
Abstract
Humic acids were divided into several fractions using buffer solutions as extraction agents with different pH values. Two methods of fractionation were used. The first one was subsequent dissolution of bulk humic acids in buffers adjusted to different pH. The second one was sequential dissolution in buffers with increasing pH values. Experimental data were compared with hypothesis of partial solubility of humic acids in aqueous solutions. Behaviour of humic fractions obtained by sequential dissolution, original bulk sample and residual fractions obtained by subsequent dissolution at pH 10 and 12 agrees with the hypothesis. Results demonstrated that regardless the common mechanism, solubility and dissociation degree of various humic fractions may be very different and can be estimated using parameters of the model based on the proposed mechanism. Presented results suggest that dissolving of solid humic acids in water environment is more complex than conventional solubility behaviour of sparingly soluble solids.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Humic acids are composed of natural polydispersed polyelectrolyte bio-colloids occurring in water as dissolved organic matter or in sediments and soil (Bratskaya et al. 2008; Davies et al. 2001; Koopal et al. 2001; Michalowski and Asuero 2012; Steelink 2002). A major fraction of organic matter in natural ecosystems is in fact formed by humic acids, which are characterized by the presence of several functional groups with labile protons such as carboxylic acids, phenols and amines. These moieties are able to bind protons and metal ions that not only affect the geochemistry of natural systems, but also regulate the buffer capacity of waters, soils and sediments, and metal speciation and transport (Fernandes et al. 2009; Masini et al. 1998; Ritchie and Perdue 2003). Complexation of toxic metal ions by humic acids plays a major role in maintaining them in a bioavailable state in environmental waters. It also plays a prominent role in controlling metal speciation and influences the mobility of metal ions in soil and aqueous environments and hence plays a vital function in the environmental fate, bioavailability, toxicity and mobility of heavy metals in the biosphere (Dumat et al. 2000; Michalowski and Asuero 2012; Pinheiro et al. 2000).
Humic acids participate in both nutrient and contaminant transport, and they support the soil structure and its pH. Transport of the contaminants due to ground- and fresh-water dynamics is directly related to the risks associated with contaminants. The solubility, mobility and complexation ability of humic acids are related to their interactions with soil mineral particles and depend on pH and other environmental conditions. Since the humic substance is not a single well-defined molecule but a mixture of various heterogeneous particles or aggregates containing a variety of functional groups resulting in polyelectrolyte and polyfunctional properties, it cannot be treated as an ordinary complex-forming ligand in the interaction with ions. They have a colloidal character whose size and negative charge are strictly dependent on surface functional groups (Angelico et al. 2014; Fernandes et al. 2009; Kirishima et al. 2010; Koopal et al. 2001; Tan et al. 2011).
The acid–base chemistry of humic acids is an important component of metal speciation models in natural systems. The acidic functional groups of humic acids (mainly carboxylic and phenolic) determine their binding characteristics for protons and other major ions (Atalay et al. 2009; Tipping 2002). The commonly encountered ionizable functional groups include carboxylic acids, phenols, alcohols, ammonium ions and thiols and to a lesser extent, sulfonic acids and “active methylene” compounds (with CO–CH2–CO structural moiety) are also encountered. The major binding sites of these functional groups are attributed to the oxygen-containing groups. These are carboxylic and phenolic groups, which contribute to the total acidity of humic substances. Phenolic groups are usually assumed to be the only weak acidic groups, while the contribution of alcoholic groups in carbohydrate entities and enols is usually neglected (Badr et al. 2012; Perdue 1985).
