
Abstract
The sigma-2 (σ2) receptor has been validated as a biomarker of the proliferative status of solid tumors. Therefore, radiotracers having a high affinity and high selectivity for σ2 receptors have the potential to assess the proliferative status of human tumors using noninvasive imaging techniques such as Positron Emission Tomography (PET). Since the σ2 receptor has not been cloned, the current knowledge of this receptor has relied on receptor binding studies with the radiolabeled probes and investigation of the effects of the σ2 receptor ligands on tumor cells. The development of the σ2 selective fluorescent probes has proven to be useful for studying subcellular localization and biological functions of the σ2 receptor, for revealing pharmacological properties of the σ2 receptor ligands, and for imaging cell proliferation. Preliminary clinical imaging studies with [18F]ISO-1, a σ2 receptor probe, have shown promising results in cancer patients. However, the full utility of imaging the σ2 receptor status of solid tumors in the diagnosis and prediction of cancer therapeutic response will rely on elucidation of the functional role of this protein in normal and tumor cell biology.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


Keywords
1 Introduction
Sigma (σ) receptors represent a class of proteins that were initially identified as a subtype of the opiate receptors. Subsequent studies revealed that they represented a distinct class of receptors that are located in a variety of tissues and organs including the central nervous system (Hellewell et al. 1994; Walker et al. 1990). Two separate σ receptors, σ1 and σ2 receptors, were distinguished based on differences in the binding profile of benzomorphan compounds and respective molecular weights based on photoaffinity labeling. σ1 receptors have a molecular weight of ~25 kDa, whereas σ2 receptors have a molecular weight of ~21.5 kDa (Rothman et al. 1991). The σ1 receptor has been cloned and displays a 30% sequence homology with the enzyme, yeast C8-C7 sterol isomerase (Seth et al. 1997; Hanner et al. 1996), but this receptor lacks C8-C7 isomerase activity. Studies have shown that some neuroactive steroids bind with moderate affinity to σ1 sites and suggested that σ1 receptors may modulate the activity of GABA and NMDA receptors in the CNS (Maurice et al. 1996, 1997; Romieu et al. 2003). More recently, the s1 receptor was classified as a receptor chaperone which forms a complex with the inositol triphosphate receptor at the endoplasmic reticulum (ER) membrane, thereby regulating ER-mitochondrial Ca2+ signaling and cell survival (Tsai et al. 2009). Since the σ2 receptor has not been cloned, most of what is known regarding the biology, function, and subcellular distribution of this receptor has been obtained through the use of in vitro receptor binding studies in tissues having a high receptor expression, and fluorescent microscopy studies in cancer cells under cell culture conditions.
2 Relationship Between the Density of σ2 Receptors and Measures of Cell Proliferation
The first group to report that sigma receptors were overexpressed in cancer cells was Bem et al. (1991), who demonstrated that there is a twofold higher expression of σ receptors in tumor cells than in nonmalignant tissue. Vilner et al. (1995) later reported a high density of σ2 receptors in a panel of murine and human tumor cells grown under cell culture conditions. The density of σ2 receptors in the cancer cells was generally much higher than that of the σ1 receptor. These results suggested that the σ2 receptor may function as a biomarker for differentiating solid tumors from the surrounding, normal tissues. However, this study did not explore the relationship between the density of σ2 receptors and the two key properties of cell proliferation, proliferative status and proliferation rate. The proliferative status of cancer cells is defined as the ratio of proliferating (P) cells to quiescent (Q) cells in either a solid tumor or cell culture dish (i.e., the P:Q ratio). Proliferating cells are driven into a quiescent state by either nutrient deprivation and/or hypoxia in a solid tumor or by nutrient deprivation or contact inhibition under cell culture conditions. The second property of cell proliferation, proliferation rate, refers to the time it takes a proliferating (P) cell to pass through the four different phases of the cell cycle, G1, S, G2, and M phase. Since quiescent cells are not dividing, they are typically described as being in G0 phase.
In order to determine the relationship between the density of σ2 receptors and the proliferative status of cancer cells, Wheeler et al. used the mouse mammary adenocarcinoma cell line 66 (Wallen et al. 1984a, b) to determine if there was a difference in the density of σ2 receptors between proliferating (66P) and quiescent (66Q) tumor cells under cell culture conditions. They observed that the density of σ2 receptors in 66P cells was ~10 times greater than the density measured in 66Q cells (Fig. 1a) (Mach et al. 1997). The density of σ2 receptors in 66P cells was ~900,000 copies/cell versus ~90,000 receptors/cell in the 66Q cells. This group also reported that the expression kinetics of σ2 receptors follows the growth kinetics of the 66 cells (Al-Nabulsi et al. 1999). Since it took ~4 days for the σ2 receptor to downregulate in the 66Q cells, the density of the σ2 receptor appears to be independent of the phase of the cell cycle. Using a graphical method that correlates the density of σ2 receptors with the P:Q ratio of a tumor determined by flow cytometry (i.e., the Wheeler equation), a similar P:Q ratio of σ2 receptor density was observed in solid tumor xenografts of 66 tumors (Fig. 1b) (Wheeler et al. 2000; Shoghi et al. 2013). These results indicate that the σ2 receptor is a biomarker of the proliferative status of cancer cells and the density of the receptor is independent of the proliferation rate. Therefore, σ2 receptor radiotracers have the potential to measure the proliferative status of human breast tumors using noninvasive imaging techniques such as Positron Emission Tomography (PET) and Single Photon Emission Computed Tomography (SPECT). Since many cancer cells of different origin have a high density of σ2 receptors (Vilner et al. 1995), it is likely that this approach can be extended to assess the proliferative status of other human tumors, including head and neck, melanoma, and lung tumors.

