
Abstract
Introduction
While prediction of short versus long term survival from lung cancer is clinically relevant in the context of patient management and therapy selection, it has proven difficult to identify reliable biomarkers of survival. Metabolomic markers from tumor core biopsies have been shown to reflect cancer metabolic dysregulation and hold prognostic value.
Objectives
Implement and validate a novel ensemble machine learning approach to evaluate survival based on metabolomic biomarkers from tumor core biopsies.
Methods
Data were obtained from tumor core biopsies evaluated with high-resolution 2DLC-MS/MS. Unlike biofluid samples, analysis of tumor tissue is expected to accurately reflect the cancer metabolism and its impact on patient survival. A comprehensive suite of machine learning algorithms were trained as base learners and then combined into a stacked-ensemble meta-learner for predicting “short” versus “long” survival on an external validation cohort. An ensemble method of feature selection was employed to find a reliable set of biomarkers with potential clinical utility.
Results
Overall survival (OS) is predicted in external validation cohort with AUROCTEST of 0.881 with support vector machine meta learner model, while progression-free survival (PFS) is predicted with AUROCTEST of 0.833 with boosted logistic regression meta learner model, outperforming a nomogram using covariate data (staging, age, sex, treatment vs. non-treatment) as predictors. Increased relative abundance of guanine, choline, and creatine corresponded with shorter OS, while increased leucine and tryptophan corresponded with shorter PFS. In patients that expired, N6,N6,N6-Trimethyl-L-lysine, L-pyrogluatmic acid, and benzoic acid were increased while cystine, methionine sulfoxide and histamine were decreased. In patients with progression, itaconic acid, pyruvate, and malonic acid were increased.
Conclusion
This study demonstrates the feasibility of an ensemble machine learning approach to accurately predict patient survival from tumor core biopsy metabolomic data.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
The 5-year survival rate for lung cancer remains < 5% for all stages combined (Howlader et al. 2020, based on November 2020 SEER data submission, posted to the SEER web site, April 2021.), despite improvements in survival since the early 1990’s. Identification of patients likely to experience short or long survival has clinical utility by helping to minimize over- or under-treatment, potentially leading to improved clinical decision making. Staging relying on tumor (T) size, location of cancerous lymph nodes (N), and presence of metastasis (M) has been the gold standard for stratifying survival probability in the clinical setting (Lim et al., 2018), with mixed results. Linear methods such as Cox proportional hazards regression and logistic regression (e.g., nomograms) have seen routine use for diagnostic and prognostic applications (e.g., (Liang et al., 2015)). However, these linear methodologies may be unable to capture complex hidden features within high-dimensional data sets (Randall & Cable, 2016), leading to unreliable biomarker selection. In particular, nomograms applied to non-small cell lung cancer (NSCLC) and small cell lung cancer (SCLC) for predicting overall survival (OS) using clinical features such as pathologic stage, presence of lymph node metastasis and histology have yielded fair to poor accuracy, even with large sample sizes (Pan et al., 2017; Zhang et al., 2019).
More complex analyses such as machine learning have been employed to improve upon these linear methods, yielding a variety of results that have yet to find clinical application. A deep neural network (DNN) was combined with gene expression and clinical data in (Lai et al., 2020) to predict 5-year survival of NSCLC patients (AUC = 0.8163, accuracy = 75.44%). In (She et al., 2020) a deep learning model trained and externally validated with patient clinical data outperformed prediction of lung cancer–specific survival via Cox proportional hazard regression (C statistic = 0.739 vs. 0.716). A deep learning model using pretreatment and follow-up time series computed tomography (CT) images of NSCLC tumors was applied in (Xu et al., 2019) to predict survival. Another deep learning model was trained to extract prognostic information from pre-operative CT examinations in (Kim et al., 2020). In (Doppalapudi et al., 2021), deep learning models (artificial neural networks (ANN), recurrent neural networks (RNN), and convolutional neural networks (CNN)) were applied to the SEER database to predict lung cancer survival, obtaining 71.18% accuracy when survival periods were segmented into three classes (less than 6 months, 6–24 months, and over 24 months). An ensemble data mining approach was applied to the SEER database in (Agrawal et al., 2012) to predict survival for 6-month, 9-month, 1-year, 2-year and 5-years.
