol 6(4):159–184
Samsel A, Seneff S (2013b) Glyphosate’s suppression of cytochrome P450 enzymes and amino acid biosynthesis by the gut microbiome: pathways to modern diseases. Entropy 15(4):1416–1463
Samsel A, Seneff S (2016) Glyphosate, pathways to modern diseases V: amino acid analogue of glycine in diverse proteins. J Biol Phys Chem 16:9–46
Sazonova OV, Blishchenko EY, Tolmazova AG, Khachin DP, Leontiev KV, Karelin AA, Ivanov VT (2007) Stimulation of fibroblast proliferation by neokyotorphin requires Ca2+ influx and activation of PKA, CaMK II and MAPK/ERK. FEBS J 274(2):474–484
Schinasi L, Leon ME (2014) Non-Hodgkin lymphoma and occupational exposure to agricultural pesticide chemical groups and active ingredients: a systematic review and meta-analysis. Int J Environ Res Public Health 11(4):4449–4527
Slaninova A, Smutna M, Modra H, Svobodova Z (2009) A review: oxidative stress in fish induced by pesticides. Neuroendocrinol Lett 30:2–12
Suzuki Y, Inoue T, Ra C (2010) L-type Ca2+ channels: a new player in the regulation of Ca2+ signaling, cell activation and cell survival in immune cells. Mol Immunol 47(4):640–648
Thongprakaisang S, Thiantanawat A, Rangkadilok N, Suriyo T, Satayavivad J (2013) Glyphosate induces human breast cancer cells growth via estrogen receptors. Food Chem Toxicol 59:129–136
US EPA (1993) U.S. Environmental Protection Agency Reregistration Eligibility Decision (RED) Glyphosate. USEPA, Washington, DC (EPA-738-R-93-014)
US EPA (2002) U. S. Environmental Protection Agency. Memorandum: glyphosate in/on pasture and rangeland grasses, Roundup ready® wheat, and nongrass animal feeds. Health Effects Division (HED) Risk Assessment. 37 pages: Tolerance, Vol. 69. Number 217. USEPA, Washington, DC https://www3.epa.gov/pesticides/chem_search/cleared_reviews/csr_PC-103601_20-Feb-02_a.pdf
US EPA (2004) U. S. Environmental Protection Agency. Glyphosate: pesticide tolerance, vol. 69. Number 217. USEPA, Washington, DC
US EPA (2011) Edition of the drinking water standards and health advisories. Office of Water USEPA, Washington, DC (EPA 820-R-11-002)
Vasiluk L, Pinto LJ, Moore MM (2005) Oral bioavailability of glyphosate: studies using two intestinal cell lines. Environ Toxicol Chem 24(1):153–160
White CD, Sacks DB (2010) Regulation of MAP kinase signaling by calcium. Methods Mol Biol 661:151–165
WHO (2015) IARC monographs volume 112: evaluation of five organophosphate insecticides and herbicides
Wood CE, Hukkanen RR, Sura R, Jacobson-Kram D, Nolte T, Odin M, Cohen S.M (2015) Scientific and Regulatory Policy Committee (SRPC) review*: interpretation and use of cell proliferation data in cancer risk assessment. Public Health Resources. Paper 318. http://digitalcommons.unl.edu/publichealthresources/318
Wright DC, Geiger PC, Han DH, Jones TE, Holloszy JO (2008) Calcium induces increases in peroxisome proliferator-activated receptor γ coactivator-1α and mitochondrial biogenesis by a pathway leading to p38 mitogen activated protein kinase activation. J Biol Chem 282:18793–18799
Xu J, Li G, Wang Z, Si L, He S, Cai J, Huang J, Donovan MD (2016) The role of L-type amino acid transporters in the uptake of glyphosate across mammalian epithelial tissues. Chemosphere 145:487–494
Zhang W, Liu HT (2002) MAPK signal pathways in the regulation of cell proliferation in mammalian cells. Cell Res 12:9–18
Zhang YP, Wang WL, Liu J, Li WB, Bai LL, Yuan YD, Song SX (2013) Plasminogen activator inhibitor-1 promotes the proliferation and inhibits the apoptosis of pulmonary fibroblasts by Ca(2+) signaling. Thromb Res 131(1):64–71
Acknowledgments
This work was financially supported by Project No. 8366 Organic Pollutants in Environment—Markers and Biomarkers of Toxicity (OPENTOX), funded by the Croatian Science Foundation.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Responsible editor: Philippe Garrigues
Electronic supplementary material
Supplementary Table 1
(DOCX 15 kb).
Rights and permissions
About this article

Cite this article
Kašuba, V., Milić, M., Rozgaj, R. et al. Effects of low doses of glyphosate on DNA damage, cell proliferation and oxidative stress in the HepG2 cell line. Environ Sci Pollut Res 24, 19267–19281 (2017). https://doi.org/10.1007/s11356-017-9438-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11356-017-9438-y