The σ2 receptor densities in proliferating and quiescent 66 tumor cells under cell culture conditions (a) or in solid tumor xenografts (b)
3 Identification of σ2 Receptor Selective Ligands
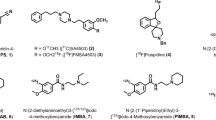
Although a number of structurally diverse compounds have been shown to possess a high affinity to σ receptors (Walker et al. 1990), most of these compounds bind selectively to the σ1 receptor or have similar affinities to both σ1 and σ2 receptors. The first σ2 selective ligand reported was the benzomorphan-7-one analog, CB-64D (Fig. 2) (Bowen et al. 1995a). This compound was synthesized as part of a structure–activity relationship (SAR) study aimed at improving the affinity of (−)-2-methyl-5-(3-hydroxyphenyl)morphan-7-one for μ versus κ opioid receptors (Bertha et al. 1994). A second class of compounds having a high affinity for σ2 receptors are the 3-(ω-aminoalkyl)-1H-indole analogs (Perregaard et al. 1995; Moltzen et al. 1995). These compounds were originally designed to be serotonin 5-HT1A agonists, but SAR studies resulted in the identification of Lu 28-179 (Soby et al. 2002), also known as siramesine, which has a high affinity for σ2 receptors and a 140-fold selectivity for σ2 versus σ1 receptors. Other compounds that were reported to have a higher affinity for σ2 versus σ1 receptors are: (1) the hallucinogen, ibogaine (Bowen et al. 1995b; Mach et al. 1995), (2) the mixed serotonin 5-HT3 antagonist/5-HT4 agonist BIMU-1 (Bonhaus et al. 1993), (3) the tropane analog SM-21 (Mach et al. 1999; Ghelardini et al. 2000), (4) the trishomocubane analog ANSTO-19 (Nguyen et al. 1996), (5) the piperazine analog PB28 (Azzariti et al. 2006), and (6) the conformational-flexible benzamide analog YUN252 (Mach et al. 2004). Many of these structures have served as the lead compounds for fluorescent probes and radioligands for imaging σ2 receptors both in vitro and in vivo.

Structures of σ2 receptor ligands in the literature
4 Development of Fluorescent Probes for Studying the Subcellular Distribution of σ2 Receptors
A number of SAR studies using BIMU-1 as the lead compound have resulted in the identification of high affinity, high selectivity σ2 receptor ligands (Vangveravong et al. 2006; Ariazi et al. 2006; Arttamangkul et al. 2000). The most promising analog from the initial SAR study was the compound SV-119 which had a σ2 receptor affinity of 5.2 nM and a σ2:σ1 selectivity of ~275 (Fig. 3a) (Ariazi et al. 2006). SV-119 was used as a lead compound for the development of the fluorescent probes, K05-138, SW120, SW107, and SW116 that have proven useful in two-photon and confocal microscopy studies of σ2 receptors in tumor cells growing under cell culture conditions (Zeng et al. 2007, 2011). This was accomplished by the recognition that introduction of a spacer group onto the bridgehead nitrogen group in SV-119 allowed for the introduction of bulky large fluorescent moieties with only a modest loss in affinity for the σ2 receptor.

Structures of SV-119 and the fluorescent probes, K05-138, SW120, SW107, and SW116 (a). Structures of the fluorescent probes based on PB28 and the 3,4-dihydroisoquinoline-1-one scaffold (b)
A second compound which has proven to be useful in developing fluorescent probes for σ2 receptors is PB28. In this case, introduction of a spacer group into the 5-methoxy group enabled the introduction of fluorescent moieties having a high affinity for σ2 receptors and a good σ1:σ2 selectivity ratio (Abate et al. 2011). Another promising fluorescent probe for studying the σ2 receptor utilized the 3,4-dihydroisoquinoline-1-one scaffold (Niso et al. 2015) (Fig. 3b). Examples of fluorescent probes based on SV-119, PB28, and the 3,4-dihydroisoquinoline-1-one scaffold are shown in Fig. 3.
The fluorescent probes shown in Fig. 3 have provided valuable information regarding the localization of σ2 receptors in breast tumor cells. Zeng et al. (2007, 2011) conducted a series of confocal and two-photon microscopy studies in MDA-MB-435 cells incubated with 30 nM K05-138 (confocal microscopy studies) and SW120 (two-photon microscopy studies) and five subcellular organelle markers: the mitochondria tracker, MitoTracker Red CMXRos (20 nM), the endoplasmic reticulum tracker, ER-Tracker™ Red (500 nM), the lysosome tracker, LysoTracker Red DND-99 (50 nM), the nuclear marker, DAPI (300 nM), or the plasma membrane tracker, FM 4-64FX (5 μg/mL). The results showed that SW120 and K05-138 partially co-localized with MitoTracker (Fig. 4a), ER-Tracker (Fig. 4b), LysoTracker, and the plasma membrane tracker, suggesting that σ2 receptors may localize in mitochondria, endoplasmic reticulum, lysosomes, and the plasma membrane. The data also showed that SW120 and K05-138 did not co-localize with the nuclear marker, DAPI, suggesting that either the σ2 receptor does not exist in the nucleus or SW120 does not enter the nucleus. Similar results were obtained for the other σ2 fluorescent probes (SW116 and SW107) (Zeng et al. 2007, 2011). This subcellular distribution of σ2 receptors was recently confirmed with confocal microscopy studies by Abate et al. using the fluorescent probes based on PB28 (Abate et al. 2011) and the 3,4-dihydroisoquinoline-1-one scaffold (Niso et al. 2015) (Fig. 3b).

Subcellular distribution of SW120 in MDA-MB-435 cells with and without MitoTracker (a), and ER-Tracker (b). Ki-67 expression and SW120 fluorescent intensity in solid mouse breast tumors and peripheral blood mononuclear cells (PBMC) of mice by flow cytometric analysis (c)
The microscopy studies conducted by Zeng et al. (2007) and Abate et al. (2011) have provided useful information for the interpretation of studies evaluating σ2 receptor ligands as potential cancer chemotherapeutic agents. Mitochondria are a key organelle to regulate the intrinsic pathway of apoptosis. Apoptotic signals such as UV irradiation or treatment with chemotherapeutic agents cause the release of cytochrome C from the mitochondria and the subsequent activation of caspase-3 leading to an apoptotic cell death (Jiang and Wang 2004). The subcellular localization of σ2 ligands in mitochondria is consistent with previous studies demonstrating that σ2 ligands trigger apoptosis in tumor cells by acting on mitochondria (Balakumaran et al. 2009). The endoplasmic reticulum (ER) serves as a dynamic Ca2+ storage pool (Berridge 2002). σ2 selective ligands have been reported to induce transient Ca2+ release from the ER, which may be a mechanism for σ2 ligand-induced cell death (Vilner and Bowen 2000). The presence of the σ2 fluorescent probes in the ER is consistent with these results. Lysosomal proteases, such as cathepsins, calpains, and granzymes, have been reported to contribute to apoptosis (Chwieralski et al. 2006). Under physiological conditions, these proteases are found within the lysosomes but are released into the cytoplasm upon exposure to cell damaging agents, thereby triggering a cascade of intracellular events leading to cell death. The σ2 selective ligand siramesine has been reported to cause lysosomal leakage and induce cell death by caspase-independent mechanisms (Ostenfeld et al. 2005, 2008). The localization of fluorescent σ2 receptor probes in the lysosomes is consistent with the premise that siramesine induces cell death partially by targeting lysosomes to cause lysosomal damage, the release of proteases, and eventually cell death. Evidence has also been reported that σ2 receptors exist in lipid rafts which are mainly found in the plasma membrane (Gebreselassie and Bowen 2004). Lipid rafts play an important role in the signaling associated with a variety of cellular events including adhesion, motility, and membrane trafficking (Brown and London 1998; Simons and Toomre 2000). The observation that σ2 fluorescent ligands are co-localized with cytoplasmic membrane markers, and undergo receptor mediated endocytosis, is consistent with their localization in lipid rafts.
