
Abstract
Background
Load-relaxation under a constant state of deformation is a common characteristic of hydrated materials, including hydrogels and biological tissues. Overall, mechanical response in such materials is a strong function of underlying structure, which in hydrogels depends on whether the gel is formed through physical or chemical cross-linking. In order to use hydrogels in biomedical applications where their properties are matched to those of native tissues, it is critical to understand these underlying structure-properties relationships.
Objective
The objective of current work is to quantitatively characterize the load-relaxation behavior of physical and chemical gels and perform a comparative analysis with several biological tissues.
Methods
Microindentation-based load-relaxation experiments were performed on three physical (agar, alginate, and gelatin) gels and one chemical (polyacrylamide) gel with a range of experimental time frames.
Results
All three physical gels exhibit strong time-dependent load-relaxation behavior where faster indentation leads to pronounced load-relaxation over short time-scales. The polyacrylamide gel is largely time-independent and exhibits negligible relaxation within short time-scales. The material property intrinsic permeability, which relates to underlying pore structure, was time-independent for both physical and chemical gels.
Conclusions
A comparative analysis reveals that different aspects of the time-dependent properties of biological tissues are captured by physical and chemical hydrogels, with implications for tissue engineering applications.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Hydrogels are ideal materials for biomedical applications due to their biocompatibility, hydrophilicity, and tissue-like water contents [1]. They are being studied as artificial tissue scaffolds to facilitate regeneration of damaged tissue [2, 3]; as drug carriers to provide targeted drug release [4]; and as synthetic extra-cellular matrices to study cell-matrix interactions [5]. Success of hydrogel biomaterials largely depends on how closely they can imitate the mechanical properties of native tissues. Historically, tissue stiffness has been the primary design variable for fabricating hydrogel biomaterials, although biological tissues are much more complex than a linear elastic material. Most biological tissues deform in a time-dependent manner and undergo load-relaxation under constant deformation. Several studies have characterized load-relaxation behavior of both soft and stiff tissues [6,7,8,9], and demonstrated its critical role to regulate tissue function and cell mechanics [5, 10]. Recreating load-relaxation characteristics of native tissues is therefore an important prerequisite for hydrogel biomaterials.
The load-relaxation behavior of biological tissues involves coupled viscoelastic and poroelastic relaxation. Topological reorganization of the biopolymer networks of cell cytoskeleton [11] and extra-cellular matrix (ECM) [12] induces viscoelastic relaxation in tissues. Poroelastic relaxation occurs due to intrinsic fluid migration within the porous ECM structure [13]. Similarly, hydrogels also undergo coupled viscoelastic and poroelastic relaxation where the dominant relaxation mechanism largely depends on whether the gel is formed through physical or chemical cross-links [14]. Physical gels formed by reversible and non-covalent cross-links predominantly undergo viscoelastic relaxation whereas covalently cross-linked chemical gels exhibit poroelastic relaxation.
Load-relaxation also critically affects the effective elastic properties of hydrogels and biological tissues. Most biological tissues exhibit significantly greater effective modulus at faster loading than slower loading [6, 15,16,17]. Such time-dependent stiffness is essential to prevent excessive tissue deformation and damage specially during impact conditions. Hence, hydrogels to be used as artificial tissue replacements must closely mimic this time-dependent effective tissue modulus as well. In addition to elastic modulus, load-relaxation is also linked to microscopic porous structure since fluid-flow induced poroelastic relaxation is a function of gel or tissue permeability [13]. If the permeability of hydrogel scaffolds is not comparable to native tissues, it will not be able to capture the tissue-like poroelasticity which is a critical requirement for tissue engineering of fibrous connective tissues like cartilage and tendon [18].
Mimicking time-dependent mechanics of native tissues in hydrogels has been an active research field for more than a decade. Several physical and chemical hydrogels have been developed to mimic some aspects of native tissue load-relaxation behavior by tailoring gel composition parameters using trial-and-error method [19,20,21]. However, a rigorous understanding of the structural basis for hydrogel’s time-dependent mechanics has not been established. It remains poorly understood whether physical or chemical hydrogels are more representative of native tissues in terms of load-relaxation characteristics. More importantly, it remains unclear how comparable are the time-dependent effective modulus and permeability of different hydrogels to those of various biological tissues.
Indentation testing has been shown to be a powerful technique for the mechanical analysis of compliant, hydrated materials including biological tissues [13] and hydrogels [1]. Using indentation techniques, the viscoelastic [8, 22], poroelastic [23, 24] or poroviscoelastic [25] material parameters can be deconvoluted from creep or load-relaxation data. An advantage of using indentation for characterization of such materials is the simple execution of mechanical tests, without the need to “grip” the samples, and relatively forgiving requirements for sample preparation in terms of flat surfaces. All of the analyses best suited for these materials are based on the same underlying elastic Hertzian contact mechanics for contact between a spherical probe and a sample half-space [26]. These existing analyses allow for studies of structure-properties relationships in hydrated materials using straightforward methods for data analysis based on curve-fitting, not requiring inverse finite element analysis or other computationally intensive techniques for analysis of large quantities of experimental data.
