
Abstract
Purpose
Sigma-1 receptors (S1Rs) are overexpressed in almost all human cancers, especially in breast cancers. 1-(4-Iodophenyl)-3-(2-adamantyl)guanidine (IPAG) is a validated high-affinity S1R antagonist. The objective of the current study is to evaluate the potential of iodine-124-labeled IPAG ([124I]IPAG) to image S1R-overexpressing tumors.
Procedures
[124I]IPAG was synthesized from a tributyltin precursor dissolved in ethanol using chloramine-T as oxidant. Purity was analyzed using HPLC. In vitro and in vivo studies were performed using the breast cancer cell line MCF-7. Competitive inhibition studies were performed using haloperidol and cold IPAG. Tumors were established in athymic nude mice by injecting 107 cells subcutaneously. Mice were imaged on micro-positron emission tomography (PET) at 4, 24, 48, 72, and 144 h post i.v. injection. Biodistribution studies were performed at same time points. In vivo tracer dilution studies were performed using excess of IPAG and haloperidol. The efficacy of [124I]IPAG to image tumors was evaluated in LNCaP tumor–bearing mice as well.
Results
[124I]IPAG was synthesized in quantitative yield and in vitro studies indicated that [124I]IPAG binding was specific to S1R. PET imaging studies in MCF7 tumor–bearing mice reveal that [124I]IPAG accumulates in tumor and is preferentially retained while clearing from non-target organs. The tumor to background increases with time, and tumors could be clearly visualized starting from 24 h post administration. Similar results were obtained in mice bearing LNCaP tumors. In vivo tracer dilution studies showed that the uptake of [124I]IPAG could be competitively inhibited by excess of IPAG and haloperidol.
Conclusions
[124I]IPAG was synthesized successfully in high yields, and in vitro and in vivo studies demonstrate specificity of [124I]IPAG. [124I]IPAG shows specific accumulation in tumors with increasing tumor to background ratio at later time points and therefore has high potential for imaging S1R-overexpressing cancers.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Sigma-1 receptor (S1R) is a 223-amino acid protein associated with the endoplasmic reticulum and cell surface and is present throughout the central nervous system, liver, kidney, and endocrine glands such as the ovary, adrenal, testis, and pituitary gland [1, 2]. Sigma-1 receptors play an important role in several biological processes and diseases including cancers [3]. Due to their critical role in various processes, S1R expression appears to be upregulated in various types of cancers most prominently prior to mitosis, especially in breast cancers [4, 5].
Previous work in our lab on several cancer cell lines including prostate (LNCaPs) and breast cancers (MCF-7) revealed that S1Rs are involved in protein homeostasis, one of the important pathways involved in cell death and cancers. Disruption of S1R signaling using S1R antagonist such as 1-(4-iodophenyl)-3-(2-adamantyl)guanidine (IPAG) leads first to unfolded protein response (UPR) followed by autophagy [3]. We also demonstrated that continuous S1R antagonism for an extended period of time forces cancer cells to undergo apoptosis [3]. Due to S1R upregulation in several cancers, they have been pursued as a target for imaging tumors [6]. Though the precise role of S1R in cancer cells remains inconclusive, it appears to upregulate anti-apoptotic pathways and aid cancer cell survival. Therefore, the upregulation of S1R in various cancers presents S1R as a compelling potential target for imaging cancers using radiolabeled ligands, especially using positron emission tomography (PET) due to high sensitivity and quantitative nature of the imaging technique [7].
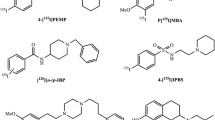
Because S1Rs play a key role in neurological diseases, initial attempts to image S1R using PET were focused on studying their expression levels in the brain. Very few attempts were made to image S1R for cancers [6, 8, 9]. One of the earlier examples of S1R imaging for melanomas using 1-(3-[18F]fluoropropyl)-4-(4-cyanophenoxymethyl)piperidine ([18F]FPS, 1, Fig. 1) was reported by the Waterhouse group [9]. Similar piperazine-based molecules (2 and 3, Fig. 1) were also synthesized and used for imaging S1R expression in brains for correlating neurological anomalies [10]. [18F]Fluspidine (4, Fig. 1) is of particular interest as it is in clinical translation for imaging S1Rs in humans [11]. All the PET active molecules synthesized so far have been labeled either with carbon-11 of fluorine-18, which have very short half-lives. Slow ligand binding kinetics of S1Rs [12] necessitate sigma-1 ligands labeled with longer-lived isotopes to achieve equilibrium.