Since the σ2 receptor has been validated as a proliferation marker in cell culture and in solid tumors, it is possible that σ2 fluorescent probes could preferentially label proliferating cells versus nonproliferating cells and serve as agents to image cell proliferation in vivo. To test this hypothesis, nude mice implanted with murine mammary adenocarcinoma 66 cells (Zeng et al. 2011) were treated with SW120 (50 μg in 100 μL PBS) for 1 h. Both peripheral blood mononuclear cells (PBMC), which are commonly used as controls for nonproliferative cells, and tumor cells were isolated from the mice. These cells were analyzed by flow cytometry for SW120 uptake and for Ki67 expression, a commonly used proliferation marker. The data showed that PBMC were Ki67 negative, whereas a large portion of the tumor cells were Ki-67 positive (Fig. 4c). The data also showed that PBMC were not labeled by SW120, whereas a portion of the tumor cells were labeled with SW120. The trend of the positive correlation between Ki67 expression and SW120 labeling indicates that the fluorescent probe may possess in vivo selectivity toward proliferating cells versus nonproliferative cells. The data also suggest that σ2 receptor ligands hold a potential to serve as cancer chemotherapeutics since they may selectively target tumor cells in vivo.
5 In Vivo Imaging Studies of σ2 Receptors
The conformationally flexible benzamide analog YUN252 has been used in the development of PET radiotracers for imaging the σ2 receptor status of solid tumors. The first PET radiotracers prepared were the 11C-labeled analogs shown in Fig. 5a (Tu et al. 2005). MicroPET and tumor uptake studies were conducted with [11C]1-4; the most promising analog proved to be [11C]4. Although all four analogs had a high affinity for σ2 receptors, the optimal lipophilicity of [11C]4 played an important role for the high tumor uptake and suitable signal:normal tissue ratios for imaging (Fig. 5b). These data indicate that both receptor affinity and lipophilicity are important properties for successful receptor-based tumor imaging agents. MicroPET/CT imaging studies in EMT-6 breast tumors show the potential of [11C]4 as a radiotracer for imaging the σ2 receptor status of breast tumors with PET.

Structure of YUN252 and 11C-labeled radiotracers, [11C]1–4 (a). The relationship between tumor uptake and log P is shown in (b)
Although [11C]4 demonstrated feasibility in the microPET imaging studies described above, the short half-life of carbon 11 (t 1/2 = 20.4 min) is not ideal for the utilization of PET radiotracers in multicenter clinical PET imaging studies. The longer half-life of 18F (t 1/2 = 109.8 min) compared to 11C places fewer time constraints on tracer synthesis, allows imaging studies to be conducted up to 2 h after injection of the radiotracers, and often results in higher tumor:normal tissue ratios relative to their 11C-labeled analogs. A number of 18F-labeled conformationally flexible benzamide analogs (Fig. 6a) have been evaluated in murine breast tumor models (Tu et al. 2007). The design of these analogs involved replacement of the 2-methoxy group in the benzamide ring with a 2-fluoroethoxy group. The 2-fluoroethoxy- for methoxy-substitution is a common strategy used in the development of 18F-labeled radiotracers. Biodistribution studies in female Balb/c mice bearing EMT-6 tumor allografts demonstrated that all four 18F-labeled compounds had a high tumor uptake (2.5–3.7% ID/g) and acceptable tumor:normal tissue ratios at 1 and 2 h post-i.v. injection. The moderate to high tumor/normal tissue ratios and the rapid clearance from the blood for [18F]ISO-1 and [18F]ISO-2 suggest that these radiotracers are likely the best candidates for imaging of solid tumors with PET. MicroPET imaging studies indicate that [18F]ISO-1 and [18F]ISO-2 are suitable probes for imaging the σ2 receptor status of solid tumors with PET (Fig. 6b).

Structures of [18F]ISO-1 and [18F]ISO-2 (a) and microPET imaging studies of these radiotracers in a murine model of breast cancer (b). Structures of the PET radiotracers based on PB28 and the 3,4-dihydroisoquinoline-1-one scaffold (c)
A number of 18F- and 11C-labeled analogs of the σ2 receptor ligands developed in the Abate lab have been reported in the literature. The 3,4-dihydroisoquinoline-1-one analog, [18F]5, which can be considered to be a rigid analog of the conformationally flexible benzamide derivatives described above, was found to have a high affinity for σ2 receptors and an excellent selectivity for σ2 versus σ1 receptors (Fig. 6c). This compound was developed as a radiotracer for imaging σ2 receptors in the CNS; unfortunately, the radiotracer has very low brain uptake, which may be a consequence of it being a substrate for P-glycoprotein. The aminotetralin analog, [11C](S)-6, which is structurally related to PB28 and has a good σ2 affinity and reasonable selectivity versus σ1 receptors, was evaluated in a similar manner and was found to have a high uptake in rodent brain. Surprisingly, this radiotracer had only a modest uptake in EMT6 mouse breast tumor allografts. However, the tumors used in this imaging study were quite large and likely had regions of tumor necrosis, and possibly quiescence, which could reduce the uptake of a σ2 receptor imaging agent.
A second technique which could be used in molecular imaging studies of tumors is Single Photon Emission Computed Tomography or SPECT. This technique was the Nuclear Medicine imaging procedure of choice prior to the emergence of clinical PET imaging studies. Although it does not have the prominence it once had, SPECT still represents a sensitive molecular imaging technique and would benefit from the development of receptor-targeting radiotracers for tumor imaging studies. One advantage SPECT has over PET is that it does not require a cyclotron to produce the radionuclides used in the radiolabeling studies; the longer half-lives of the SPECT radionuclides (e.g., 6 h for 99mTc and 13.2 h for 123I) make it more accessible for delayed imaging studies. A limited number of 123I- and 99mTc-labeled σ2 receptor probes have been reported in the literature. The presence of an iodo group in compound ISO-2 indicates that it can be readily labeled with 123I, and gammaSPECT/CT images of [123I]ISO-2 in a murine model of breast cancer indicate that it has potential as an SPECT radiotracer for translational imaging studies (Fig. 7a). A 99mTc-labeled analog of SV-119 (Fig. 7b) has also been prepared and evaluated in a murine model of breast cancer (Mach et al. 2001). Since there are no stable isotopes of Tc, in vitro binding studies to determine the σ2 receptor affinity and selectivity versus σ1 receptors were conducted with the corresponding rhenium (Re) analog. Re-7 was found to have a reasonable σ2 affinity and good selectivity versus σ1 receptors. In vivo studies with [99mTc]7 demonstrated a clear visualization of 66 murine breast tumor xenografts in nude mice (Fig. 7b). These data suggest that [99mTc]7 may also be a useful radiotracer for SPECT imaging studies of breast cancer patients.