The objective of current work is to quantitatively characterize load-relaxation behavior of hydrogels with different gel structures and demonstrate how closely they compare with native tissue time-dependent properties (i.e. effective elastic modulus and permeability). Three physically cross-linked gels, namely agar, alginate, and gelatin, and one chemical gel, polyacrylamide, are considered as model systems. All four gels consist of distinct gel structures (Fig. 1) with different gelation mechanisms. Agar is a polysaccharide extracted from seaweeds with a sugar-based skeleton that forms thermo-reversible physical gel. Randomly coiled polymer chains of agar transform into double helices on cooling that entangle with each other to form the gel network (Fig. 1(a)) [27]. Alginate is also polysaccharide in which sugar units contain guluronate-mannuronate blocks. The gulurontate blocks of adjacent polymer chains form ionic cross-links in presence of divalent cations (for example, \(\text {Ca}^{2+}\)) to form the alginate gel (Fig. 1(b)) [28]. Gelatin is a protein derived from collagenous tissues of animals (for example, skin, tendon, and bone). Here, gelation occurs due to rearrangement of polymer chains into ordered triple helix configurations (Fig. 1(c)) upon cooling which act as physical cross-links in gelatin gels [29]. By contrast, polyacrylamide is a synthetic polymer which is transformed into gel by chemically joining acrylamide chains with covalent cross-linkers such as bisacrylamide (Fig. 1(d)) [30].

Schematics of different types of hydrogels studied in this work. (a) In agar gel, polymer chains with double helical configurations form the gel through physical entanglements. (b) alginate gel is formed by ionic cross-links (blue circles) between gulurontate blocks of adjacent polymer chains. (c) gelatin gel is obtained through the formation of ordered triple helical junctions among polymer chains. (d) in polyacrylamide gel, polymer chains are chemically bonded by covalent cross-links (red squares)
Load-relaxation behavior of all four gels have been independently characterized in the literature [14, 23, 31], but an in-depth and systematic comparison of their time-dependent properties in close relevance to native tissue properties is not established. Such comparative analysis will not only elucidate the structural basis of hydrogels time-dependent mechanics but also establish fundamental guidelines to design hydrogel biomaterials with native tissue-like time-dependent properties. Here, spherical microindentation experiments are used to quantify the load-relaxation behavior of all four hydrogels. Systematic load relaxation tests as a function of experimental time-scale are performed to reveal how nature of cross-linking and gel structure affect time-dependent load-relaxation behavior of hydrogels. Experimental findings are further analyzed theoretically and compared with various biological tissues, providing insight into comparative advantages of physical and chemical gels for tissue engineering applications.
Materials and Methods
Hydrogel Preparation
Agar, sodium alginate, calcium chloride, and porcine gelatin (300 g bloom) were obtained from Sigma-Aldrich Inc. (NC, USA).
Agar (3 \(\%\) w/w) and gelatin (10\(\%\) w/w) gels were prepared by mixing the solid polymer with distilled water at room temperature. The mixtures were stirred with a magnetic bar in hot plates at 90 \(^{\circ }\)C (agar) or 65 \(^{\circ }\)C (gelatin) until clear solutions were obtained. The polymer solutions were poured into glass petri dishes (50 mm diameter) and allowed to cool for four hours. The petri dishes were filled with distilled water and stored in a 4 \(^{\circ }\)C refrigerator for 24 hours.
Alginate gel (4\(\%\) w/w) was prepared by mixing sodium alginate with distilled water at room temperature. The solutions were poured into 30 mm diameter polystyrene dishes and covered with filter paper. The dishes were then submerged into 200 mM \(\text {CaCl}_2\) solution for 24 hours to induce ionic cross-linking. The gels were removed from the plastic dishes and placed in distilled water for 24 hours while refrigerated.
Polyacrylamide gels (Petrisoft, \(\text{Easycoat}^\circledR\)) were obtained from Matrtigen LLC (Brea, CA, USA). The manufacturer used bisacrylamide as cross-linker to fabricate the polyacrylamide gel. A gel with polyacrylamide concentration of 10.9 wt% (0.2 wt% biacrylamide) was used here. Gels were obtained in 100 mm polystyrene dishes and were immersed in distilled water for 24 hours before performing indentation tests.
All the gels were thawed for one hour before testing and all the tests were performed under fully hydrated conditions (distilled water).
Indentation Experiments
Microindentation experiments were conducted using a TA Electroforce 5500 low-force mechanical tester (TA instruments, New Castle, DE, USA) equipped with a 22 N load cell. Stainless steel spherical probes of radii (R) 3 mm (agar and alginate) or 6 mm (gelatin and polyacrylamide) were used for indentation tests. The indentation depth (\(\delta _0\)) was fixed at 0.5 mm for agar gel and at 1 mm for other three gels. The combination of indenter radius and depth were selected to ensure the measured forces were within the resolution of a 22 N load cell. For each test, the probe was manually brought in contact with the gel surface and subsequently, the machine was programmed to indent the gel to a depth (\(\delta _0\)) within a prescribed ramp time (\(t_\text {r}\)) from 0.4 s to 100 s. After the indentation ramp to peak depth, the probe was held at fixed position for specified time (100 s) to capture load-relaxation behavior of the gel. In addition, relaxation tests with longer hold time (11000 s) were performed for polyacrylamide gel. For each ramp time, three indentation tests were performed and results were presented as the average of the three measurements.