Interaction between humic acids and metals ions is of great importance for nutritional, transport and sedimentary processes. However, the understanding of physical-chemical processes associated with chelation reactions still remains a challenge, fundamentally due to the dynamic nature and complexity of organic matter (Fernandes et al. 2009). Since the anionic charge of humic substances plays a major role in their interactions with other components, pH and ionic strength are the most important variables controlling metal complexation (Liu and Gonzalez 2000; Michalowski and Asuero 2012; Tan et al. 2011; Yonebayashi and Hattori 1990), many authors hope that fractionation of humic matter by extractions at different pH values would be useful in reducing their complexity and detail understanding of the interactions of natural organic matter with other components. Baalousha et al. (2006) studied the influence of pH on size, molecular weight, conformation and aggregation of humic acids as important aspects to control their physicochemical reactions in aquatic systems. Curtis et al. (1981) fractionated humic acids using a pH gradient method (universal buffer mixed with sodium hydroxide) as a fingerprint technique for humic acids and as a means for fractionating these materials prior to further characterization. Fujitake et al. (1998, 1999, 2003) isolated soil humic acids by the sequential extraction with pyrophosphate solutions at different pH values. Their analysis suggested that the proportion of components with a larger particle size increased gradually and regularly with the increase of the pH value (Fujitake et al. 1998). The H/C ratios increased gradually with increasing pH values, while the O/C and O/H ratios decreased. The results suggested that decarboxylated humic acids were extracted at the higher pH values and the humic acids with a higher content of unsaturated bonds and a higher degree of oxidation were extracted at the lower pH values within the range from 5 to 13 (Fujitake et al. 1999). As the pH values of extractant increased the absorption strength of the bands in FT-IR spectra attributed to aliphatic and amide groups increased and those to carboxylic groups decreased. The 1H-NMR spectra confirmed that the proportion of aromatic structures decreased with the increase of the pH values (Fujitake et al. 2003). Yonebayashi and Hattori (1990) reported that humic acids soluble in universal buffer adjusted to pH 7 consist of a few structural subunits assumed to be condensed aromatic rings with short aliphatic substituents and many carboxyl groups, while fraction soluble at pH 11 was characterized by phenolic groups. Dai et al. (2006) used the sequential extraction with pH buffers for fractionation and characterization of humic and fulvic acids from paddy soils. They showed that fulvic acids (in contrast to humic acids) contained an easily dissociable fraction in acidic solution. You et al. (2006) extracted humic substances from soil organic matter using base with subsequent pH lowering and sequential pH extraction. They obtained a significant correlation between the ratio of organic carbon associated with humic acids and fulvic acids and the NaOH extractable organic carbon content of soils. Between pH 5 and 7, which is a typical soil solution pH, a significant amount of humic acids associated organic carbon was soluble. Kipton et al. (1992) fractionated humic acids by dissolution in several media as a function of pH and ionic strength. The solubility of humic acids and the proportion of large molecules in solution increased with increase in pH and decreased with increase in ionic strength. Bakina and Orlova (2012) studied the regularities of extracting humic acids from soils of different types with solutions of sodium pyrophosphate at the equilibrium pH values of 5–13. The increase in the humic acids yield with the increasing alkalinity of the solution applied was directly related to the capability of acid functional groups (carboxyl and phenol hydroxyl) for dissociation at definite pH values. Recent work of Jovanovic et al. (2013) is focused on aggregation of humic acids in the 2–11 pH range. They concluded that humic acids behave as macromolecular aggregates or supramolecular structures, formed from small individual moieties (sizes < 10 nm) at higher pH values. The dependence of zeta potential on pH revealed the minimum in the 5–7 pH range, caused most likely by dissociation of acidic functional groups prevailing at lower pH values and deaggregation predominating over dissociation at higher pH values. Leenheer et al. (2003) showed that only a small fraction of carboxylic functional groups in humic substances are exceptionally acidic with pK a values as low as 0.5. Their average structural model of the most acidic fraction derived from the characterization data indicated a high density of carboxyl groups clustered on oxygen-heterocycle alicyclic rings. Intramolecular H-bonding between adjacent carboxyl groups in these ring structures enhanced stabilization of the carboxylate anion which results in low pK a values.
The solubility and dissociation of humic acids in water and aqueous solutions play a crucial role in their interactions and function in natural systems in general.
The complex structure of humic acids with the great diversity of functional groups makes an exact understanding of mechanisms of their behaviour almost impossible. Schnitzer and Monreal (2011) stated that despite utilizing different chemical and physicochemical techniques, even after more than two centuries of substantial research, the basic chemical nature and reactivity of humic substances and soil organic matter are still poorly understood. This complex nature of humic acids makes it difficult to obtain precise information on its chemical structure and properties. One of the ways of overcoming these difficulties is to separate humic acids into several fractions which reduces their heterogeneity. This work is focused on solubility and dissociation behaviour of humic acids in buffer solution adjusted to different pH values and basic chemical and physicochemical properties of obtained fractions.
Our previous works on the solubility of humic acids in aqueous environments (Klucakova and Kolajova 2014; Klucakova and Pekar 2005, 2008) demonstrated that their solubility is substantially different from the common solubility of sparingly soluble substances (salts) and is inseparably connected with their dissociation. The following model of dissolving and dissociation behaviour of humic acids was developed on the basis of experimental data:

According to scheme (1) the solid humic acid is in water or aqueous solution either dissolved without dissociation (step 1) or dissociates releasing the proton into the water while the anion remains in the solid state (step 3). The dissolved humic molecule can undergo usual dissociation (step 2). In this work, the hypothesis of mechanism (1) is further tested for the bulk humic sample and their fractions obtained using the both ways of extraction of humic acids by buffer solutions at the pH region 4–12.