Structure of [123I]ISO-2 and gammaSPECT imaging in a murine model of breast cancer (a). Structure of [99mTc]7 and microSPECT studies in the same murine model of breast cancer (b). The arrow points to the tumor in each image
6 The Importance of Imaging Proliferative Status in Cancer Patients
Treatments that specifically target proliferation and produce a cytostatic response are not well evaluated by traditional imaging methods such as MRI, CT, and bone scan (Marinovich et al. 2013). Measures of cancer metabolism using the radiolabeled glucose analog, [18F]FDG, provide information about cancer response to cytotoxic and endocrine agents, but cannot measure proliferation (Avril et al. 2001), proliferation rate, or tumor proliferative status (Shoghi et al. 2013). Many researchers have noted that there is a need for new biomarkers to enable patient selection for new cell-cycle drugs and sophisticated methods need to be developed to measure the effect of these drugs on cellular proliferation (Migliaccio et al. 2014).
An excellent method of imaging cell proliferation would be to develop a tracer that closely mimics the expression of Ki-67, the “gold standard” method for measuring proliferation in histology studies. Ki-67 is expressed in all phases of the cell cycle and has low expression in quiescent tumor cells and senescent normal tissue. While the radiolabeled thymidine analog, [18F]FLT, is well established as a proliferation marker, there are two key features that create the need to find an alternative agent to image proliferation: (1) [18F]FLT has high background uptake in bone marrow, making it impossible to monitor bone metastasis and (2) [18F]FLT is taken up only during S phase, measuring proliferation rate, and not during G1, M, and G2, like Ki-67, which measures proliferative status.
While Ki-67 is a good biomarker for measuring proliferative status in biopsy specimens, there are no small molecules that could be used in the development of PET radiotracers for imaging Ki-67. The σ2 receptor behaves exactly like Ki-67 and has small molecules that bind with high affinity, allowing PET radiotracer development. Therefore, the σ2 receptor radiotracers described above provide the only means to date to image the proliferative status of solid tumors with PET.
The only σ2 selective PET radiotracer that has been used in human studies is [18F]ISO-1 (Fig. 6). There is a high correlation between uptake of [18F]ISO-1 and the P:Q ratio of 66 solid mammary tumors (Fig. 8a)

Correlation of [18F]ISO-1 uptake and P:Q ratio in a murine model of breast cancer (a) and with Ki-67 score in a heterogeneous population of cancer patients (b). PET imaging studies of [18F]ISO-1 in a patient with lymphoma having a bone metastasis (c)
(Shoghi et al. 2013). These data indicate that [18F]ISO-1 images both proliferating and quiescent tumor cells. An early validation study in a dichotomous group of patients with head and neck, lymphoma, and breast cancer demonstrated specific uptake in cancer with ability to stratify patients into high or low Ki-67 scores based on [18F]ISO-1 uptake (Fig. 8b) (Dehdashti et al. 2013). The results of this study provided the foundation for an expanded clinical trial in breast cancer patients which is currently ongoing at the University of Pennsylvania. Additionally, bone marrow uptake was at a low level, making this a possible imaging agent for bone metastasis (Fig. 8c), in contrast to what has been reported for [18F]FLT (Mankoff et al. 2005; Shields et al. 1998).
In addition to identifying primary tumors and metastases, imaging the proliferative status with [18F]ISO-1 could be used to guide cancer therapy. An example of this is the therapies targeting the cyclin-dependent kinase 4/6 (CDK4/6). The current indication for the newly approved CDK 4/6 inhibitor, palbociclib, is for primary therapy of ER-expressing metastatic breast cancer, where the addition of palbociclib has been shown to provide an average clinical benefit (Beaver et al. 2015). Currently, the use of palbociclib is guided by the same marker used to direct endocrine therapy, namely the presence of hormone receptors. However, it is difficult to predict which individual patients will benefit from this intervention (Carey and Perou 2015). There is evidence that proliferative measures, like Ki-67, provide an early indication of response to endocrine therapy. In the Immediate Preoperative Anastrozole, Tamoxifen, or Combined with Tamoxifen (IMPACT) trial, pretreatment Ki-67 levels were prognostic for endocrine response and the change in Ki-67 from baseline to 2 weeks after therapy initiation was predictive; in multivariate analysis only the 2-week value was an independent predictor of risk and long-term outcome (Klintman and Dowsett 2015). Therefore, a PET study with [18F]ISO-1 to measure proliferative status is expected to be useful in identifying patients likely to have a favorable response to CDK4/6 inhibitors.
7 Molecular Identification of the σ2 Receptor
Although the above preclinical and clinical imaging studies have identified the σ2 receptor as a biomarker for imaging solid tumors, the clinical significance of this protein as a receptor-based marker of cell proliferation has been impeded by the fact that the molecular identity of this protein was not known. The early work of Bowen et al. yielded valuable information regarding the molecular weight of the σ2 receptor, its localization in lipid rafts, and its involvement as a mediator of cell death (Bowen 2000; Gebreselassie and Bowen 2004). However, these studies did not lead to either the sequencing of the σ2 receptor protein or the identification of the gene encoding this protein.
In 2011, the Mach group reported their work to determine the molecular identity of the σ2 receptor. This group developed a strategy to utilize a σ2 selective photoaffinity probe, WC-21, to irreversibly label σ2 receptors in rat liver membrane homogenates (Xu et al. 2011). WC-21 contains an azide moiety for the photoaffinity tagging of the protein and an FITC group for protein visualization (Fig. 9a)

Photoaffinity labeling of the PGRMC1 using the σ2 receptor photoaffinity probe WC-21 (a). Co-localization of the PGRMC1 with the σ2 fluorescent probe SW120 (b)
. WC-21 exhibits high binding affinity for sigma-2 receptors (Ki = 8.7 nM) and relatively low binding affinity for sigma-1 receptors (Ki > 4,000 nM). The rat liver membrane homogenates were photolabeled with 100 nM WC-21 and then separated by gel electrophoresis. The western blot analysis using anti-FITC antibodies showed that WC-21 labeled a protein band at ~24 kD. Labeling of this protein band with WC-21 could be blocked by DTG and haloperidol, which are ligands with high affinities to σ1 and σ2 receptors, as well as RHM-1, which is the σ2 ligand. These data suggest that WC-21 labeled proteins are σ2 receptors. Proteomic studies of the protein in the ~24 kD band labeled by WC-21 identified progesterone receptor membrane component 1 (PGRMC1). A review of the literature revealed a number of similarities between PGRMC1 and sigma-2 receptors: (1) both PGRMC1 and σ2 receptors are cancer biomarkers and therapeutic targets, (2) both are found in microsomal membranes and have similar subcellular localization, (3) both are associated with cytochrome P-450 proteins, and (4) progesterone binds to both PGRMC1 and σ2 receptors with the modest affinity. It is also important to note that the molecular weight of the protein sequence of the PGRMC1 is 21.4 kDa, which is virtually identical to the 21.5 kDa of the σ2 receptor identified previously (Hellewell et al. 1994). Therefore, PGRMC1 was chosen for further validation.