. Chemical structures of some of the known radioactive ligands for sigma-1 receptors.
To achieve such equilibrium, sigma-1 ligands labeled with longer-lived isotopes of radioactive iodine were developed. Figure 1 provides some of the radioiodinated tracers used for imaging S1R in melanoma and breast cancers which include N-[2-(diethylamino)ethyl]-4-[123I]iodobenzamide ([123I]IDAB, 5) and N-[2-(piperidinylylamino)ethyl]-4-[125I]iodobenzamide ([125I]IPAB, 6) [13, 14], N-(2-diethylaminoethyl)3-[131/123I]iodo-4-methoxybenzamide (IMBA, 7,), N-[2-(1′-piperidinyl)ethyl]-3-[125I]iodo-4-methoxybenzamide (PIMBA, 8), and N-(N-benzylpiperidin-4-yl)-4-([125I]iodobenzamide (9) [15,16,17]. One big disadvantage of using benzamide derivatives is their binding to melanin, and therefore benzamide derivatives are not exclusive S1R ligands.
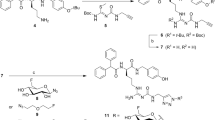
To address the need of a longer-lived PET agent to image S1R expression in cancers, we report synthesis and evaluation of I-124-labeled 1-(4-iodophenyl)-3-(2-adamantyl)guanidine ([124I]IPAG, 10, Fig. 2). IPAG binds to S1R with high affinity (Kd = 9 nM) and high selectivity. Taking advantage of novel palladium-catalyzed method developed in our lab, we synthesized trimethyltin precursor of IPAG 1-(4-tributylstannly)-3-(2-adamantyl)guanidine without using any protecting groups; using this precursor we have developed a high-yielding and reliable radiosynthetic method for the synthesis of [124I]IPAG [18]. The long half-life of iodine-124 (t1/2 = 4.2 days) allows for following the kinetics and receptor occupancy for an extended period (days) which we have demonstrated as essential for inducing apoptosis in cancer cells. Therefore, we believe this agent will be useful for determining the efficacy of novel S1R inhibitors in animal models and clinical trials. We had shown previously that hormonal sensitive breast cancer (MCF-7) and prostate cancer (LNCaP) cells express S1R natively and hence, the cell lines were used to perform these experiments [3]. Other groups have independently verified the expression of S1R’s in MCF-7 cell lines [17]. The distribution of [125I]IPAG in the mouse brain has been independently studied, but so far, no reports on its use as a tumor imaging agent have been reported [19]. Herein, we report in vivo PET imaging and biodistribution studies of S1R-specific imaging agent [124I]IPAG, in MCF-7 and LNCaP tumor xenograft models.

. a Radiochemical synthesis of [124I]IPAG (10) by iododestannylation reaction from the corresponding tributyltin precursor (11) using [124I]I-NaI and chloramine-T in ethanol. b HPLC chromatogram of the crude reaction mixture during the synthesis of [124I]IPAG. Both radioactive signal (blue) and UV signal at 254 nm (red) are shown. c Quality control analysis of [124I]IPAG on HPLC showing co-injection with IPAG. Both radioactive signal (blue) and UV signal at 254 nm (red) are shown.
Materials and Methods
All reagents and solvents were obtained from Sigma-Aldrich or Fisher Scientific Company and used without further purification. Carrier-free [131I]NaI in 0.05–0.1 N NaOH was obtained from Nuclear Diagnostic Products (Rockaway, NJ, USA), and carrier-free [124I]NaI in 0.1 N NaOH was obtained from the Radioisotopes and Molecular Imaging Probes (RMIP) core facility at MSKCC. Haloperidol hydrochloride (Cat. No. 0931) and IPAG (Cat. No. 0544) were purchased from Tocris Bioscience, Pittsburgh, PA. Isoflurane USP was purchased from Baxter Healthcare (Deerfield, IL, USA). To perform purification and quality control of radioactive products, an HPLC system was employed. The system used was manufactured by Shimadzu Scientific Corporation (Columbia, MD) and contained a binary high-pressure solvent delivery module LC-20AB and SPD-20A UV-Vis detector (dual wavelength capable) connected to a Phenomenex® Gemini 5 μm, 250 × 4.6 mm 110 Å (reversed-phase C18 column, part number: 00G-4435-E0). The radioactive signal was detected using Bioscan Flow-Count (Eckert and Ziegler Radiopharma Inc. Hopkinton, MA) detector connected to an FC3200 Na/I PMT–based detector. For both purification and analysis, an isocratic solvent system of 40 % acetonitrile in 0.1 % trifluoroacetic acid at a flow rate of 1 ml/min was used.