In our previous works (Klucakova and Kolajova 2014; Klucakova and Pekar 2005, 2008) we introduced an assumption that only a specific fraction of complex humic mixture is soluble (under given conditions) and if this fraction is removed, residual humic sample remains practically insoluble. Assumption of only partial solubility of humic acids can be expressed as:
where [HA(aq)] + [A− (aq)] is the total molar concentration of dissolved humic acids, k b is the proportionality constant and m is the total weight of humic acids in 1 dm3 of dispersion. Constant k b is product of the dimensionless fraction of soluble humic acids κ and the amount of functional groups able to eliminate H+ ion in a mass unit of humic acids, which is represented by the total acidity α divided by the standard concentration c st (1 mol⋅dm−3 in this case). Parameter ψ in is the proportionality coefficient, transforming units from [g.dm−3] to dimensionless relative concentration.
Using balance from Table 1, the equilibrium of step 1 in scheme (1) is expressed as
The equilibrium constant of step 2 in scheme (1) can be written as follows:
Supposing w t << x (acidity in system is caused mainly by H+ ions formed during dissociation of dissolved HAs), Eq. (4) can be simplified to:
and linearized as:
Following an additional condition resulting from the balance given in Table 1 was derived as:
Used symbols are summarized in the material balance of system humic acids - water in Table 1 (relative concentrations for the standard state c st = 1 mol⋅dm−3 are used). The activity of H+ ions formed by dissociation of water (w 0 or w t) is included in this balance, but we suppose that the amount of H+ ions formed by dissociation of water is insignificant. Another assumption is that x >> y (cf. Table 1) and therefore the acidity in the system is caused mainly by the H+ ions formed during dissociation of dissolved HAs, i.e. by x.
The equilibrium of step 3 in scheme (1) can be described by the equilibrium constant K 3:
Substituting x + y + w t = 10− pH = h, we can write:
Comparing the material balance in Table 1 with Eq. (9) we see that the denominator should be the expression (ψ − k b) m − y. But values of y cannot be experimentally determined and we assume that they are much lower than the value of x. Because values of (ψ − k b) m are much higher than x, the value of y was disregarded also in this case. Calculating the value of K 3 is therefore very difficult because y cannot be directly measured and its value is much lower than that of h or x (Klucakova and Kolajova 2014; Klucakova and Pekar 2005, 2008). The material balance in system humic acids–water is summarized in Table 1.
Experimental
Preparation of humic samples
Humic acids, studied in this work, were isolated by means of alkaline extraction from South-Moravian lignite. The procedure represents our own modification of a method published by Piccolo et al. (1999) and has been previously utilized in our other experimental works (Klucakova and Kolajova 2014; Klucakova 2010; Klucakova and Pekar 2008, 2009). More details on the chemical structure of both the original lignite matrix and the isolated humic sample can be found in previously published papers (Barancikova et al. 2003; Klucakova 2010; Klucakova and Pekar 2002, 2003a, b, 2004, 2005).
Universal buffer solutions composed of NaOH, H3PO4, CH3COOH and H3BO3 adjusted to different pH values (from 4 to 12) were used for the preparation of fractionated humic acids. Two ways of fractionation (the pH sequential extraction and the subsequent dissolution in buffers adjusted to different pH values) described below were used.
Sequential extraction in buffers with increasing pH values (procedure A)
The finely ground humic sample was mixed with the buffer solution at the lowest pH value (pH = 4) in the ratio 2 g/100 cm3 and stirred for 24 h. The dissolved fraction was precipitated from the filtrate by the concentrated HCl solution to pH = 1. The clot was washed by deionized water and dried (50 °C). The humic fractions obtained using the procedure A were denoted as A-HA-X, where X is the pH value of used buffer solution. The insoluble residue was (partially) dissolved in the buffer solution with the higher pH value and the whole procedure was repeated. The residue insoluble in the buffer solution with the highest pH value (pH = 12) was denoted as A-HA-R.