Receptor binding studies showed that the PGRMC1 ligand, AG-205, and the known σ2 ligands, DTG, siramesine, SV119, and WC-26, readily displaced σ2 radioligand, [125I]RHM-4, binding in HeLa cell membrane homogenates. Knockdown of PGRMC1 using a PGRMC1-specific siRNA reduced the binding of [125I]RHM-4 to HeLa cells (Fig. 9a), whereas the overexpression of PGRMC1 in HeLa cells increased the binding of [125I]RHM-4. Knocking down PGRMC1 in HeLa cells decreased caspase-3 activation induced by WC-26, suggesting that σ2 ligand-induced caspase-3 activation is mediated by PGRMC1. Taken collectively, the results of these studies suggest that the PGRMC1 protein complex is the putative σ2 receptor binding site.
Recently, Izzo and colleagues reported the link between the σ2 receptor and PGRMC1 during the discovery and development of small molecule therapeutics targeting beta amyloid (Aβ) 1-42 oligomers for treating Alzheimer’s disease (AD) (Izzo et al. 2014). Soluble oligomers of Aβ have been recognized to be early and key intermediates in AD related synaptic dysfunction. Soluble Aβ oligomers caused synaptic dysfunction and impaired performance in memory tasks. Alterations in membrane trafficking induced by Aβ oligomers are believed to mediate synaptic dysfunction. By screening a library of central nervous system drug-like small molecules for their abilities to reverse Aβ-induced membrane trafficking deficit in primary neurons, the lead compounds were identified. These compounds can displace Aβ oligomer binding to synaptic puncta, and prevent and reverse Aβ oligomer-induced synapse loss in primary neuronal culture, as well as reverse the memory loss in mouse models of AD. In order to identify the molecular target of the lead compounds, these compounds were examined for their activities to displace radioligand binding to receptors or for their effects on enzyme activities for 100 receptors and enzymes in the central nervous system. As a result, these compounds were found to be σ2 receptor ligands. Since PGRMC1 has been reported to be associated with the σ2 receptor, validation of PGRMC1 as a potential molecular target of the σ2 ligands was conducted. The data showed that knockdown of PGRMC1 with increasing doses of siRNA decreased Aβ binding to neurons up to 91% in a dose dependent manner. Incubation of PGRMC1 antibody against the C-terminal amino acids 185–195 of PGRMC1 in live cells for 30 min significantly reduced Aβ oligomer binding to hippocampal and cortical neurites. These data suggest that PGRMC1 is a key mediator in Aβ oligomer-induced synaptic dysfunction. That both σ2 ligand treatment and knockdown of PGRMC1 can reduce Aβ oligomer binding to neurons strongly supports the concept that PGRMC1 or its closely associated proteins contain the σ2 receptor binding site.
In another recent report, the Mach group demonstrated the positive correlation between PGRMC1 protein expression and σ2 fluorescent probe binding activity in rat hippocampal cell culture (Zeng et al. 2015). In this study, the PGRMC1 protein levels were examined in rat primary cultures of neurons, astrocytes, oligodendrocytes, and microglia cells by immunohistochemistry. The σ2 receptor binding activities of SW120, a σ2 fluorescent probe, were also examined in the aforementioned cell types. The data showed that the PGRMC1 is expressed in all brain cell types but with different expression levels. The expression level of PGRMC1 in neurons is consistently higher than that in astrocytes, oligodendrocytes, and microglia. Similarly, SW120 binding activity is also high in neurons and relatively low in astrocytes, oligodendrocytes, and microglia. In order to study whether PGRMC1 and SW120 co-localize in the cells, double staining of a rat hippocampal cell mixture for PGRMC1 with anti-PGRMC1 antibodies and the σ2 receptor with SW120 was performed. The results showed that PGRMC1 and SW120 partially co-localized in the same subcellular organelles of cells and PGRMC1 protein levels and SW120 binding activity were highly correlated (the Pearson correlation coefficient is 0.818) (Fig. 9b). These results are consistent with our previous report that the PGRMC1 protein complex is the putative σ2 receptor binding site.
Two recent reports have challenged the concept that the σ2 receptor binding site resides within the PGRMC1 protein complex. Abate and colleagues (Abate et al. 2015) stably silenced PGRMC1 with shRNA targeting PGRMC1 or overexpressed PGRMC1 in human MCF7 adenocarcinoma cells. Western blot analyses showed that PGRMC1 protein level was reduced by about 80% in PGRMC1 knockdown cells, whereas PGRMC1 protein level is increased by about twofold in PGRMC1 overexpressed cells. Scatchard analyses with radioligand [3H]DTG showed that the σ2 receptor densities in wild-type, PGRMC1 knockdown and PGRMC1 overexpressed cells are essentially the same. They also showed that AG205, a known PGRMC1 ligand, did not displace [3H]-DTG in the competition binding assay in rat liver membranes (Ki > 10,000 nM). The data suggest that PGRMC1 is not the binding site of DTG and does not affect the DTG binding site either.
In another report, the Ruoho group knocked out or overexpressed PGRMC1 using CRISPR/cas9 inhibition technology in a mouse motor neuron-like cell line, NSC34 cells (Chu et al. 2015). Western blot analysis showed that PGRMC1 expression was reduced by more than 90% in knockdown cells and dramatically increased in overexpression cells. Scatchard analysis using [3H]DTG showed that the maximal σ2 receptor densities (Bmax) and the binding affinities (Kd) of [3H]DTG were similar in the wild-type, PGRMC1 knockout and overexpression cells. In addition, using a σ2 photoaffinity probe [125I]-iodoazido-fenpropimorph ([125I]IAF) developed in this group (Fontanilla et al. 2008), they showed that the intensities of the [125I]IAF photolabeled protein band (~18 kDa) on an SDS gel were similar in the wild-type and PGRMC1 knockout cells, and this band could be protected by DTG but not (+)-pentazocine, a specific σ1 ligand. These results suggested that PGRMC1 and the σ2 receptor are distinct binding sites. However, the molecular weight of the protein labeled by [125I]IAF (~18 kDa) is significantly lower than the 21.5 kDa molecular weight of the σ2 receptor reported previously (Hellewell et al. 1994).