Part of the challenge in finding a reliable method to improve upon current clinical measures is the identification of a consistent set of molecular signatures as biomarkers for the prediction of survival (Riley et al., 2009). In this regard, metabolomics holds promise as a method for generating high-dimensional molecular data, from which a more accurate prognosis can be made (Collino et al., 2013). Although previous work has contributed to the understanding of lung cancer metabolism, the use of metabolomic data for survival prediction remains relatively unexplored (Bamji-Stocke et al., 2018). We have recently shown that tumor core biopsy-derived metabolomic data is capable of discriminating patients based on therapy outcome (Miller et al., 2021), where a support vector machine (SVM) trained and validated on features selected by partial least squares discriminant analysis (PLS-DA) performed best at predicting disease control vs. progressive disease groups (AUC = 0.970).
This study implements and validates a novel ensemble machine learning approach to predict overall survival (OS) and progression-free survival (PFS) in lung cancer patients by analyzing tumor core biopsy-derived metabolomic data. Unlike samples obtained from biofluids, analysis of the tumor tissue itself is expected to accurately reflect the cancer metabolism and its impact on patient survival. Our hypothesis is that an ensemble machine learning analysis of metabolomic data from lung tumor core biopsies is able to predict short vs. long survival in terms of OS and PFS. We test this hypothesis by implementing a comprehensive suite of machine learning algorithms that are trained as base learners and then combined into a stacked-ensemble meta-learner for predicting “short” vs. “long” survival on an external validation cohort. Further, an ensemble method of feature selection is employed to identify a reliable set of biomarkers with potential clinical utility.
2 Methods
2.1 Data pre-processing and statistical analysis
Metabolomic data from NSCLC patient tumor core biopsies were previously obtained from an ongoing study as described in (Miller et al., 2021), for which sample processing and metabolite extraction, 2DLC-MS/MS analysis and data pre-processing, and organization of MS peak intensity data were already performed. As reported previously, 2DLC-MS/MS data of unlabeled samples were first matched to an in-house database that contains parent ion m/z, MS/MS spectra, and retention time of authentic standards (MSI Level 1 identification). Data without a match (MSI Level 2 identification) were analyzed using Compound Discoverer software v2.0 (Thermo Fisher Scientific, Germany).
In the study in (Miller et al., 2021), informed consent was obtained to participate, and all specimens were collected following approved Internal Review Board protocols at University of Louisville Hospital (IRB 05.0523) and Norton Hospital (IRB 18.0264) from patients with known or suspected NSCLC. Demographics including sex, race, age, personal history of malignancy, smoking history, and relevant family history were recorded. Samples were collected by the clinical team, which was blinded to the data analysis. Only patients with confirmed primary histology of Non-small cell and histology subtypes of adenocarcinoma or squamous cell carcinoma were included. Data were split into internal and external validation sets while ensuring both data sets were balanced in terms of major clinical features such as age, gender, pathological staging, histology subtype, treatment versus non-treatment, status (alive/expired; progression/no progression) and survival outcome (short vs. long OS/PFS).
For this study, the MS data were log-transformed and imputed by probabilistic principal component analysis (PPCA) prior to analysis. Patients were categorized into “short” and “long” survival groups based on the mean OS and PFS across both data sets (29.21 months for OS; 21.11 months for PFS). A preliminary step to handle missing values by removing metabolic features with more than 50% was performed, resulting in a data set of approximately 25% missing values. Imputation was chosen over limit of detection (LOD) substitution as it was clear that some data was missing at random (MAR) and some was missing not at random (MNAR). Further details on the data pre-processing can be found in (Miller et al., 2022), in which we showed that the missingness of the data (categorized as missing or not missing) was not significantly associated with the events of death or progression in this patient cohort.. Significant differences in relative abundance were evaluated either by an unpaired T-test assuming equal variance or unpaired Wilcoxon rank-sum test, depending on normality of the data, and correlations between survival as a continuous outcome and metabolite levels were found via Pearson or Spearman method.