Subsequent dissolution in buffers adjusted to different pH values (procedure B)
The bulk humic sample was individually dissolved in the buffer solutions adjusted to different pH values in the same ratio as in the case of Procedure A. The dissolved humic fraction was precipitated from the filtrate by the concentrated HCl solution to pH = 1. The clot was washed by deionized water and dried (50 °C). The humic fractions obtained using the procedure B were denoted as B-HA-X, where X is the pH value of used buffer solution. The insoluble residues were denoted as B-HA-R-X, where X is the pH value of used buffer solution.
Characterization of humic samples
The obtained humic fractions as well as the bulk humic sample were characterized by the elemental analysis (CHNSO Micro-analyzer Flash 1112, Carlo Erba), and the content of carboxylic groups. Carboxylic acidity was determined using standard acetate method (Klucakova and Pekar 2005, 2008; Schnitzer and Khan 1972; Stevenson 1994).
The dissociation behaviour of prepared samples in water was investigated according to our previous works (Klucakova and Pekar 2005, 2008). Samples of bulk humic acids or their fraction were suspended (and partially dissolved) in deionized water in the ratios 0.4–4 g/100 cm3 and stirred. The pH values (pH-meter Sentron Titan) and the UV/VIS spectra (Hitachi U-3300) of solution above the solid (undissolved) humic particles were measured in the equilibrium (after 24 h).
Results and discussion
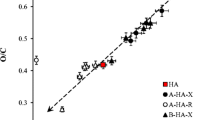
Results of basic characterization of obtained fractions and the bulk humic sample are summarized in Table 2. We can see that the fractions prepared by both procedures and the bulk sample differ in elemental composition as well as in content of COOH groups. The N content in bulk humic acids is higher than this content in any other A-fraction. However, it does not mean that the total N content decreased. If we calculate the N contents in individual A-fractions and take account of different yields at different pH values (see Fig. 2a), we obtained the same total N content in A-fractions (sum of N contents in A-fractions) as in the bulk humic sample. In case of COOH contents, the reason is similar. If we use the buffer solution with a low pH value, we are able to extract only the fractions very rich in strong carboxylic groups therefore the yield of extraction is low but the COOH content in the extracted fraction is high and the COOH content in the residual fraction is only little lower in comparison with the bulk humic sample. If the pH value of used buffer solutions increased we are able to extract higher amounts of humic acids but with lower COOH contents and lower COOH contents have also residual fractions because the extraction yields are higher. The total COOH content expressed as the sum of soluble and residual B-fractions are constant (considering different extraction yields). Comparing the carboxylic acidity with the O/C ratio we obtained a relatively good linear dependence (R 2 > 0.95) for all studied samples (Fig. 1a). Similar dependence was determined for the O/H ratio (Fig. 1b). The first hypothesis could be that the strong correlation is probably related to the increasing solubility with increasing initial pH values of used buffers, because the content of COOH groups decreases with increasing initial pH values in all cases. The good linear correlation between the COOH content and O/C ratio is probably caused by the fact that this radio changes mainly as a result of extraction of some humic fractions rich in COOH content. This linear correlation is therefore caused mainly by “quality” of extracted humic fractions and less by the extraction efficiency because a maximum for yield at pH = 8 was detected and no maximum for this dependence was observed. The good linear correlation for O/H ratio and O/C ratio has the similar reason. The extraction of humic fraction rich in COOH groups causes changes in C, O, and H contents which are to a certain extent interdependent. As we can see, yields obtained for the sequential extraction (Procedure A) showed a maximum at pH = 8 (Fig. 2a). As estimated, the equilibrated pH values are much lower for the Procedure B which corresponds with the obtained contents of dissolved humic fractions. Comparing the dissolved contents obtained for the sequential extraction with those of the subsequent dissolution we can state that the sequential extraction gives lower amounts of narrower fractions but the cumulative yields are higher. The results obtained for the buffer solutions with the pH value equal to 4 are identical for both used procedures because the initial step of Procedure A is the same as that of Procedure B. Another interesting result is that if we use the subsequent dissolution (Procedure B) buffer solutions with alkaline pH values (≥8), their equilibrium leachates are neutral with pH near 7 (Fig. 2b). Results of both used procedures indicate that the solubility and dissociation behaviour of humic acids are more complex and it is not determined only by their carboxylic acidity but also by strength of the functional groups participating on their behaviour in aqueous solutions (pK a). Humic acids are not a chemical individual but the mixture of many different substances. Every substance of this mixture has a different solubility, a different strength of its functional groups and different dissociation ability. In water and aqueous solutions, humic acids are soluble only partially, thus equilibrium between solid undissolved particles, protonated dissolved particles and dissociated ones are established. It is generally accepted that the solubility of humic acids in aqueous solutions is supported mainly by the dissociation of carboxylic functional groups. Although, their typical average values for humic acids are between 3 and 5, some fractions can have the pK a values very low, which enable them to dissociate in relatively acidic solutions (Dai et al. 2006; Kipton et al. 1992; You et al. 2006). On the other hand, Campitelli et al. (2006) determined five types of carboxylic groups in structure of humic acids with pK a values for between 3 and 7.