Estrogen-sensitive human breast cancer cell lines MCF7 and human androgen-dependent human prostate adenocarcinoma LNCaP were obtained from American Type Culture Collection (ATCC, Manassas, VA) and cultured in media suggested by the supplier obtained from media preparation core at MSKCC. Cells were cultured at 37 °C in a humidified 5 % CO2 atmosphere. 17β-Estradiol pellets (0.75 mg/pellet, Cat. No. SE-121) were purchased from Innovative Research of America (Sarasota, FL, USA.) RIPA lysis buffer was purchased from Thermo-Fisher (Cat. No. 89900). Disposable Cell culture tubes were purchased from Kimble (Cat. No. 51012-100). Matrigel was purchased from Becton Dickinson through Fischer Scientific.
Six-to-eight-week-old female athymic nude mice (Hsd:Athymic Nude-Foxn1nu) were obtained from Envigo RMS, Inc. (Indianapolis, IN), and were allowed to acclimatize at least for 1 week. Mice were provided with food and water ad libitum. All animal studies were reviewed and approved by MSKCC’s Institutional Animal Care and Use Committee (protocols 86-02-020 and 08-07-013) and followed the National Institute of Health guidelines for animal welfare. PET images were obtained either on microPET Focus scanner (Concorde Microsystems) or Inveon PET/CT scanner (Siemens Healthcare Global).
Radioactivity was quantified with a WIZARD™ 3″ 1480 gamma counter (Perkin Elmer, Waltham, MA, USA) or a dose calibrator (Capintec® CRC-30BC, Ramsey, NJ, USA). The average and standard deviation of the %ID/g was determined using GraphPad Prism version 6.07 for Windows (GraphPad PRISM software, San Diego, CA, USA, www.graphpad.com). The images were analyzed using ASIPro VM software or Inveon Research Workplace software and optimized to show localization of PET signal. Using ASIPro, maximum pixel value was recorded for tumors. Post-reconstruction smoothing was applied for visual representation of images in the figures.
Synthesis of Iodine-124- and Iodine-131-Labeled 1-(4-Iodophenyl)-3-(2-adamantyl)guanidine
[124I]IPAG and iodine-131-labeled 1-(4-iodophenyl)-3-(2-adamantyl)guanidine ([131I]IPAG) were synthesized by using well-established iodo-destannylation protocol as described earlier (Fig. 2) [18]. Briefly, to a solution of 25 μg stannylated precursor (Sn(Bu)3-IPAG, 11, Fig. 2) in dry ethanol (50 μl), [124I]NaI (1.08 mCi (40 MBq) in 18 μl 0.1 N NaOH) or [131I]NaI (1–2 mCi (40–80 MBq) in 15–20 μl 0.05–0.1 N NaOH) and chloramine-T (2 μl, 2 mg/ml solution in glacial acetic acid) were added. The solution was stirred gently, diluted with 200 μl deionized water, and purified on HPLC (Fig. 2). The eluent from the HPLC corresponding to the radioactive peak was collected. Product identity was confirmed by co-injecting non-radioactive IPAG with the radioactive fraction onto HPLC. After confirming the identity and purity of product to be [124I]IPAG or [131I]IPAG, it was further diluted in PBS to be used for further in vitro experiments. For in vivo studies, the excess solvent was removed under reduced pressure, reconstituted in 5 % ethanol (in 0.9 % saline) and injected into mice intravenously.