One possible explanation for these discordant findings is that since the PGRMC1 is a protein complex containing one or more partner proteins, the ligands used in the above studies label different members of the PGRMC1 protein complex. That is, the azide group of WC-21 comes in contact with PGRMC1, but the DTG binding site resides on one of the partner proteins making up the PGRMC1 protein complex. Studies aimed at identifying the different partner proteins that bind to the PGRMC1 and form the σ2 receptor binding site should clear up the discordant observations described above.
8 Conclusions
The σ2 receptor continues to be an important protein in the field of tumor biology. The high expression of this receptor in proliferating versus quiescent breast tumors indicates that the σ2 receptor is an important clinical biomarker for determining the proliferative status of solid tumors using the functional imaging techniques PET and SPECT. The σ2 receptor fluorescent probes identified the subcellular localization of σ2 receptors using confocal and two-photon microscopy techniques, and this information has proven useful in identifying the mechanism of action of σ2 receptor ligands as cancer chemotherapeutics. The full utility of the σ2 receptor in the diagnosis, prediction of therapeutic response, and treatment of cancer will rely on additional studies clarifying the functional relationship between the σ2 receptor binding site and PGRMC1, and the functional role of this protein complex in normal and tumor cell biology.
References
Abate C, Hornick JR, Spitzer D, Hawkins WG, Niso M, Perrone R, Berardi F (2011) Fluorescent derivatives of sigma receptor ligand 1-cyclohexyl-4-[3-(5-methoxy-1,2,3,4-tetrahydronaphthalen-1-yl)propyl]piperazine (PB28) as a tool for uptake and cellular localization studies in pancreatic tumor cells. J Med Chem 54(16):5858–5867. doi:10.1021/jm200591t
Abate C, Niso M, Infantino V, Menga A, Berardi F (2015) Elements in support of the ‘non-identity’ of the PGRMC1 protein with the sigma2 receptor. Eur J Pharmacol 758:16–23. doi:10.1016/j.ejphar.2015.03.067
Al-Nabulsi I, Mach RH, Wang LM, Wallen CA, Keng PC, Sten K, Childers SR, Wheeler KT (1999) Effect of ploidy, recruitment, environmental factors, and tamoxifen treatment on the expression of sigma-2 receptors in proliferating and quiescent tumour cells. Br J Cancer 81(6):925–933
Ariazi EA, Ariazi JL, Cordera F, Jordan VC (2006) Estrogen receptors as therapeutic targets in breast cancer. Curr Top Med Chem 6(3):181–202
Arttamangkul S, Alvarez-Maubecin V, Thomas G, Williams JT, Grandy DK (2000) Binding and internalization of fluorescent opioid peptide conjugates in living cells. Mol Pharmacol 58(6):1570–1580
Avril N, Menzel M, Dose J, Schelling M, Weber W, Janicke F, Nathrath W, Schwaiger M (2001) Glucose metabolism of breast cancer assessed by 18F-FDG PET: histologic and immunohistochemical tissue analysis. J Nucl Med 42(1):9–16
Azzariti A, Colabufo NA, Berardi F, Porcelli L, Niso M, Simone GM, Perrone R, Paradiso A (2006) Cyclohexylpiperazine derivative PB28, a sigma2 agonist and sigma1 antagonist receptor, inhibits cell growth, modulates P-glycoprotein, and synergizes with anthracyclines in breast cancer. Mol Cancer Ther 5(7):1807–1816. doi:10.1158/1535-7163.MCT-05-0402
Balakumaran BS, Porrello A, Hsu DS, Glover W, Foye A, Leung JY, Sullivan BA, Hahn WC, Loda M, Febbo PG (2009) MYC activity mitigates response to rapamycin in prostate cancer through eukaryotic initiation factor 4E-binding protein 1-mediated inhibition of autophagy. Cancer Res 69(19):7803–7810
Beaver JA, Amiri-Kordestani L, Charlab R, Chen W, Palmby T, Tilley A, Zirkelbach JF, Yu J, Liu Q, Zhao L, Crich J, Chen XH, Hughes M, Bloomquist E, Tang S, Sridhara R, Kluetz PG, Kim G, Ibrahim A, Pazdur R, Cortazar P (2015) FDA approval: palbociclib for the treatment of postmenopausal patients with estrogen receptor-positive, HER2-negative metastatic breast cancer. Clin Cancer Res 21(21):4760–4766. doi:10.1158/1078-0432.ccr-15-1185
Bem WT, Thomas GE, Mamone JY, Homan SM, Levy BK, Johnson FE, Coscia CJ (1991) Overexpression of sigma receptors in nonneural human tumors. Cancer Res 51(24):6558–6562
Berridge MJ (2002) The endoplasmic reticulum: a multifunctional signaling organelle. Cell Calcium 32(5-6):235–249
Bertha CM, Mattson MV, Flippen-Anderson JL, Rothman RB, Xu H, Cha XY, Becketts K, Rice KC (1994) A marked change of receptor affinity of the 2-methyl-5-(3-hydroxyphenyl)morphans upon attachment of an (E)-8-benzylidene moiety: synthesis and evaluation of a new class of sigma receptor ligands. J Med Chem 37(19):3163–3170. doi:10.1021/jm00045a022
Bonhaus DW, Loury DN, Jakeman LB, To Z, DeSouza A, Eglen RM, Wong EH (1993) [3H]BIMU-1, a 5-hydroxytryptamine3 receptor ligand in NG-108 cells, selectively labels sigma-2 binding sites in guinea pig hippocampus. J Pharmacol Exp Ther 267(2):961–970
Bowen WD (2000) Sigma receptors: recent advances and new clinical potentials. Pharm Acta Helv 74(2–3):211–218
Bowen WD, Bertha CM, Vilner BJ, Rice KC (1995a) CB-64D and CB-184: ligands with high sigma 2 receptor affinity and subtype selectivity. Eur J Pharmacol 278(3):257–260
Bowen WD, Vilner BJ, Williams W, Bertha CM, Kuehne ME, Jacobson AE (1995b) Ibogaine and its congeners are sigma 2 receptor-selective ligands with moderate affinity. Eur J Pharmacol 279(1):R1–R3