Analysis
Linear elastic model
Apparent elastic modulus of different gels was measured based on linear elastic Hertzian contact model [26]. The Hertz model was originally developed to describe the contact behavior of two ellipsoidal bodies. For spherical indentation, the Hertz model can be applied assuming the spherical indenter to be a rigid sphere (significantly larger than gel stiffness) and the gel to be an elastic half-space (elastic sphere of zero curvature). The corresponding load-displacement relation during ramp loading (\(t \le t_\text {r}\)) is given as:
where \(\delta (t)\) is the applied indentation depth and \(E_\text {a}\) is the apparent elastic modulus for a given ramp time (\(t_\text {r}\)) and \(\nu\) is the Poisson’s ratio. \(E_\text {a}\) was determined by fitting equation (1) to the load-displacement curve from the ramp phase. A Poissons ratio (\(\nu\)) of 0.5 was assumed, which is a common assumption for macro-scale hydrated testing of hydrogels where the water migration is negligible during ramp loading [24, 32].
Poroelastic model
The empirical poroelastic model of Hu et al. [24] was used to determine the intrinsic permeability of hydrogels based on a poroelastic (PE) material assumption. The theory assumes the hydrogel to behave as an incompressible material at the beginning of relaxation since water does not have time to migrate and reach a new equilibrium state. As such, the force at the beginning of relaxation can be calculated from elastic Hertzian contact as \(P_0=16/3G_aR^{1/2}\delta _0^{3/2}\), where \(G_a\) is the apparent shear modulus of the hydrogel. When the gel reaches equilibrium state due to complete solvent migration, it acts as a compressible material. The equilibrium force (\(P_\infty\)) is then given by \(P_{\infty }=P_0/2(1-\nu _\text {d})\), where \(\nu _\text {d}\) is the drained Poisson’s ratio. Based on these assumptions, an empirical equation for load-relaxation was derived [24] based on finite element simulations:
where D is the diffusion coefficient and a is the contact radius (\(a=\sqrt{R\delta _0}\)). Load relaxation curves for different ramp times were fitted to equation (2) to determine the diffusion coefficient (D). Intrinsic permeability (k) was then calculated as \(k=D\mu (1-2\nu )/2G(1-\nu _\text {d})\), where \(\mu\) is the solvent viscosity (\(\mu =0.89 \times 10^{-3}\) \(\text {Pa.s}\)).
Poroviscoelastic model
In hydrogels, both poroelastic and viscoelastic relaxations can occur concurrently resulting in a poroviscoelastic (PVE) relaxation. It has been shown that the coupled effect of poroelasticity and viscoelasticity can be presented as [25]:
where \(P_{\text {PE}}\) and \(P_{\text {VE}}\) are the poroelastic and viscoelastic responses of the gel. The poroelastic (\(P_{\text {PE}}\)) response can be described based on the empirical model of equation (2). The viscoelastic response can be represented based on the Generalized Maxwell Model [8]. It consists of a linear spring connected in parallel with n number of Maxwell units (series connection of a linear spring and a dashpot). The model defines the relaxation modulus E(t) using a series of exponentials as:
where \(E_\infty\) is the elastic modulus of the linear spring; \(E_n\) is the modulus of the spring in \(n^{th}\) Maxwell element and \(\tau _n\) is the characteristic relaxation time of \(n^{th}\) Maxwell element (\(n=2\)). The corresponding viscoelastic load relaxation (\(P_{\text {VE}}\)) can be derived as:
The original Maxwell model is derived based on step loading assumption. To account for finite ramp time effect, a ramp correction factor (\(X_\text {n}\)) is incorporated which is defined as [8]:
The poroviscoelastic model of equation (3) with the expressions of \(P_{\text {PE}}\) and \(P_{\text {VE}}\) from equations (2) and (5) was fitted to the load-relaxation curves using a least-square optimization algorithm [25].
Results
Load-Relaxation Behavior
The load-relaxation curves of four gels for two ramp times (\(t_\text {r}\) = 0.4 s and 100 s) are illustrated in Fig. 2. The horizontal axis corresponds to the hold time (i.e. total time (t) minus ramp time (\(t_\text {r}\))) where the spherical probe was held at fixed position. All three physical gels exhibited considerable load-relaxation where the degree of load-relaxation was much greater in agar and alginate gels compared to gelatin gels. Larger load-relaxation occurred in all three physical gels for faster initial indentation, but the relaxation curves with different ramp times roughly converged together after around 40 s (Fig. 2(a)–(c)). This demonstrates that ramp time largely affected the gel relaxation at short time-scales, whereas the long-term gel response was mostly ramp time-independent. In contrast to physical gels, polyacrylamide gel showed a roughly elastic response with negligible drop in indentation load (P) within an identical hold time of 100 s (Fig. 2(d)).

Load-relaxation curves of different gels for two different ramp times (\(t_\text {r}\) = 0.4 & 100 s) (a) agar gel, (b) alginate gel, (c) gelatin gel, and (d) polyacrylamide gel
The effects of gel structure and type of cross-linking on gel relaxation behavior were observed to be more evident when load-relaxation curves were normalized by \(P_0\) (Fig. 3). All four gels exhibited distinct rate of load-relaxation as a function of ramp time (\(t_\text {r}\)). For \(t_\text {r} = 0.4\) s (Fig. 3(a)), both polysaccharide gels (agar and alginate) showed rapid load-relaxation where the indentation load dropped below 30% of \(P_0\) within just 100 s, where the drop in indentation load for the protein (gelatin) gel was less than 10%. For significantly slower indentations with \(t_\text {r} = 100\) s (Fig. 3(b)), agar and alginate gels also exhibited significant load-relaxation where the percentile drop in indentation load (P) was around 60%. The gelatin gel showed around 7% drop in indentation load for \(t_\text {r} = 100\) s. However, the chemically crosslinked polyacrylamide gel demonstrated less than 1% drop in load in the same time period for both slow and fast indentations.