Subcutaneous Xenograft Model
For breast cancer models, female athymic nude mice were implanted with sustained release 17β-estradiol pellets subcutaneously 72 h prior to tumor cell implantation. MCF7 cells (1 × 107) were suspended in 1:1 mixture (100 μl) of Matrigel and cell culture medium and were injected subcutaneously (s.c.) on right forelimb of mice. To establish prostate cancer xenografts, LNCaP cells (5 × 106) were suspended in 1:1 mixture (100 μl) of Matrigel and cell culture medium and were injected subcutaneously (s.c.) on hind forelimb of mice. Tumors were allowed to grow for 3–5 weeks to reach a minimum size of 100 mm3 before any studies were performed.
PET Imaging
Athymic nude mice (n = 5) bearing s.c. tumors (MCF-7 or LNCaP, 100–300 mm3) were injected with 100 μl of 0.1 % solution of KI (intraperitoneal (i.p.)) 1 h prior to the injection of radioactivity to block thyroid uptake. The mice were injected with [124I]IPAG intravenously (i.v.) (200 μCi (7.4 MBq) in 150 μl 0.5 % ethanol in 0.9 % sterile saline), anesthetized with 2 % isoflurane gas in medical air at a rate of 2 l/min for 15 min, and imaged under PET scanner at 4, 24, 48, and 72 h post-injection.
Ex Vivo Biodistribution
Athymic nude mice (n = 5) bearing s.c. tumors (100–300 mm3) were injected with 0.1 % solution of KI (intraperitoneal (i.p.)) 1 h prior to the injection of radioactivity. Mice injected with [124I]IPAG (20–30 μCi in 5 % ethanol in 0.9 % saline solution) i.v. were euthanized by CO2 asphyxiation at pre-determined time points (4, 24, 48, and 72 h), and organs were harvested. For tracer dilution studies, [124I]IPAG formulation for injection was made such that the final volume contained 3.5–100-fold excess (by mass) of either IPAG or haloperidol hydrochloride (calculated using the specific activity of [124I]IPAG). Activity in syringe prior to injection and after injection was used to determine the total activity administered referred to as injected dose (ID). Activity in the collected organs was measured, count data was background and decay corrected to the time of injection, and %ID/g for all the organs was calculated by weighing the organs. The average and standard deviation of the %ID/g was determined.
Results
Radioiodine-labeled IPAG ([124I]IPAG and [131I]IPAG) compounds were synthesized and isolated in quantitative radiochemical yields starting from tributyltin precursor (Fig. 2a). The radioactive compound was purified and isolated on an HPLC with high radiochemical and chemical purity (Fig. 2b, c). The apparent specific activity of the product is about 2–170 mCi/μmol. Binding of radiolabeled IPAG analogs [131I]IPAG was successfully inhibited using IPAG and haloperidol. The half maximal inhibitory concentration (IC50) was determined to be in the range of 4.7–14.15 nM (95 % confidence intervals (CI), 0.98 to 22.45 nM) for IPAG (Suppl. Fig. 1, see Electronic Supplementary Material (ESM)) calculated using Prism.
In vivo pharmacokinetic properties of [124I]IPAG were evaluated by PET imaging and ex vivo biodistribution. PET imaging studies in MCF7 tumor–bearing mice reveal that [124I]IPAG accumulates in the tumor and is preferentially retained while clearing from non-target organs (Fig. 3). At 4 h, the significant amount of activity remains bound to the blood pool and therefore appears as non-specific uptake in most of the organs. Tumor visualization by PET is difficult at 4 h post-injection of [124I]IPAG because of high background activity in non-target organs, which eventually clears with time. At the end of 144 h post-injection, only S1R-expressing tissues tumor, liver, and salivary glands retain the radioactivity as evidenced by PET images while clearing from non-target organs.

. PET images showing transaxial (top) and coronal (bottom) slices of mice bearing MCF7 tumor xenografts (s.c., dotted white circle) on right forelimb administered with [124I]IPAG at 4-, 30-, 72-, and 144-h time points. It is clear from the images that the activity clears from the circulation at later time points providing high contrast images of the tumor.