2.2 Machine learning methods (base learners)
Out of 44 total patients, 2/3 were kept for training as the internal validation set and the remaining 1/3 were reserved as the external validation set (Table 1). The data were randomly split using function createDataPartition from caret package in R, where short vs. long OS was the grouping factor. The internal validation set was used for parameter tuning with each base learner using all possible combinations of 10 values for each tuning parameter and Cohen’s kappa as the optimization metric for determining the optimal tuning parameter set for each individual model. Kappa represents classification accuracy for data with imbalanced classes and is a superior metric over accuracy alone. The approach used for estimating model performance within the internal validation set on unseen data was repeated k-fold cross-validation where k = 5 and 10 resampling iterations were performed for each base learner. The same tuning process was applied to meta learners after formation of each stacked ensemble model. The receiver operating characteristic curve (ROC) was plotted from the sensitivity and false positive rate (1-specificity). The Area under the Receiver Operating Characteristic curve (AUROC) was then calculated for model evaluation. An AUROC of 1.0 represents a perfect prediction, while an AUROC of 0.5 is equivalent to random chance showing no discriminatory power of the model.
2.3 Feature selection and variable importance
For each predictive model, the relative contribution of each feature was determined from the variable importance scores which were calculated from the varImp function in the caret package. The variable importance scores were used to rank key features for each model during feature selection. Some prediction models such as Random Forest, Neural Network (single-layer), and Nearest Shrunken Centroids have specific variable importance methods used while other prediction models such as k-Nearest Neighbors and Naïve Bayes use a generic ROC curve analysis method. Forward feature selection was employed after ranking features from each model by re-training the models on every feature subset incrementing by one predictor.
2.4 Ranking of most important predictors
An ensemble feature selection method was used to determine the top key features from the variable importance ranking and classification performance in each predictive model. For each outcome, the highest AUROC from each predictive model was weighted with an AUROC of 1 being 1 and an AUROC of 0.5 or less being 0. Similar to Shahrjooihaghighi et al. (Shahrjooihaghighi et al., 2017) which found that ensemble approaches of feature selection outperformed individual feature selection algorithms, we chose to maximize the stability (i.e., reliability) of biomarkers by finding the weighted sum of ranks by each variable importance feature selection method (a modified version of the Borda count), with weights assigned proportionally to the performance of each machine learning algorithm. All feature selection methods were weighted proportionally by their performance with an AUROC of 1 weighted as one and an AUROC of 0.5 or less weighted as zero. The ranked features were found by the weighted sum of ranks of all feature selection:
where \(R({f}_{i})\) is the overall sum of ranks of feature i, \({w}_{k}\) is the weight coefficient assigned to method k, \({r}_{i,k}\) is the rank of feature i by method k, and num is the total number of methods used to rank the features.
2.5 Ensemble machine learning method
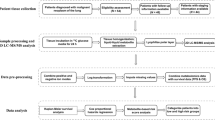
After individual predictive models were trained using cross-validation, an ensemble machine learning approach was employed to arrive at the optimal predictive model (Fig. 1). Ensemble methods are powerful machine learning tools which combine the predictions of several base learning models. By taking advantage of the unique strengths present in each base learner, ensemble methods typically achieve better prediction accuracy than any single base learner. Here, we employed a stacked generalization technique using the caretEnsemble package. First, a list of models with each base learner is produced, and the models are then stacked into a meta-model which uses a base learner as the aggregating method (i.e. meta learner). We filtered the base learners by choosing only those that had a maximum AUROC of 0.7 or higher in the internal validation set results during feature selection. Using the key predictors identified by feature selection, the stacked ensemble models were trained and validated using forward feature selection where variable importance was calculated as in Eq. 1. The same models, including base-learners, meta-learner, feature subset and hyper-parameters were used when evaluating performance on both internal and external validation sets to ensure no data leakage from the external validation set.