Normalized load-relaxation curves of different gels for two different ramp times (a) \(t_\text {r}\) = 0.4 s and (b) \(t_\text {r}\) = 100 s. The load (P) is normalized by the peak indentation load (\(P_0\)) at maximum depth (\(\delta _0\))
Time-dependent relaxation behavior of the gels was further investigated by systematically varying ramp time from 0.4 s to 100 s (Fig. 4). The ramp time effect was monotonic for all three physical gels where degree of load relaxation within a fixed hold phase (100 s) increased with decrease in ramp time (Fig. 4(a)–(c)). For agar gel, degree of load-relaxation was equivalent for \(t_\text {r} \le\) 10 s (Fig. 4(a)), indicating ramp time effect was negligible beyond this limit. Alginate showed the most pronounced time-dependent load-relaxation where the ramp time effect diminished for \(t_\text {r} \le\) 2 s (Fig. 4(b)). In contrast, degree of relaxation in gelatin showed negligible ramp time-sensitivity beyond \(t_\text {r} <\) 50 s (Fig. 4(c)). Polyacrylamide gel demonstrated roughly elastic response with negligible relaxation over 100 s for all ramp times (Fig. 4(d)). Load-relaxation tests with longer hold time (11000 s) were also performed for polyacrylamide gel (Fig. 4(d), inset). It was observed that load relaxation curves of polyacrylamide gel for different ramp times did not exhibit any consistent correlation with ramp time and the associated variability was also small (\(\sim\) 5%).

Normalized load-relaxation curves of four gels for a broad range of ramp times (\(t_\text {r}\))- (a) agar gel, (b) alginate gel, (c) gelatin gel, and (d) polyacrylamide gel. The load (P) is normalized by the peak indentation load (\(P_0\)) at maximum depth (\(\delta _0\)). The inset in (d) corresponds to long relaxation tests (11000 s) of polyacrylamide gel. Each curve represents the average of three indentation measurements and the error bars represent the corresponding standard deviation
Apparent Elastic Modulus
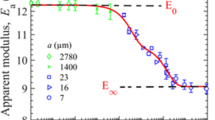
The apparent elastic modulus (\(E_\text {a}\)) was observed to be strongly dependent on the experimental ramp time (Fig. 5) for each material. Agar gel showed marked increases in \(E_\text {a}\) with decreasing ramp time. Alginate and gelatin gels demonstrated moderate ramp time dependency and \(E_\text {a}\) of polyacrylamide gel remained approximately constant for varying ramp times. For agar gels, \(E_\text {a}\) increased from 109 kPa to 399 kPa as the ramp time was varied from 100 s to 0.4 s. Alginate and gelatin gel moduli roughly varied from 100 kPa to 60 kPa in the same range of ramp time. The apparent modulus of polyacrylamide gel was around 50 kPa irrespective of fast or slow indentations.

(a) Variation of apparent elastic modulus (\(E_\text {a}\)) with ramp time (\(t_\text {r}\)) for all four gels. Each data point represents the average of three indentation measurements and the error bars (comparable to the size of the symbols) represent the corresponding standard deviation
Intrinsic Permeability
The intrinsic permeability (k) of all four gels were observed to be largely time-independent. As illustrated in Fig. 6, k was roughly constant for various ramp times in the range from 0.4 s to 100 s for all four gels. While k was time-independent for all four gels, it was observed to strongly vary with gel structure. The permeability of the chemical gel (polyacrylamide) was smaller than all three physical gels by at least two orders of magnitude. Specifically, intrinsic permeability of polyacrylamide was found to be in the order of 3.3 - 3.8 \(\text {nm}^2\) which was in good agreement with the values reported in literature [32, 33]. Gelatin and agar gels showed intrinsic permeability in the order of 200-400 \(\text {nm}^2\) which were in the same order of other similar studies [25]. Alginate demonstrated the largest permeability values in the order of 1000-3000 \(\text {nm}^2\) which was comparable to literature values [34].

Intrinsic permeability (k) of different hydrogels for various ramp times (\(t_\text {r}\)) as measured using poroelastic (open symbols) and poroviscoelastic (filled symbols) models
Discussion
The time-dependent load-relaxation behavior of four hydrogels with distinct microscopic gel structures has been investigated in this work. Experimental findings demonstrate that load-relaxation characteristics of hydrogels is a strong function of the gel structure. Pronounced relaxation occurs at shorter ramp times for physical gels whereas chemically cross-linked polyacrylamide demonstrates negligible load-relaxation for both fast and slow indentations (Figs. 2–3). The effect is largely determined by the relative competition of intrinsic viscoelastic and poroelastic relaxation times of the gels. For all three physical gels, the degree of load-relaxation roughly time-independent beyond \(t_\text {r} < 2\) s (Fig. 4(a)–(c)). Polyacrylamide gel shows roughly elastic response in the limit of 100 s, implying its intrinsic relaxation time-scale is larger than 100 s.