COOH content (a) and O/H ratio (b) of obtained fractions in dependence on their O/C ratio

Yield of fractionation (a) and equilibrium pH value (b) in dependence of initial pH value of used buffer solutions (dashed lines are guides to the eyes)
In order to study dissociation mechanism of prepared humic fractions they were equilibrated with water in various ratios. Measured pH values of the aqueous leachates were used for the data fitting according the mathematical model described in previous paragraphs. Results obtained on the basis of the model are listed in Table 3. As we can see in Fig. 3, the model cannot be universally applied on the behaviour of obtained humic fractions in water. Although measured pH values of solutions above solid particles in prepared suspensions are strongly dependent on humic content, the dependence agrees with Eq. 6 only in several cases. The applicability of the model on bulk humic acids were expected (Klucakova and Pekar 2005, 2008) and confirmed experimentally for the sample used in this work. A good agreement between the model and data obtained for the fractions prepared using procedure A (A-HA-X) was found and parameters of the mathematical model (except K 3 value) were calculated (Table 3). Small amount of A-HA-4 fraction does not allow carrying out dissociation experiments with this sample.

Experimental data obtained for B-HA-R-X fractions in water fitted using Eq. (6) (dashed lines are guides to the eyes)
As we can see the values of the equilibrium constants K 1 and K 2 of A-HA-X humic fractions are higher than those of the bulk sample. While the values of dissociation constant K 2 of dissolved humic fractions increase with increasing pH values of buffer solutions used for fractionation, the equilibrium constant K 1 shows a maximum for pH = 8. It corresponds also with the highest value of dimensionless soluble fraction κ indicating relatively high solubility. This confirms that the solubility of humic fractions is not dependent only on the content of carboxylic groups but also on their dissociation ability and the presence of other structural units. It corresponds with the fact that the dissociation ability of the bulk humic sample is much lower than the ability of A-HA-12 fraction (see Fig. 4a) contrary to the practically the same value of the equilibrium constant K 1. The difference in the dissociation behaviour of these two samples is thus caused mainly by the values of dissociation constant K 2. On the other hand, the significance of content of carboxylic groups cannot be underestimated, because their content decreases with increasing initial pH values of buffer solution used in both fractionation procedures. The decreasing content of carboxylic groups which was extracted with the increasing pH value of used buffer solution caused the decrease of the strength of functional groups in extracted fractions. Humic acids contain functional groups with wide scale of their pK a and the amount of functional groups of given strength is limited. Therefore the change of extraction efficiency and extraction yield is necessarily connected with the change of the dissociation ability of extracted fraction. The residual sample A-HA-R can dissociate only weekly and its dissociation ability decreases with its increasing content in suspension. This fact could support the assumption that only a specific fraction of humic acids is soluble (under given conditions) and if this fraction is removed, residual humic sample remains practically insoluble.

Equilibrium pH values measured for aqueous leachates of humic fractions obtained by procedure A (a) and B-HA-R-X (b)
Humic fractions prepared by subsequent dissolution of bulk humic sample in buffers adjusted to different pH (B-HA-X and B-HA-R-X) show different properties than fractions obtained by procedure A. The majority of experimental data cannot be fitted by derived dissociation model (see Fig. 3b). The model is usable only for the residual humic fractions B-HA-R-10 and B-HA-R-12. Although these fractions contain the lowest amounts of carboxylic functional groups (Table 2), their solubility is surprisingly high. The high solubility of these fractions was indicated on the basis of their dark colourings, mainly in case of B-HA-R-12. The sample B-HA-R-12 had to be diluted for measurement of UV/VIS spectra for that reason (Fig. 5). The values of equilibrium constants K 1 and K 2 determined for the B-HA-R-10 sample are comparable with the results obtained for the A-HA-12 fraction and the value of dimensionless soluble fraction κ is higher for the B-HA-R-10 sample. The reason could be based on some destruction of structure caused by use of very alkaline buffer solutions in fractionation procedure. This possible destruction is more noticeable if we compare the dissociation ability of B-HA-R-12 fraction represented by values of K 1 and K 2 with Figs. 4b and 5. As we can see the values of equilibrium constants K 1 and K 2 are very low in comparison with the bulk humic sample and other humic fractions. It corresponds also with value of dimensionless soluble fraction κ and constants k b and ψ. Measured pH values of aqueous leachates are relatively high and close to neutral region. On the other hand, the solubility of the B-HA-R-12 sample detected by UV/VIS spectrometry is very high. The leachate of this fraction has to be strongly diluted in order to measure spectra with values of absorbance comparable with other studied fractions (Fig. 5). It indicates that some humic fractions poor in carboxylic groups can be (under certain circumstances) relatively easily dissolved. The humification index E46 (Schnitzer and Khan 1972) of individual studied fractions is showed in Fig. 6, we can see that its value obtained for the B-HA-R-12 sample is very high (around 7), thus its particles should have low molar mass. Practically the same E46 value was determined for the B-HA-12 sample. The A-HA-12 sample has the humification index much lower in comparison with the corresponding B-fractions. A clarification is not easy. It seems that dissolving in very alkaline buffer solution could destruct both soluble and residual fraction but only in the case of the bulk humic sample. If the sample was subjected to sequential extraction in buffers with increasing pH values, humification index was much lower and no destruction was detected.