[124I]IPAG uptake in MCF7 tumor–bearing mice was quantified by performing a biodistribution study. Five mice were sacrificed for each time point of 4, 24, 48, and 72 h post-injection, and the uptake in each organ was normalized to the weight of each organ. Biodistribution studies indicate the %ID/g uptake in MCF7 tumors is 1.53 ± 0.27, 1.09 ± 0.24, 0.94 ± 0.23, and 0.15 ± 0.15 ID/g at 4, 24, 48, and 72 h respectively (Fig. 4a, for the complete list of organs see Suppl. Table 1 (ESM)). The relative distribution of [124I]IPAG in organs matches the spatial distribution of [124I]IPAG as observed by PET images. The %ID/g values from biodistribution studies are higher than those obtained from drawing ROI obtained using ASIPro software.

. a Biodistribution data of [124I]IPAG in mouse bearing MCF-7 tumor xenografts (s.c.) at 4, 24, 48, and 72 h post-injection in selected organs. b Tumor to organ ratio of [124I]IPAG uptake in various organs showing clearance from selected organs.
The tumor to background ratio increases with time, and tumors could be clearly visualized starting from 24 h post administration, showing that radioactivity cleared slowly from the blood pool while specifically accumulating in tumor and organs expressing higher levels of S1R including salivary glands and liver (Fig. 4b, for the complete list of organs see Suppl. Table 2 (ESM)). Using biodistribution data and applying exponential one-phase decay to study the kinetics (GraphPad Prism) of elimination of drug from selected organs, it was found that [124I]IPAG cleared rapidly from all the organs while the amount of activity in the tumor remained almost constant for 72 h (Suppl. Fig. 2 (ESM)).
PET images obtained when the mice bearing LNCaP tumors were imaged using [124I]IPAG were comparable with the mice bearing MCF-7 (Fig. 5a). When mice bearing LNCaP tumors were injected with 100-fold excess of cold IPAG along with [124I]IPAG, the calculated %ID/g values were significantly lower than tumor-bearing mice injected with [124I]IPAG alone (Fig. 5b). %ID/g values were calculated by drawing regions on interest on PET images using ASIPro software.

. a PET images showing transaxial (top) and coronal (bottom) slices of mice bearing LNCaP tumor xenografts (s.c., dotted white circle) on the right hind limb administered with [124I]IPAG at 4, 24, 53, and 122 h. b Blocking study: PET images showing transaxial (top) and coronal (bottom) slices of mice bearing LNCaP tumor xenografts (s.c., dotted white circle) on the right hind limb administered with [124I]IPAG and 100-fold excess of IPAG at 4, 24, and 72 h showing clear decrease in the %ID/g values.
In vivo tracer dilution studies showed that the uptake of [124I]IPAG could be competitively inhibited by 100-fold excess of cold IPAG at 24 and 48 h (Fig. 6a, for the complete list of organs, see Suppl. Table 3 (ESM)), showing that the uptake of [124I]IPAG was specific. With a 100-fold excess of IPAG, the tumor uptake is reduced by 81.14 % and 88.74 % at 24 and 48 h, respectively. At 24 h post-injection (p.i.), the highest inhibition was observed in salivary glands followed by the small intestine and bone. While the uptake is blocked in salivary glands at 48 h, the inhibition drops dramatically in small intestines and bones. The tracer uptake in the heart (blood and blood pool) and muscle was comparable in both blocked and unblocked mice. The blocking of > 60 % was only observed in the tumor, spleen, stomach, brain and salivary glands. Similar inhibition of about 3-fold was observed in tumor at 72 h p.i. when 3.5-fold excess of haloperidol, a known sigma-1 antagonist, was administered intravenously along with [124IIPAG (Fig. 6b, for the complete list of organs, see Suppl. Table 4 (ESM)). Similar to tracer dilution studies using cold IPAG, the highest inhibition was observed in salivary glands, brain, and tumors.

. a Biodistribution data of [124I]IPAG in mouse bearing MCF-7 tumor xenografts (s.c.) at 24 and 48 h post administration. The corresponding blocking studies at 24 and 48 h with 100-fold excess of IPAG are also shown, which clearly demonstrated significantly reduced uptake in the tumor, liver, and salivary glands. b Biodistribution data of [124I]IPAG in mouse bearing MCF-7 tumor xenografts (s.c.) at 72 h post administration both at a tracer level and with a 3.5-fold excess of haloperidol (Haldol) block (n = 3 for tumors).