Poroelastic relaxation time (\(T_\text {p}\)) is commonly determined as \(T_\text {p} \approx a^2/D\) [13]. \(T_\text {p}\) represents the time required by the liquid to migrate over a characteristic distance equal to the contact radius (\(a=\sqrt{R\delta _0}\)) within the porous gel structure with diffusivity, D. To achieve reliable poroelastic properties, the ramp time must be sufficiently small compared to poroelastic relaxation time (\(T_\text {p}\)). Based on the microindentation results, \(T_\text {p}\) of the gels is in the order of \(10^4-10^5\) s which is two orders of magnitude larger than the longest ramp time (\(t_\text {r}\)= 100 s). Therefore, it is reasonable to assume that negligible fluid migration occurs during the ramp phase. However, for nanoindentation testing with \(a \sim\) 1 \(\mu \text {m}\), \(T_\text {p}\) is in the order of 1-10 s and significant poroelastic relaxation can occur during ramp phase. This duality of ramp time and experimental length-scale associated with hydrogel poroelasticity has been studied in ref. [35]. In contrast to poroelasticity, significant viscoelastic relaxation can occur during ramp loading. The ramp correction factor (\(X_\text {n}\)) of equation (6) accounts for this ramp time effect on gel viscoelasticity.
Poroelastic relaxation time varies quadratically with contact radius (a) whereas viscoelastic relaxation is largely size-independent. Several earlier studies performed multi-scale indentation experiments on polyacrylamide gel to explore this length-scale effect [23, 32]. However, degree of micro-scale load-relaxation in the polyacrylamide gel was not observed to be as strong as that of agar or alginate gel. For example, the drop in indentation load (P) was reported to be around 20-25% of peak indentation load (\(P_0\)) for polyacrylamide gel [32]. In contrast the relative drop in agar and alginate gels can be as large as 70-80% (Fig. 4(a)–(c)). In general, a poroelastic relaxation limit (1-\(P_\infty /P_0\)) is restricted by the ratio of undrained (\(\nu\)) and drained (\(\nu _\text {d}\)) Poisson’s ratios of the gel through \(1-\frac{1-\nu }{1-\nu _\text {d}}\) [36]. Based on incompressibility assumption (\(\nu\) = 1/2), the poroelastic relaxation limit of polyacrylamide gel is around 30% which is close to the gel response in this work (Fig. 4(d), inset).

Comparison of poroelastic (\(P_\text {PE}\)), viscoelastic (\(P_\text {VE}\)), and poroviscoelastic (\(P_\text {PVE}\)) relaxations with experimental load-relaxation curves for (a) agar and (b) polyacrylamide hydrogels. The ramp time was 2 s
In hydrogels, viscoelastic and poroelastic deformations are coupled. While the poroelastic relaxation time (\(T_p \sim 10^4 - 10^5\) s) was much larger than the experimental time-scale, it was still active during load-relaxation of the hydrogels. In practice, the poroviscoelastic model is more representative of the hydrogel responses compared to pure poroelastic assumption. In Fig. 7, the poroelastic, viscoelastic, and poroviscoelastic contributions to the load-relaxation responses of agar and polyacrylamide gels have been compared for a ramp time of 2 s. The short-term load-relaxation response of the agar gel is dominated by viscoelasticity (Fig. 7(a)). The poroelastic effect is only evident beyond 50 s when the viscoelastic relaxation is approximately complete. Similar poroviscoelastic behavior has also been observed for the other two physical gels. Hence, the strong load-relaxation response observed over short time-scales for physical gels is due to gel viscoelasticity. Indeed, for slower indentations (\(t_\text {r}>\) 10 s) where indentation ramp time is comparable to viscoelastic relaxation times of physical gels, significant relaxation occurs during indentation (\(0< t < t_\text {r}\)), even before the initiation of experimental relaxation regime (\(t > t_\text {r}\)). Consequently, the gels exhibit a softer response during the ramp phase followed by reduced load-relaxation under constant displacement (Fig. 4(a)–(c)). In contrast, the response of the chemically cross-linked polyacrylamide gel is entirely dominated by poroelasticity as shown in Fig. 7(b).

Load-relaxation behavior of various biological tissues and four hydrogels studied in this work. The load (P) is normalized by the peak indentation load (\(P_0\)) at maximum depth (\(\delta _0\)). Data for various biological tissues are compiled from refs. [8, 37,38,39,40,41]. Hydrogel data are replotted from Fig. 3(a)
It is of interest to compare the load-relaxation behavior of hydrogels with various biological tissues. In Fig. 8, normalized load-relaxation curves (similar to Fig. 3) for six different tissues are compared with the hydrogels studied in the current work. Data are presented for human cornea [37] , murine heart [38], porcine brain [39], porcine liver [40], lapine meniscus [41], and porcine cartilage [8]. All the data were obtained using microindentation tests with contact radius(a) in the order of 0.3 - 1 mm similar to current work. Interestingly, all six tissues exhibited rapid load-relaxation behavior such that the relative drop in the indentation load was 40-80% within just 100 s of hold time. The variability in load-relaxation behavior among different tissues is also characteristic of tissue microstructure and function. For example, the strong load-relaxation as observed for cartilage is important to support dynamic compressive loads. The comparison of tissues and hydrogels indicates that both agar and alginate gels can closely mimic the load-relaxation behavior of meniscus and cartilage. In contrast, gelatin and polyacrylamide gels are unable to recreate the extent of load-relaxation observed in different tissues.