UV/VIS spectra of aqueous leachates prepared from B-HA-R-10 and B-HA-R-12 fractions (28 g dm−3)

Humification index (E46) of aqueous leachates (20 g dm−3) in dependence of initial pH values of used buffer solutions (dashed lines are guides to the eyes)
Conclusion
The bulk humic sample isolated from lignite was subdivided into several fractions by two different ways: the subsequent dissolution in buffers adjusted to different pH and the sequential dissolution in buffers with increasing pH values. It was found that extracted humic fractions were rich in carboxylic groups in comparison with residual ones. The increase of pH of used buffer solutions caused the decrease of strength and dissociation ability of functional groups in extracted fractions. Prepared fractions were studied with respect to their solubility in water. Experimental data obtained for individual fractions were compared with hypothesis of partial solubility of humic acids in aqueous solutions developed humic fractions in our previous works. It was found that humic fractions obtained by the sequential dissolution in buffers with increasing pH values (procedure A) and residual fractions from the subsequent dissolution of bulk humic acids in buffers adjusted to pH 10 and 12 are in very good agreement with the dissociation model. These two fractions had very low COOH contents but their solubility was relatively high which was indicated by dark colouring and measured UV/VIS spectra. The dissociation of other humic fractions cannot be described by the model and their behaviour is more complex. The humification index E46 of A-fractions was lower in comparison with B-fractions which can indicate higher molar mass of extracted humic particles without their destruction in the extraction procedure.
References
Angelico R, Ceglie A, He JZ, Liu YR, Palumbo G, Colombo C (2014) Particle size, charge and colloidal stability of humic acids coprecipitated with ferrihydrite. Chemosphere 99:239–247. doi:10.1016/j.chemosphere.2013.10.092
Atalay YB, Carbonaro RF, Di Toro DM (2009) Distribution of proton dissociation constants for model humic and fulvic acid molecules. Environ Sci Technol 43:3626–3631. doi:10.1021/es803057r
Baalousha M, Motelica-Heino M, Le Coustumer P (2006) Conformation and size of humic substances: effects of major cation concentration and type, pH, salinity, and residence time. Colloid Surface A 272:48–55. doi:10.1016/j.colsurfa.2005.07.010
Badr MH, El-Halafawi MH, Zeid ERAE-A (2012) Comparison between the effect of ionic strength on acidity and dissociation constants of humic acids extracted from sewage sludge and Nile water hyacinth composts. Glob J Environ Res 6:36–43. doi:10.5829/idosi.gjer.2012.6.1.64117
Bakina LG, Orlova NE (2012) Special features of humus acids extraction from soils by sodium pyrophosphate solutions of different alkalinity. Eurasian Soil Sci 45:392–398. doi:10.1134/S1064229312040023
Barancikova G, Klucakova M, Madaras M, Makovnikova J, Pekar M (2003) Chemical structure of humic acids isolated from various soil types and lignite. Humic Subst Environ 3:3–8
Bratskaya S, Golikov A, Lutsenko T, Nesterova O, Dudarchik V (2008) Charge characteristics of humic and fulvic acids: comparative analysis by colloid titration and potentiometric titration with continuous pK-distribution function model. Chemosphere 73:557–563. doi:10.1016/j.chemosphere.2008.06.014
Campitelli PA, Velasco MI, Ceppi SB (2006) Chemical and physicochemical characteristics of humic acids extracted from compost, soil and amended soil. Talanta 69:1234–1239. doi:10.1016/j.talanta.2005.12.048
Curtis MA, Witt AF, Schram SB, Rogers LB (1981) Humic-acid fractionation using a nearly linear pH gradient. Anal Chem 53:1195–1199. doi:10.1021/ac00231a014