Discussion
Sigma-1 receptor presents a wonderful opportunity for serving as a common marker for imaging tumors as its overexpression has been observed in most of the tumors. Taking advantage of S1R overexpression, we have developed S1R-specific PET imaging probe [124I]IPAG. I-124-labeled IPAG offers the advantage of long half-life (4.18 days) ligand that is chemically identical to IPAG, a well-known substrate for S1Rs but has slow clearance from background tissues.
Good specific activities and high radiochemical yields of [124I]IPAG were attained as a result of our access to tributyltin precursor. The presence of tributyltin as a leaving group affords a clean and simple reaction with quantitative yields and minimal amounts of radiochemical impurities or free radioactive iodide. Apart from tributyltin being a very good leaving group for electrophilic substitution by iodine-124, synthesizing [124I]IPAG in high chemical and radiochemical purity was possible because of the difference in the retention times of tributyltin precursor and [124I]IPAG product.
[124I]IPAG shows high affinity for sigma-1 as evidenced by nanomolar inhibitory constants for cold IPAG and haloperidol in vitro. Despite repeated attempts, the variability in the uptake of radioiodinated-IPAG in competitive binding assays could not be reduced. The variability can be partially attributed to the poor solubility of IPAG in both cell media and PBS. Specific uptake of [124I]IPAG is also observed in S1R-expressing tissues in vivo. Imaging data shows that, at 4 h post-injection, the activity is in the blood as well as other organs, distributed non-specifically. Tumor could not be delineated from the background organs at 4 h as evidenced from the PET images. Tumor outlines became apparent at 24 h post-injection suggesting that [124I]IPAG cleared from the non-target organs where S1R expression is minimal. The long clearance time of IPAG from background organs (around 24 h) further strengthens the argument for using long-lived I-124 for imaging of SIRs using PET as opposed to short-lived isotope such as flourine-18 or carbon-11 for compounds with slow clearance. Clearance of [124I]IPAG from other organs while being retained in the tumor shows its specificity for S1R and strengthens the case for using [124I]IPAG for imaging various cancers that overexpress S1R.
The specificity of [124I]IPAG for S1R is strengthened by its high uptake in the liver, which is known to express higher levels of S1Rs. In vivo tracer dilution studies utilizing [124I]IPAG were performed using excess of haloperidol and IPAG at various time intervals (Fig. 6b). When the blocking experiments were performed using 100-fold cold IPAG in vivo, it was observed that, along with tumor and salivary glands, the uptake could be specifically blocked in the liver as well. High uptake inhibition is seen in most of the organs at 24 h p.i, when mice were co-injected with cold IPAG. The inhibition of uptake in the heart, lungs, liver intestines, muscle, and bone decreased significantly at 48 h p.i. of IPAG. When the blocking experiment was repeated with a different antagonist haloperidol (3.5-fold excess), the 72-h biodistribution study revealed reduced uptake not only in tumor but also in salivary glands (Fig. 6). A comprehensive literature search revealed no previous exclusive reports on S1R expression in the salivary glands. However, the presence of S1R in salivary glands can be indirectly inferred by observation of the fact that S1R antagonists such as haloperidol cause dry mouth, and administration of S1R agonists can stimulate salivary glands [20]. In addition, Waarde et al. reported significantly higher uptake of the S1R-specific ligand based PET tracer in the salivary gland in comparison with the tumor [21]. However, the liver uptake was not diminished as significantly but the exact reasons for such an anomaly are not clear. We suspect this could be the result of lower amounts of blocking agent (3.5 vs 100-fold) and different plasma half-lives and metabolic clearance patterns of IPAG and haloperidol.
We hypothesize that [124I]IPAG could be potentially used as an imaging agent to image brain tumors because of its high lipophilicity, where most of the other tracers have shown little or no ability to cross the blood-brain barrier. Our belief is further strengthened by the ability of IPAG to target various neurological receptors, for which it has to cross the blood-brain barrier. While we see the potential of [124I]IPAG to image brain tumors in mice models, the image analysis might be complicated by the tumor’s proximity to salivary glands and thyroid, which are known targets for [124I]IPAG and free radioactive iodine respectively.
In summary, we (1) reinforce sigma-1 receptor’s potential as a target to image various cancers, (2) synthesize a novel PET imaging agent, [124I]IPAG, and (3) demonstrate the utility of [124I]IPAG to image sigma-1 receptors in breast and prostate cancer.