Apparent elastic modulus of all three physical gels decreases gradually for slower indentation due to pronounced load-relaxation within the ramp phase whereas the variation of gel modulus with ramp time is negligible for the chemical gel (Fig. 5). The effect is more pronounced for agar gel compared to alginate and gelatin gels although all three gels are physically cross-linked gels. The observed differences can be understood based on the cross-linking mechanism of the gels. Agar gels are formed through physical entanglements which are primarily located at the helical chain ends (Fig. 1(a)). Faster indentation induces faster disassociation of these easy-to-break physical cross-links, which requires higher mechanical load, resulting in stiffer gel response. Cross-links in alginate gels are formed over finite regions through ionic bonds (Fig. 1(b)). Gelatin gel is cross-linked through the triple helical junctions which also expand into finite regions (Fig. 1(c)). Since breaking these finite cross-links is more expensive, faster indentation does not entail as strong effect as agar gel. Similarly, presence of rigid chemical cross-links makes the polyacrylamide gel modulus relatively ramp time-independent as well.

Variation of apparent elastic modulus (\(E_\text {a}\)) with ramp time (\(t_\text {r}\)) for different hydrogels and biological tissues. \(E_\text {a}\) is normalized by the modulus values for the largest ramp time, denoted by \(E^*\) to compare different materials. Data for murine brain tissue (white matter) are obtained from [6] where \(E^*\) = 1.25 kPa for a ramp time, \(t_\text {r}\) = 160 s. Data for bovine meniscus are compiled from [16] for which \(E^*\) = 1.4 MPa and \(t_\text {r}\) = 100 s. Modulus values for bovine cartilage are taken from [17] where \(E^*\) = 1.02 MPa for a ramp time, \(t_\text {r}\) = 240 s. Hydrogel data are replotted from Fig. 5
In Fig. 9, the variation of apparent elastic modulus (\(E_\text {a}\)) with ramp time for three tissues is compared with the hydrogels studied in current work. Data are presented for murine brain tissue (white matter) [6], bovine meniscus [16], and cartilage [17] as measured by microindentation tests with contact radius, \(a = 0.3 - 0.6\) mm comparable to current work. The vertical axis is normalized with modulus values (\(E^*\)) corresponding to largest ramp times to compare ramp time sensitivity of various materials. For brain tissue, \(E^*\) is 1.4 kPa for \(t_\text {r}\) = 160 s whereas \(E^*\) of meniscus is 1.4 MPa for \(t_\text {r}\) = 100 s. For cartilage, \(E^*\) is 1.4 MPa for \(t_\text {r}\) = 240 s. Hydrogel data are replotted from Fig. 5. Interestingly, the ramp time sensitivity of cartilage closely follows agar gel whereas both brain tissue and meniscus demonstrate similar ramp time sensitivity to alginate and gelatin gels. The alginate and gelatin gels demonstrate comparable ramp time sensitivity since they have similar elastic modulus over short time-scales (\(t_\text {r}\) = 0.4 s). More importantly, the chemical polyacrylamide gel is inadequate to recreate the ramp time-dependent elastic modulus of the three biological tissues, which is an important limitation for designing engineering tissue scaffolds.
Permeability of hydrogels also critically affects their ability to transport nutrients or release drug molecules under physiological environments. In contrast to elastic modulus, intrinsic permeability (k) of both physical and chemical gels do not vary significantly with ramp time (Fig. 6), indicating transport properties of the gels are not affected by loading time-scales. However, permeability of the hydrogels strongly depends on gel structures where physical gels are found to be more permeable than chemical gels. Intrinsic permeability (k) is also indicative of effective pore size since \(k \sim \xi ^2\), where \(\xi\) is the average pore size in the polymer network of hydrogels [13]. Based on the permeability results (Fig. 6), the pore size of alginate gel is around 30 - 60 nm whereas the pore size of agar and gelatin gels is around 14-18 nm. In contrast, the polyacrylamide gel has a pore size around 2 nm. The permeability values as calculated from pure poroelastic and poroviscoelastic models were comparable (Fig. 6). It confirms that pure poroelastic analysis can also be used to obtain a reasonable estimate of gel permeability.

Comparison of intrinsic permeability (k) for different hydrogels and biological tissues. Data for physical gels are compiled from current work and ref. [31]. Permeability values of polyacrylamide gel with different concentrations are replotted from ref. [42]. Data for heart, kidney and liver tissues are obtained from ref. [9]. Cartilage data is obtained from ref. [36]. The solid lines indicates the theoretical scaling between intrinsic permeability and elastic modulus (\(k \propto E^{-3/4}\))
Intrinsic permeability of physical and chemical hydrogels is compared with various biological tissues in Fig. 8. Permeability values of physical gels are taken from current work and ref. [31]. To represent chemical gel, data for polyacrylamide gel with different concentrations are obtained from ref. [42]. Data for heart, kidney and liver tissues are obtained from ref. [9]. Cartilage data is taken from ref. [36]. It is noted that all the permeability values of different hydrogels and tissues were measured using the same poroelastic model (equation (2)) to ensure accurate comparison. As observed in Fig. 9, permeability of physical gels is generally higher than chemical gels due to relatively larger pore sizes and open configurations. The permeability of chemical gels decreases gradually with increase in elastic modulus, indicating the permeability can be controlled systematically in terms of polymer or cross-link concentration. Previous theoretical studies have shown that elastic modulus varies quadratically (\(E \propto c^2\)) and permeability varies as \(k \propto c^{-3/2}\) with gel concentration (c) [1, 43]. It leads to a scaling relation between intrinsic permeability and elastic modulus as \(k \propto E^{-3/4}\). The chemical gels closely follow this scaling relationship as indicated by the solid line in Fig. 10. In contrast, physical gels show large scatter in permeability values with respect to elastic modulus since the gel structures are highly dynamic due to mobile cross-links. Biological tissues demonstrate a broad range of permeability where permeability also meets the function. For example, both liver and kidney tissues demonstrate large permeability in the order of 300-400 \(\text {nm}^2\) since fluid transport is critical for blood filtration (liver) or waste product removal from body (kidney). Heart tissue exhibits average permeability in the order of 10 \(\text {nm}^2\), comparable to polyacrylamide gel with similar elastic modulus. Cartilage has a small permeability in the order of 0.01 - 0.1 \(\text {nm}^2\), representative of very complex and dense structure of cartilage.