Dai J, Ran W, Xing B, Gu M, Wang L (2006) Characterization of fulvic acid fractions obtained by sequential extractions with pH buffers, water, and ethanol from paddy soils. Geoderma 135:284–295. doi:10.1016/j.geoderma.2006.01.003
Davies G, Ghabbour EA, Steelink C (2001) Humic acids: marvelous products of soil chemistry. J Chem Educ 78:1609–1614. doi:10.1021/ed078p1609
Dumat C, Quiquampoix H, Staunton S (2000) Adsorption of cesium by synthetic clay-organic matter complexes: effect of the nature of organic polymers. Environ Sci Technol 34:2985–2989. doi:10.1021/es990657o
Fernandes AN, Giacomelli C, Giovanela M, Vaz DO, Szpoganicz B, Sierra MMD (2009) Potentiometric acidity determination in humic substances influenced by different analytical procedures. J Br Chem Soc 20:1715–1723. doi:10.1590/s0103-50532009000900021
Fujitake N, Kusumoto A, Tsukamoto M, Kawahigashi M, Suzuki T, Otsuka H (1998) Properties of soil humic substances in fractions obtained by sequential extraction with pyrophosphate solutions at different pHs: I. yield and particle size distribution. Soil Sci Plant Nutr 44:253–260. doi:10.1080/00380768.1998.10414446
Fujitake N, Kusumoto A, Tsukamoto M, Noda Y, Suzuki T, Otsuka H (1999) Properties of soil humic substances in fractions obtained by sequential extraction with pyrophosphate solutions at different pHs: II. Elemental composition and UV–VIS spectra of humic acids. Soil Sci Plant Nutr 45:349–358. doi:10.1080/00380768.1999.10409349
Fujitake N, Kusumoto A, Yanagi Y, Suzuki T, Otsuka H (2003) Properties of soil humic substances in fractions obtained by sequential extraction with pyrophosphate solutions at different pHs: III. FT-IR and H-1 NMR spectra of humic acids. Soil Sci Plant Nutr 49:347–353. doi:10.1080/00380768.2003.10410019
Jovanovic UD, Markovic MM, Cupac SB, Tomic ZP (2013) Soil humic acid aggregation by dynamic light scattering and laser Doppler electrophoresis. J Plant Nutr Soil Sci 176:674–679. doi:10.1002/jpln.201200346
Kipton H, Powell J, Town RM (1992) Solubility and fractionation of humic acid; effect of pH and ionic medium. Anal Chim Acta 267:47–54. doi:10.1016/0003-2670(92)85005-Q
Kirishima A, Ohnishi T, Sato N, Tochiyama O (2010) Simplified modelling of the complexation of humic substance for equilibrium calculations. J Nucl Sci Technol 47:1044–1054. doi:10.1080/18811248.2010.9711669
Klucakova M (2010) Adsorption of nitrate on humic acids studied by flow-through coulometry. Environ Chem Lett 8:145–148. doi:10.1007/s10311-009-0201-6
Klucakova M, Kolajova R (2014) Dissociation ability of humic acids: spectroscopic determination of pKa and comparison with multi-step mechanism. React Funct Polym 78:1–6. doi:10.1016/j.reactfunctpolym.2014.02.005
Klucakova M, Pekar M (2002) Study of structure and properties of humic and fulvic acids. II. Complexation of Cu2+ ions with humic acid extracted from lignite. J Polym Mater 19:287–294
Klucakova M, Pekar M (2003a) Study of structure and properties of humic and fulvic acids. III. Study of complexation of Cu2+ ions with humic acid in sols. J Polym Mater 20:145–154
Klucakova M, Pekar M (2003b) Study of structure and properties of humic and fulvic acids. IV. Study of interactions of Cu2+ ions with humic gels and final comparison. J Polym Mater 20:155–162
Klucakova M, Pekar M (2004) Study of diffusion of metal cations in humic gels. In: Ghabbour EA, Davies G (eds) Humic substances: nature’s most versatile materials. Taylor & Francis, New York, pp 263–273
Klucakova M, Pekar M (2005) Solubility and dissociation of lignitic humic acids in water suspensions. Colloid Surface A 252:157–163. doi:10.1016/j.colsurfa.2004.10.019
Klucakova M, Pekar M (2008) Behaviour of partially soluble humic acids in aqueous suspension. Colloid Surface A 318:106–110. doi:10.1016/j.colsurfa.2007.12.023