Conclusion
[124I]IPAG was synthesized in high yields and good specific activity. In vitro and in vivo studies demonstrate both specificity and affinity of [124I]IPAG as a tracer for S1R which can be competitively blocked by known S1R antagonists. The longer-lived isotope allows PET imaging for extended time period post-injection (144 h, ~ 0.17 % ID/g) that facilitates clearance of activity from the background and thereby improves tumor delineation. Therefore, [124I]IPAG has high potential for imaging a large fraction of human cancers where S1Rs is overexpressed.
References
Hanner M, Moebius FF, Flandorfer A, Knaus HG, Striessnig J, Kempner E, Glossmann H (1996) Purification, molecular cloning, and expression of the mammalian sigma1-binding site. Proc Natl Acad Sci U S A 93:8072–8077
Schmidt HR, Zheng S, Gurpinar E, Koehl A, Manglik A, Kruse AC (2016) Crystal structure of the human σ1 receptor. Nature 532:527–530
Kim FJ, Schrock JM, Spino CM, Marino JC, Pasternak GW (2012) Inhibition of tumor cell growth by sigma1 ligand mediated translational repression. Biochem Biophys Res Commun 426:177–182
Van Waarde A, Rybczynska A, K Ramakrishnan N et al (2010) Sigma receptors in oncology: therapeutic and diagnostic applications of sigma ligands. Curr Pharm Des 16:3519–3537
Aydar E, Palmer CP, Djamgoz MB (2004) Sigma receptors and cancer possible involvement of ion channels. Cancer Res 64:5029–5035
van Waarde A, Rybczynska AA, Ramakrishnan NK, Ishiwata K, Elsinga PH, Dierckx RAJO (2015) Potential applications for sigma receptor ligands in cancer diagnosis and therapy. Biochim Biophys Acta Biomembr 1848:2703–2714
Gambhir SS (2002) Molecular imaging of cancer with positron emission tomography. Nat Rev Cancer 2:683–693
James ML, Shen B, Zavaleta CL, Nielsen CH, Mesangeau C, Vuppala PK, Chan C, Avery BA, Fishback JA, Matsumoto RR, Gambhir SS, McCurdy CR, Chin FT (2012) New positron emission tomography (PET) radioligand for imaging σ-1 receptors in living subjects. J Med Chem 55:8272–8282
Waterhouse RN, Collier TL (1997) In vivo evaluation of [18F]1-(3-fluoropropyl)-4-(4-cyanophenoxymethyl)piperidine: a selective sigma-1 receptor radioligand for PET. Nucl Med Biol 24:127–134
Toyohara J, Sakata M, Ishiwata K (2009) Imaging of sigma1 receptors in the human brain using PET and [11C]SA4503. Cent Nerv Syst Agents Med Chem 9:190–196
Kranz M, Sattler B, Wüst N, Deuther-Conrad W, Patt M, Meyer P, Fischer S, Donat C, Wünsch B, Hesse S, Steinbach J, Brust P, Sabri O (2016) Evaluation of the enantiomer specific biokinetics and radiation doses of [18F] fluspidine—a new tracer in clinical translation for imaging of σ1 receptors. Molecules 21:1164
Kovács KJ, Larson AA (1998) Up-regulation of [3H]-DTG but not [3H](+)-pentazocine labeled σ sites in mouse spinal cord by chronic morphine treatment. Eur J Pharmacol 350:47–52
Everaert H, Bossuyt A, Flamen P et al (1997) Visualizing ocular melanoma using iodine-123-N-(2-diethylaminoethyl) 4-iodobenzamide SPECT. J Nucl Med 38:870
John C, Bowen W, Saga T, Kinuya S, Vilner BJ, Baumgold J, Paik CH, Reba RC, Neumann RD, Varma VM (1993) A malignant melanoma imaging agent: synthesis, characterization, in vitro binding and biodistribution of iodine-125-(2-piperidinylaminoethyl) 4-iodobenzamide. J Nucl Med 34:2169–2175
Nicholl C, Ashour M, Hull WE et al (1997) Pharmacokinetics of iodine-123-IMBA for melanoma imaging. J Nucl Med 38:127–133
John CS, Bowen WD, Fisher SJ, Lim BB, Geyer BC, Vilner BJ, Wahl RL (1999) Synthesis, in vitro pharmacologic characterization, and preclinical evaluation of N-[2-(1′-piperidinyl) ethyl]-3-[125I]iodo-4-methoxybenzamide (P[125I]IMBA) for imaging breast cancer. Nucl Med Biol 26:377–382