Brown DA, London E (1998) Functions of lipid rafts in biological membranes. Annu Rev Cell Dev Biol 14:111–136
Carey LA, Perou CM (2015) Palbociclib – taking breast-cancer cells out of gear. N Engl J Med 373(3):273–274. doi:10.1056/NEJMe1506680
Chu UB, Mavlyutov TA, Chu ML, Yang H, Schulman A, Mesangeau C, McCurdy CR, Guo LW, Ruoho AE (2015) The sigma-2 receptor and progesterone receptor membrane component 1 are different binding sites derived from independent genes. EBioMedicine 2(11):1806–1813. doi:10.1016/j.ebiom.2015.10.017
Chwieralski CE, Welte T, Buhling F (2006) Cathepsin-regulated apoptosis. Apoptosis 11(2):143–149
Dehdashti F, Laforest R, Gao F, Shoghi KI, Aft RL, Nussenbaum B, Kreisel FH, Bartlett NL, Cashen A, Wagner-Johnston N, Mach RH (2013) Assessment of cellular proliferation in tumors by PET using 18F-ISO-1. J Nucl Med 54(3):350–357. doi:10.2967/jnumed.112.111948
Fontanilla D, Hajipour AR, Pal A, Chu UB, Arbabian M, Ruoho AE (2008) Probing the steroid binding domain-like I (SBDLI) of the sigma-1 receptor binding site using N-substituted photoaffinity labels. Biochemistry 47(27):7205–7217
Gebreselassie D, Bowen WD (2004) Sigma-2 receptors are specifically localized to lipid rafts in rat liver membranes. Eur J Pharmacol 493(1–3):19–28
Ghelardini C, Galeotti N, Bartolini A (2000) Pharmacological identification of SM-21, the novel sigma(2) antagonist. Pharmacol Biochem Behav 67(3):659–662
Hanner M, Moebius FF, Flandorfer A, Knaus HG, Striessnig J, Kempner E, Glossmann H (1996) Purification, molecular cloning, and expression of the mammalian sigma1-binding site. Proc Natl Acad Sci U S A 93(15):8072–8077
Hellewell SB, Bruce A, Feinstein G, Orringer J, Williams W, Bowen WD (1994) Rat liver and kidney contain high densities of sigma 1 and sigma 2 receptors: characterization by ligand binding and photoaffinity labeling. Eur J Pharmacol 268(1):9–18
Izzo NJ, Xu J, Zeng C, Kirk MJ, Mozzoni K, Silky C, Rehak C, Yurko R, Look G, Rishton G, Safferstein H, Cruchaga C, Goate A, Cahill MA, Arancio O, Mach RH, Craven R, Head E, LeVine H 3rd, Spires-Jones TL, Catalano SM (2014) Alzheimer’s therapeutics targeting amyloid beta 1-42 oligomers II: sigma-2/PGRMC1 receptors mediate Abeta 42 oligomer binding and synaptotoxicity. PLoS One 9(11):e111899. doi:10.1371/journal.pone.0111899
Jiang X, Wang X (2004) Cytochrome C-mediated apoptosis. Annu Rev Biochem 73:87–106
Klintman M, Dowsett M (2015) Early surrogate markers of treatment activity: where are we now? J Natl Cancer Inst Monogr 2015(51):24–28. doi:10.1093/jncimonographs/lgv002
Mach RH, Smith CR, Childers SR (1995) Ibogaine possesses a selective affinity for sigma 2 receptors. Life Sci 57(4):PL57–PL62
Mach RH, Smith CR, al-Nabulsi I, Whirrett BR, Childers SR, Wheeler KT (1997) Sigma 2 receptors as potential biomarkers of proliferation in breast cancer. Cancer Res 57(1):156–161
Mach RH, Wu L, West T, Whirrett BR, Childers SR (1999) The analgesic tropane analogue (+/-)-SM 21 has a high affinity for sigma2 receptors. Life Sci 64(10):PL131–PL137
Mach RH, Wheeler KT, Blair S, Yang B, Day CS, Blair JB, Choi SR, Kung HF (2001) Preparation of a technetium-99m SPECT agent for imaging the sigma-2 receptor status of solid tumors. J Label Compd Radiopharm 44(13):899–908
Mach RH, Huang Y, Freeman RA, Wu L, Vangveravong S, Luedtke RR (2004) Conformationally-flexible benzamide analogues as dopamine D3 and sigma-2 receptor ligands. Bioorg Med Chem Lett 14(1):195–202
Mankoff DA, Shields AF, Krohn KA (2005) PET imaging of cellular proliferation. Radiol Clin North Am 43(1):153–167
Marinovich ML, Macaskill P, Irwig L, Sardanelli F, von Minckwitz G, Mamounas E, Brennan M, Ciatto S, Houssami N (2013) Meta-analysis of agreement between MRI and pathologic breast tumour size after neoadjuvant chemotherapy. Br J Cancer 109(6):1528–1536. doi:10.1038/bjc.2013.473
Maurice T, Roman FJ, Privat A (1996) Modulation by neurosteroids of the in vivo (+)-[3H]SKF-10,047 binding to sigma 1 receptors in the mouse forebrain. J Neurosci Res 46(6):734–743
Maurice T, Junien JL, Privat A (1997) Dehydroepiandrosterone sulfate attenuates dizocilpine-induced learning impairment in mice via sigma 1-receptors. Behav Brain Res 83(1–2):159–164
Migliaccio I, Di Leo A, Malorni L (2014) Cyclin-dependent kinase 4/6 inhibitors in breast cancer therapy. Curr Opin Oncol 26(6):568–575. doi:10.1097/cco.0000000000000129
Moltzen EK, Perregaard J, Meier E (1995) Sigma ligands with subnanomolar affinity and preference for the sigma 2 binding site. 2. Spiro-joined benzofuran, isobenzofuran, and benzopyran piperidines. J Med Chem 38(11):2009–2017
Nguyen VH, Kassiou M, Johnston GA, Christie MJ (1996) Comparison of binding parameters of sigma 1 and sigma 2 binding sites in rat and guinea pig brain membranes: novel subtype-selective trishomocubanes. Eur J Pharmacol 311(2–3):233–240
Niso M, Riganti C, Pati ML, Ghigo D, Berardi F, Abate C (2015) Novel and selective fluorescent sigma2-receptor ligand with a 3,4-dihydroisoquinolin-1-one scaffold: a tool to study sigma2 receptors in living cells. Chembiochem 16(7):1078–1083. doi:10.1002/cbic.201402712
Ostenfeld MS, Fehrenbacher N, Hoyer-Hansen M, Thomsen C, Farkas T, Jaattela M (2005) Effective tumor cell death by sigma-2 receptor ligand siramesine involves lysosomal leakage and oxidative stress. Cancer Res 65(19):8975–8983