Klucakova M, Pekar M (2009) Transport of copper (II) ions in humic gel—new results from diffusion couple. Colloid Surface A 349:96–101. doi:10.1016/j.colsurfa.2009.08.001
Koopal LK, van Riemsdijk WH, Kinniburgh DG (2001) Humic matter and contaminants. General aspects and modeling metal ion binding. Pure Appl Chem 73:2005–2016. doi:10.1351/pac200173122005
Leenheer JA, Wershaw RL, Brown BK, Reddy MM (2003) Characterization and diagenesis of strong-acid carboxyl groups in humic substances. Appl Geochem 18:471–482. doi:10.1016/S0883-2927(02)00100-2
Liu A, Gonzalez RD (2000) Modeling adsorption of copper (II), cadmium (II) and lead (II) on purified humic acid. Langmuir 16:3902–3909. doi:10.1021/la990607x
Masini JC, Abate G, Lima EC, Hahn LC, Nakamura MS, Lichtig J, Nagatomy HR (1998) Comparison of methodologies for determination of carboxylic and phenolic groups in humic acids. Anal Chim Acta 364:223–233. doi:10.1016/S0003-2670(98)00045-2
Michalowski T, Asuero AG (2012) New approaches in modeling carbonate alkalinity and total alkalinity. Crit Rev Anal Chem 42:220–244. doi:10.1080/10408347.2012.660067
Perdue EM (1985) Acidic functional groups of humic substances. In: Aiken GR, McKnight DM, Wershaw RL, MacCarthy P (eds) Humic substances in soil, sediment and water. Wiley, New York, pp 493–526
Piccolo A, Conte P, Cozzolino A (1999) Effects of mineral and monocarboxylic acids on the molecular association of dissolved humic substances. Eur J Soil Sci 50:687–694. doi:10.1046/j.1365-2389.1999.00276.x
Pinheiro JP, Mota AM, Benedetti MF (2000) Effect of aluminum competition on lead and cadmium binding to humic acids at variable ionic strength. Environ Sci Technol 34:5137–5143. doi:10.1021/es0000899
Ritchie JD, Perdue EM (2003) Proton-binding study of standard and reference fulvic acids, humic acids, and natural organic matter. Geochim Cosmochim Acta 67:85–96. doi:10.1016/S0016-7037(02)01044-X
Schnitzer M, Khan SU (1972) Humic substances in the environment. Marcel Dekker Inc, New York
Schnitzer MI Monreal CM (2011) Quo vadis Soil Organic Matter Research? A Biological Link to the Chemistry of Humification. Adv Agron 113:139–213. doi:10.1016/B978-0-12-386473-4.00008-7
Steelink C (2002) Investigating humic acids in soils. Anal Chem 74:326a–333a. doi:10.1021/ac022040m
Stevenson FJ (1994) Humus chemistry: genesis, composition, reactions, 2nd edn. Wiley, New York
Tan WF, Norde W, Koopal LK (2011) Humic substance charge determination by titration with a flexible cationic polyelectrolyte. Geochim Cosmochim Acta 75:5749–5761. doi:10.1016/j.gca.2011.07.015
Tipping E (2002) Cation binding by humic substances. Cambridge University Press, Cambridge
Yonebayashi K, Hattori T (1990) A new fractionation of soil humic acids by adsorption chromatography. Geoderma 47:327–336. doi:10.1016/0016-7061(90)90036-9
You S-J, Thakali S, Allen HE (2006) Characteristics of soil organic matter (SOM) extracted using base with subsequent pH lowering and sequential pH extraction. Environ Int 32:101–105. doi:10.1016/j.envint.2005.07.003
Acknowledgments
This work was supported by the Ministry of Education, Youth and Sports, Project LO1211.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The author declares that the manuscript complies with the ethical rules applicable for environmental science and pollution research.
Additional information
Responsible editor: Philippe Garrigues
Rights and permissions
About this article

Cite this article
Klučáková, M. Characterization of pH-fractionated humic acids with respect to their dissociation behaviour. Environ Sci Pollut Res 23, 7722–7731 (2016). https://doi.org/10.1007/s11356-015-5932-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11356-015-5932-2