John CS, Vilner BJ, Gulden ME, Efange SM, Langason RB, Moody TW, Bowen WD (1995) Synthesis and pharmacological characterization of 4-[125I]-N-(N-benzylpiperidin-4-yl)-4-iodobenzamide: a high affinity σ receptor ligand for potential imaging of breast cancer. Cancer Res 55:3022–3027
Pickett JE, Váradi A, Palmer TC, Grinnell SG, Schrock JM, Pasternak GW, Karimov RR, Majumdar S (2015) Mild, Pd-catalyzed stannylation of radioiodination targets. Bioorg Med Chem Lett 25:1761–1764
Kimes AS, Wilson AA, Scheffel U, Campbell BG, London ED (1992) Radiosynthesis, cerebral distribution, and binding of [125I]-1-(p-iodophenyl)-3-(1-adamantyl)guanidine, a ligand for sigma binding sites. J Med Chem 35:4683–4689
Choenwald RD, Barfknecht CF (1995) Use of sigma receptor ligands as salivary gland stimulants. United States Patent 387:614
van Waarde A, Jager PL, Ishiwata K, Dierckx RA, Elsinga PH (2006) Comparison of sigma-ligands and metabolic PET tracers for differentiating tumor from inflammation. J Nucl Med 47(1):150–154
Funding
The authors thank Integrative Graduate Education and Research Traineeship (IGERT 0965983 at Hunter College) by the National Science Foundation for their generous support. Technical services provided by the MSKCC Small-Animal Imaging Core Facility, supported in part by NIH Cancer Center Support Grant No. 2 P30 CA008748-48 and Radiochemistry and Molecular Imaging Probes Core Facility (RMIP) are gratefully acknowledged. Additional support was obtained from the National Institute on Drug Abuse (DA06241) and from Mr. William H. Goodwin and Mrs. Alice Goodwin and the Commonwealth Foundation for Cancer Research and The Experimental Therapeutics Center of Memorial Sloan Kettering Cancer Center to GWP.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
S.M. Larson reports receiving commercial research grants from Genentech, Inc., Wilex AG, Telix Pharmaceuticals Ltd., and Regeneron Pharmaceuticals Inc., has ownership interest (including stock, patents, etc.) in Imaginab, Inc., Samus Therapeutics Inc., Elucida Oncology Inc., Voreyda Theranostics Inc., and YMABS Therapeutic Inc., is a consultant/advisory board member for Cynvec LLC, Eli Lilly, Prescient Therapeutics Limited, Advanced Therapeutic Partners, Gerson Lehman Group, Progenics, and Janssen Pharmaceuticals Inc., and has received other remuneration from Fonde de Research Sante, Quebec. All are outside the scope of current work. G. W. Pasternak. reports personal fees and/or other fees from Novartis, Collegium, Nektar, Confo, Endo, outside the current work; He is a co-founder of Sparionbio. S. Majumdar reports other interests from Palion Therapeutics, outside the submitted work; He is a co-founder of Sparionbio. N Pillarsetty reports that he is an inventor and owner of issued patents both currently unlicensed and licensed by MSK to Samus Therapeutics, Inc., which are outside the scope of current work.
Ethics Statement
All applicable institutional and/or national guidelines for the care and use of animals were followed. This article does not contain any studies with human participants performed by any of the authors.
Additional information
This manuscript is dedicated to our colleague, mentor, and, co-author, late Dr. Gavril Pasternak.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
ESM 1
(PDF 509 kb)
Rights and permissions
About this article

Cite this article
Gangangari, K.K., Váradi, A., Majumdar, S. et al. Imaging Sigma-1 Receptor (S1R) Expression Using Iodine-124-Labeled 1-(4-Iodophenyl)-3-(2-adamantyl)guanidine ([124I]IPAG). Mol Imaging Biol 22, 358–366 (2020). https://doi.org/10.1007/s11307-019-01369-8
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11307-019-01369-8