Diagram of machine learning workflow. Baser learners are trained on the internal validation set using fivefold cross validation with 10 resampling iterations on each feature subset. Feature selection is employed by base learner variable importance. After all base learners are trained and evaluated, a stacked ensemble model is evaluated after filtering base learners which did not achieve an AUROCTRAIN of 0.7 or greater across all feature subsets in the internal validation data. The ensemble model is then evaluated on all feature subsets using an ensemble method of feature selection (Eq. 1). The classification model performance of all base-learners and meta-learners is evaluated across the feature subsets on the external validation data. EVTREE = tree models from genetic algorithms. RF random forest, NNET neural network (single layer), MLP multi-layer perceptron, NSC nearest shrunken centroids, NB naïve Bayes, BGLM boosted general linear model, KNN k-nearest neighbors, SVM support vector machine, SPLS sparse partial least squares, BLR boosted logistic regression, RLR regularized logistic regression, NNFE neural network with feature extraction, WKNN weighted k-nearest neighbors, MANN model averaged neural network, RRF regularized random forest, BGAM boosted generalized additive model. ORFSVM oblique random forest with SVM as splitting model, SVMPoly support vector machine with polynomial kernel
2.6 Evaluation of covariates
A nomogram of patient covariate data (staging, sex, age, and treatment vs. non-treatment) was created to compare to the machine learning predictions of short versus long OS and PFS. For the nomogram, staging, age, sex and treatment versus non-treatment were the predictors, and an inverse logit transformation was used to transform the linear predictors to a probability (Zhang & Kattan, 2017).
2.7 Packages and libraries
All data analyses were conducted in the R programming language version 4.1.0. The packages caret and caretEnsemble were implemented for data splitting, model hyperparameter tuning, and creating stacked ensemble models using functions createDataPartition, train, caretList, and caretStack. Feature selection and EFS score calculation was implemented via a custom written R script. Packge rms was employed for creating the nomogram with functions lrm and nomogram.
2.8 Metabolic network visualization and quantitative enrichment analysis
Quantitative enrichment analysis (QEA) was performed on unlabeled metabolite dataset with (MetaboAnalyst 5.0, RRID:SCR_015539, www.metaboanalyst.ca/) (Chong et al., 2018). The log transformed/imputed working dataset was used, obviating missing value estimation or normalization. KEGG database (RRID:SCR_012773, www.kegg.jp/) was accessed June 2022. Metabolic networks were visualized with (Cytoscape 3.8.2, RRID:SCR_003032, cytoscape.org/) and (MetScape 3.1.3, RRID:SCR_014687, metscape.ncibi.org/) using imputed and log transformed metabolite intensity values for short vs. long survival classification.
3 Results
3.1 Patient population characteristics
Patient population characteristics are summarized in Table 1, showing the division between internal validation and external validation datasets. A total of 44 patients, all of which had pathological staging information, were considered for analysis; 30 were alive and 14 had expired by the end of the respective follow-up period. Pathological staging categorized as stage I/II vs. stage III/IV was evenly distributed among all patients. The majority of patients received some form of treatment, either as chemotherapy, immunotherapy, surgery, or some combination. In the internal validation cohort, mean OS was 31.6 months, and mean PFS was 21.8 months. Primary histology of all 31 internal validation patients was classified as non-small cell. Histology subtypes included 5 squamous cell carcinoma with the remaining 26 adenocarcinoma. In the external validation cohort, mean OS was 25.8 months, and mean PFS was 20.5 months. Primary histology of all 13 external validation patients was classified as non-small cell. Histology subtypes included 2 squamous cell carcinoma with the remaining 11 as adenocarcinoma. Further details on patient information can be found in Supplementary Table 1.