In summary, this work demonstrates that indentation is a straightforward yet powerful technique to elucidate complex structure-property relationships of soft and hydrated materials such as hydrogels. By systematic microindentation experiments under identical conditions, it was quantitatively elucidated how the molecular gel structure (physical vs. chemical) affects the load-relaxation response of various hydrogels. Time-dependent mechanical and transport properties of the hydrogels have been investigated in close comparison with various biological tissues, which are highly relevant for biomedical applications. The detailed comparative analysis has provided useful insights into the relative advantages of physical and chemical hydrogels as synthetic tissue phantoms. Overall, the findings have established important design guidelines for the next generation of hydrogel biomaterials to closely mimic the time-dependent mechanics of native tissues for tissue engineering applications.
Conclusion
The time-dependent load-relaxation behavior of four different hydrogels is investigated in this work. By combining systematic micro-indentation experiments with theoretical analysis, it is demonstrated how the time-dependent characteristics of hydrogels are dictated by gel structure. Physically cross-linked gels demonstrate strong time-dependent behavior, where faster indentation entails a stiffer gel response followed by pronounced relaxation. On the contrary, chemically cross-linked polyacrylamide gel exhibits largely time-independent behavior during microindentation. The poroviscoelastic analysis reveals that the short-term load- relaxation of the physical gels is dominated by the gel viscoelasticity, where faster indentation makes the gels more viscous by exaggerating the cross-link breakage kinetics. Load-relaxation behavior of the chemical gel is largely controlled by poroelasticity. Comparison of hydrogels and various biological tissues reveal that physical gels are more representative of biological tissues in terms of load-relaxation and time-dependent elastic properties. The chemical gel can better mimic the tissue porous structure. The indentation-based analysis presented here provides a fast and robust approach to design hydrogel biomaterials with native tissue-like time-dependent characteristics.
References
Oyen ML (2014) Mechanical characterisation of hydrogel materials. Int Mater Rev 59(1):44–59
Spiller KL, Maher SA, Lowman AM (2011) Hydrogels for the repair of articular cartilage defects. Tissue Eng Part B Rev 17(4):281–299
Kumar PS, Raj NM, Praveen G, Chennazhi KP, Nair SV, Jayakumar R (2013) In vitro and in vivo evaluation of microporous chitosan hydrogel/nanofibrin composite bandage for skin tissue regeneration. Tissue Eng Part A 19(3–4):380–392
Ashley GW, Henise J, Reid R, Santi DV (2013) Hydrogel drug delivery system with predictable and tunable drug release and degradation rates. Proc Natl Acad Sci 110(6):2318–2323
Chaudhuri O, Gu L, Klumpers D, Darnell M, Bencherif SA, Weaver JC, Huebsch N, Hp Lee, Lippens E, Duda GN et al (2016) Hydrogels with tunable stress relaxation regulate stem cell fate and activity. Nat Mater 15(3):326–334
Budday S, Nay R, de Rooij R, Steinmann P, Wyrobek T, Ovaert TC, Kuhl E (2015) Mechanical properties of gray and white matter brain tissue by indentation. J Mech Behav Biomed Mater 46:318–330
Oyen ML (2008) Poroelastic nanoindentation responses of hydrated bone. J Mater Res 23(5):1307–1314
Mattice JM, Lau AG, Oyen ML, Kent RW (2006) Spherical indentation load-relaxation of soft biological tissues. J Mater Res 21(8):2003–2010
Islam MR, Virag J, Oyen ML (2020) Micromechanical poroelastic and viscoelastic properties of ex-vivo soft tissues. J Biomech 113:110090
Charrier EE, Pogoda K, Wells RG, Janmey PA (2018) Control of cell morphology and differentiation by substrates with independently tunable elasticity and viscous dissipation. Nat Commun 9(1):1–13
Kasza KE, Rowat AC, Liu J, Angelini TE, Brangwynne CP, Koenderink GH, Weitz DA (2007) The cell as a material. Curr Opin Cell Biol 19(1):101–107
Huang D, Huang Y, Xiao Y, Yang X, Lin H, Feng G, Zhu X, Zhang X (2019) Viscoelasticity in natural tissues and engineered scaffolds for tissue reconstruction. Acta Biomater 97:74–92
Oyen ML (2015) Nanoindentation of hydrated materials and tissues. Curr Opin Solid State Mater Sci 19(6):317–323
Zhao X, Huebsch N, Mooney DJ, Suo Z (2010) Stress-relaxation behavior in gels with ionic and covalent crosslinks. J Appl Phys 107(6):063509
Doehring TC, Carew EO, Vesely I (2004) The effect of strain rate on the viscoelastic response of aortic valve tissue: a direct-fit approach. Ann Biomed Eng 32(2):223–232