Ostenfeld MS, Hoyer-Hansen M, Bastholm L, Fehrenbacher N, Olsen OD, Groth-Pedersen L, Puustinen P, Kirkegaard-Sorensen T, Nylandsted J, Farkas T, Jaattela M (2008) Anti-cancer agent siramesine is a lysosomotropic detergent that induces cytoprotective autophagosome accumulation. Autophagy 4(4):487–499
Perregaard J, Moltzen EK, Meier E, Sanchez C (1995) Sigma ligands with subnanomolar affinity and preference for the sigma 2 binding site. 1. 3-(omega-aminoalkyl)-1H-indoles. J Med Chem 38(11):1998–2008
Romieu P, Martin-Fardon R, Bowen WD, Maurice T (2003) Sigma 1 receptor-related neuroactive steroids modulate cocaine-induced reward. J Neurosci 23(9):3572–3576
Rothman RB, Reid A, Mahboubi A, Kim CH, De Costa BR, Jacobson AE, Rice KC (1991) Labeling by [3H]1,3-di(2-tolyl)guanidine of two high affinity binding sites in guinea pig brain: evidence for allosteric regulation by calcium channel antagonists and pseudoallosteric modulation by sigma ligands. Mol Pharmacol 39(2):222–232
Seth P, Leibach FH, Ganapathy V (1997) Cloning and structural analysis of the cDNA and the gene encoding the murine type 1 sigma receptor. Biochem Biophys Res Commun 241(2):535–540
Shields AF, Grierson JR, Dohmen BM, Machulla HJ, Stayanoff JC, Lawhorn-Crews JM, Obradovich JE, Muzik O, Mangner TJ (1998) Imaging proliferation in vivo with [F-18]FLT and positron emission tomography. Nat Med 4(11):1334–1336. doi:10.1038/3337
Shoghi KI, Xu J, Su Y, He J, Rowland D, Yan Y, Garbow JR, Tu Z, Jones LA, Higashikubo R, Wheeler KT, Lubet RA, Mach RH, You M (2013) Quantitative receptor-based imaging of tumor proliferation with the sigma-2 ligand [(18)F]ISO-1. PLoS One 8(9):e74188. doi:10.1371/journal.pone.0074188
Simons K, Toomre D (2000) Lipid rafts and signal transduction. Nat Rev Mol Cell Biol 1(1):31–39
Soby KK, Mikkelsen JD, Meier E, Thomsen C (2002) Lu 28-179 labels a sigma(2)-site in rat and human brain. Neuropharmacology 43(1):95–100
Tsai SY, Hayashi T, Mori T, Su TP (2009) Sigma-1 receptor chaperones and diseases. Cent Nerv Syst Agents Med Chem 9(3):184–189
Tu Z, Dence CS, Ponde DE, Jones L, Wheeler KT, Welch MJ, Mach RH (2005) Carbon-11 labeled sigma2 receptor ligands for imaging breast cancer. Nucl Med Biol 32(5):423–430
Tu Z, Xu J, Jones LA, Li S, Dumstorff C, Vangveravong S, Chen DL, Wheeler KT, Welch MJ, Mach RH (2007) Fluorine-18-labeled benzamide analogues for imaging the sigma-2 receptor status of solid tumors with positron emission tomography. J Med Chem 50(14):3194–3204
Vangveravong S, Xu J, Zeng C, Mach RH (2006) Synthesis of N-substituted 9-azabicyclo[3.3.1]nonan-3alpha-yl carbamate analogs as sigma2 receptor ligands. Bioorg Med Chem 14(20):6988–6997
Vilner BJ, Bowen WD (2000) Modulation of cellular calcium by sigma-2 receptors: release from intracellular stores in human SK-N-SH neuroblastoma cells. J Pharmacol Exp Ther 292(3):900–911
Vilner BJ, John CS, Bowen WD (1995) Sigma-1 and sigma-2 receptors are expressed in a wide variety of human and rodent tumor cell lines. Cancer Res 55(2):408–413
Walker JM, Bowen WD, Walker FO, Matsumoto RR, De Costa B, Rice KC (1990) Sigma receptors: biology and function. Pharmacol Rev 42(4):355–402
Wallen CA, Higashikubo R, Dethlefsen LA (1984a) Murine mammary tumour cells in vitro. I. The development of a quiescent state. Cell Tissue Kinet 17(1):65–77
Wallen CA, Higashikubo R, Dethlefsen LA (1984b) Murine mammary tumour cells in vitro. II. Recruitment of quiescent cells. Cell Tissue Kinet 17(1):79–89
Wheeler KT, Wang LM, Wallen CA, Childers SR, Cline JM, Keng PC, Mach RH (2000) Sigma-2 receptors as a biomarker of proliferation in solid tumours. Br J Cancer 82(6):1223–1232
Xu J, Zeng C, Chu W, Pan F, Rothfuss JM, Zhang F, Tu Z, Zhou D, Zeng D, Vangveravong S, Johnston F, Spitzer D, Chang KC, Hotchkiss RS, Hawkins WG, Wheeler KT, Mach RH (2011) Identification of the PGRMC1 protein complex as the putative sigma-2 receptor binding site. Nat Commun 2:380. doi:10.1038/ncomms1386
Zeng C, Vangveravong S, Xu J, Chang KC, Hotchkiss RS, Wheeler KT, Shen D, Zhuang ZP, Kung HF, Mach RH (2007) Subcellular localization of sigma-2 receptors in breast cancer cells using two-photon and confocal microscopy. Cancer Res 67(14):6708–6716. doi:10.1158/0008-5472.CAN-06-3803
Zeng C, Vangveravong S, Jones LA, Hyrc K, Chang KC, Xu J, Rothfuss JM, Goldberg MP, Hotchkiss RS, Mach RH (2011) Characterization and evaluation of two novel fluorescent sigma-2 receptor ligands as proliferation probes. Mol Imaging 10(6):420–433
Zeng C, Garg N, Mach RH (2015) The PGRMC1 protein level correlates with the binding activity of a sigma-2 fluorescent probe (SW120) in rat brain cells. Mol Imaging Biol. doi:10.1007/s11307-015-0891-z
Acknowledgements
The authors would like to thank Dr. Hank Kung for providing the SPECT image in Fig. 8b. ESM was partially supported by Department of Energy training grant number: DE-FOA-0001075.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer International Publishing AG
About this chapter
Cite this chapter
Zeng, C., McDonald, E.S., Mach, R.H. (2016). Molecular Probes for Imaging the Sigma-2 Receptor: In Vitro and In Vivo Imaging Studies. In: Kim, F., Pasternak, G. (eds) Sigma Proteins: Evolution of the Concept of Sigma Receptors. Handbook of Experimental Pharmacology, vol 244. Springer, Cham. https://doi.org/10.1007/164_2016_96
Download citation
DOI: https://doi.org/10.1007/164_2016_96
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-65851-3
Online ISBN: 978-3-319-65853-7
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)