3.2 Classification results
Patients were stratified into “long” and “short” survival groups for classification by the mean OS and PFS across both data sets. The machine learning workflow is summarized in Fig. 1. A heatmap of the log transformed and imputed 2DLC-MS derived metabolite intensity values for short vs. long OS and PFS is shown in Supplementary Fig. 1 for the model internal validation (training) data and the external validation (test) data. PLS-DA score plots of short versus long overall and progression-free survivals based on the patient tissue-derived metabolomic data are in Supplementary Fig. 2. Long and short survival groups were separated but with some overlap for OS and PFS in the internal validation cohort. However, groups were well separated in the external validation cohort for both PFS and OS.
The maximum AUROC obtained from feature selection after internal cross-validation of all base learner models (Fig. 2) shows that the highest performance was obtained with NB (naïve Bayes) for OS (AUROCTRAIN = 0.822) and SVMPoly for PFS (AUROCTRAIN = 0.769). After filtering base learners with a max AUROC < 0.7, the AUROC with stacked ensemble meta learners (Fig. 2) increased for OS with NNFE yielding AUROCTRAIN = 0.958, while for PFS the AUROCTRAIN was 0.975 with BLR. Supplementary Fig. 3 further summarizes the AUROC obtained from all base learner models and stacked ensemble models during feature selection with predictions made on the external validation set.

Maximum AUROC obtained from feature selection after external test set validation of all base learner models and stacked ensemble meta learners for Overall Survival and Progression Free Survival. Patients were stratified into “long” and “short” survival groups for classification by the prediction models. Base learners which achieved a max AUROCTRAIN of 0.7 or above in the internal validation data (gray bars, top row) were selected for the stacked ensemble models (black bars, middle row). ROC curves of optimal stacked ensemble meta learners with repeated internal cross-validation (gray) and external validation (black) for prediction of “long” and “short” OS and PFS are shown for each case (bottom row)
ROC curves of optimal stacked ensemble meta learners with repeated internal cross-validation as well as external validation for prediction of “long” and “short” OS and PFS are further shown in Fig. 2. OS predictions achieved an AUROCTRAIN of 0.908 with SVM meta learner on the internal validation set and an AUROCTEST of 0.881 with SVM meta learner model on the external validation set, both utilizing the same feature subset of 18 metabolites. PFS predictions achieved an AUROCTRAIN of 0.924 with BLR meta learner on the internal validation set and an AUROC of 0.833TEST with BLR meta learner on the external validation set, both utilizing the same feature subset of 4 metabolites.
To compare to a linear prognostic model, a nomogram of covariate data (sex, age, staging) was implemented to predict short vs. long OS and PFS probability (Supplementary Fig. 4). The nomogram results (internal validation: OS AUROC = 0.749; PFS AUROC = 0.829; external validation: OS AUROC = 0.556; PFS AUROC = 0.738) were outperformed by the metabolomic-based ensemble machine learning approach (Fig. 2).
3.3 Key metabolites identified by variable importance
Top 25 key metabolic biomarkers identified by ensemble feature selection (EFS) are in Table 2. The EFS score was calculated as a linear weighted sum of ranks of features identified by all base learners included in each meta model (Eq. 1). The top 18 metabolites were used in the optimal stacked ensemble model for predicting OS (AUROCTEST = 0.881), while the top 4 metabolites were used for predicting PFS (AUROCTEST = 0.833). Correlations were found via Pearson or Spearman method, depending on normality of the data, between survival as a continuous outcome and metabolite level. Among the features included in the optimal meta model, those correlating with improved OS included urocanic acid, cysteine, levulinic acid, pyruvate, itaconic acid, and histamine, while glutamine correlated with improved PFS. In contrast, features correlating with decreased OS included guanine, creatine, leucine, adenosine, choline, acetyl-L-carnitine, malate, panthothenic acid, xanthine, uridine, hypoxanthine, and tryptophan, while leucine, tryptophan, and adipic acid correlated with decreased PFS.