Baro VJ, Bonnevie ED, Lai X, Price C, Burris DL, Wang L (2012) Functional characterization of normal and degraded bovine meniscus: rate-dependent indentation and friction studies. Bone 51(2):232–240
Moore A, Zimmerman B, Chen X, Lu X, Burris D (2015) Experimental characterization of biphasic materials using rate-controlled hertzian indentation. Tribol Int 89:2–8
Kuo CK, Li WJ, Mauck RL, Tuan RS (2006) Cartilage tissue engineering: its potential and uses. Curr Opin Rheumatol 18(1):64–73
Forte AE, Galvan S, Manieri F, y Baena FR, Dini D, (2016) A composite hydrogel for brain tissue phantoms. Mater Des 112:227–238
Fitzgerald MM, Bootsma K, Berberich JA, Sparks JL (2015) Tunable stress relaxation behavior of an alginate-polyacrylamide hydrogel: comparison with muscle tissue. Biomacromolecules 16(5):1497–1505
Spiller KL, Laurencin SJ, Charlton D, Maher SA, Lowman AM (2008) Superporous hydrogels for cartilage repair: Evaluation of the morphological and mechanical properties. Acta Biomater 4(1):17–25
Oyen ML (2005) Spherical indentation creep following ramp loading. J Mat Res 20(8):2094–2100
Galli M, Comley KS, Shean TA, Oyen ML (2009) Viscoelastic and poroelastic mechanical characterization of hydrated gels. J Mat Res 24(3):973–979
Hu Y, Zhao X, Vlassak JJ, Suo Z (2010) Using indentation to characterize the poroelasticity of gels. Appl Phys Lett 96(12):121904
Strange DG, Fletcher TL, Tonsomboon K, Brawn H, Zhao X, Oyen ML (2013) Separating poroviscoelastic deformation mechanisms in hydrogels. Appl Phys Lett 102(3):031913
Johnson KL (1985) Contact mechanics. Cambridge University Press, Cambridge, UK
Armisen R, Gaiatas F (2009) Agar. Handbook of hydrocolloids, 2nd edn. Elsevier, Boca Raton, pp 82–107
Rowley JA, Madlambayan G, Mooney DJ (1999) Alginate hydrogels as synthetic extracellular matrix materials. Biomaterials 20(1):45–53
Djabourov M, Leblond J, Papon P (1988) Gelation of aqueous gelatin solutions. i. structural investigation. Journal De Physique 49(2):319–332
Naghash HJ, Okay O (1996) Formation and structure of polyacrylamide gels. J Appl Polym Sci 60(7):971–979
Strange DG, Oyen ML (2012) Composite hydrogels for nucleus pulposus tissue engineering. J Mech Behav Biomed Mater 11:16–26
Kalcioglu ZI, Mahmoodian R, Hu Y, Suo Z, Van Vliet KJ (2012) From macro-to microscale poroelastic characterization of polymeric hydrogels via indentation. Soft Matter 8(12):3393–3398
Lai Y, Hu Y (2018) Probing the swelling-dependent mechanical and transport properties of polyacrylamide hydrogels through afm-based dynamic nanoindentation. Soft Matter 14(14):2619–2627
Tokita M, Tanaka T (1991) Friction coefficient of polymer networks of gels. J Chem Phys 95(6):4613–4619
Islam MR, Oyen ML (2020) A poroelastic master curve for time-dependent and multiscale mechanics of hydrogels. J Mat Res pp 1–9
Oyen ML, Shean TA, Strange DG, Galli M (2012) Size effects in indentation of hydrated biological tissues. J Mat Res 27(1):245–255
Ahearne M, Yang Y, Then KY, Liu KK (2007) An indentation technique to characterize the mechanical and viscoelastic properties of human and porcine corneas. Ann Biomed Eng 35(9):1608–1616
Rubiano A, Qi Y, Guzzo D, Rathinasabapathy A, Rowe K, Pepine C, Simmons C (2016) Stem cell therapy restores viscoelastic properties of myocardium in rat model of hypertension. J Mech Behav Biomed Mater 59:71–77
Qian L, Zhao H, Guo Y, Li Y, Zhou M, Yang L, Wang Z, Sun Y (2018) Influence of strain rate on indentation response of porcine brain. J Mech Behav Biomed Mater 82:210–217
Ahn B, Kim J (2010) Measurement and characterization of soft tissue behavior with surface deformation and force response under large deformations. Med Image Anal 14(2):138–148
Wheatley BB, Fischenich KM, Button KD, Haut RC, Donahue TLH (2015) An optimized transversely isotropic, hyper-poro-viscoelastic finite element model of the meniscus to evaluate mechanical degradation following traumatic loading. J Biomech 48(8):1454–1460
Trappmann B, Gautrot JE, Connelly JT, Strange DG, Li Y, Oyen ML, Stuart MAC, Boehm H, Li B, Vogel V et al (2012) Extracellular-matrix tethering regulates stem-cell fate. Nat Mater 11(7):642–649
De Gennes PG (1979) Scaling concepts in polymer physics. Cornell University Press, Ithaca, NY
Funding
Funding was provided by the ECU Division of Research, Economic Development and Engagement (REDE) via start-up funds to MLO, including post-doc salary support for MRI.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of Interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Islam, M.R., Oyen, M.L. Load-Relaxation Characteristics of Chemical and Physical Hydrogels as Soft Tissue Mimics. Exp Mech 61, 939–949 (2021). https://doi.org/10.1007/s11340-021-00712-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11340-021-00712-x