Figure 3 shows metabolites with a significant (p \(\le \) 0.05) or marginally significant (0.05 \(\le \) p \(\le \) 0.1) effect of group in terms of relative abundance (T-test or Wilcoxon rank-sum test) for short vs. long survival groups. Guanine, choline, and creatine were significantly increased for patients with shorter OS, while leucine and tryptophan were significantly increased for patients with shorter PFS (p \(\le \) 0.05). Additionally, leucine, acetyl-L-carnitine, adenosine, and malate trended higher for patients with shorter OS, while glutamine trended lower for patients with shorter PFS (p \(\le \) 0.1). In comparison, relative abundance analysis for event vs. non-event groups is shown in Supplementary Fig. 5. In patients that expired, N6,N6,N6-Trimethyl-L-lysine, L-pyrogluatmic acid, and benzoic acid were increased while cystine, methionine sulfoxide and histamine were decreased (p \(\le \) 0.05). Creatine, creatinine, and malate also trended higher in these patients (p \(\le \) 0.1). In patients with progression, itaconic acid, pyruvate, and malonic acid were increased (p \(\le \) 0.05) while leucine and lysine trended lower (p \(\le \) 0.1).

Relative abundance of metabolites identified as significant for “short” versus “long” OS and PFS by unpaired T-test assuming equal variance or Wilcoxon rank sum test, depending on normality of the data. Each box represents 1st and 3rd quartiles. Bands within represent the median and x is the mean. Ends of whiskers are maximum and minimum, with points outside being outliers. “Long” survival groups are in green and “Short” is in yellow (*p ≤ 0.1, **p ≤ 0.05). Color figure online
3.4 Metabolic pathway enrichment and network analysis
Top metabolic pathways identified in MetaboAnalyst 5.0 by the chosen databases are shown in Fig. 4 and listed with associated statistics in Supplementary Table 2. Enrichment ratio was determined by relative abundance differences between “short” and “long” survival groups. Significant (p \(\le \) 0.05) metabolic pathways identified by KEGG database by p-value were the following. For OS: glycine, serine and threonine metabolism; for PFS: valine, leucine and isoleucine degradation, valine, leucine and isoleucine biosynthesis, and tryptophan metabolism. Visualization of integrated metabolic networks of metabolites with significant or marginally significant differences in relative abundance for short vs. long survival groups with OS (guanine, choline, creatine, leucine, acetyl-L-carnitine, adenosine, malate) or PFS (leucine, tryptophan, glutamine) is in Supplementary Fig. 6.

Quantitative enrichment analysis. Enriched metabolic pathways were found with MetaboAnalyst 5.0 using KEGG pathway database for OS and PFS (Color figure online). KEGG database was accessed June 2022
4 Discussion
This study examined the hypothesis that an ensemble machine learning analysis of metabolomic data from lung tumor core biopsies is able to predict short vs. long survival in terms of overall survival (OS) and progression-free survival (PFS). The study adhered to REMARK criteria (McShane et al., 2005) to ensure the integrity of the modeling approach. An ensemble method of feature selection was employed to identify key metabolites associated with survival and that proved able to differentiate between short and long term survivals. A rigorous combination of feature selection and parameter tuning during training and validation of all base learner models and stacked ensemble meta-learners was performed to minimize model overfitting (Fig. 1). The same models, including base-learners, meta-learner, feature subset and hyper-parameters were used when evaluating performance on both internal and external validation sets to ensure no leakage from the external data set. OS predictions achieved AUROCTRAIN of 0.908 with SVM meta learner model on the internal validation set and AUROCTEST of 0.881 with SVM meta learner model on the external validation set (Fig. 2), where the top 18 metabolites were selected as the feature subset for training and validation (Table 2). PFS predictions achieved AUROCTRAIN of 0.924 with SVM meta-learner model on the internal validation set and AUROCTEST of 0.833 with BLR meta learner model on the external validation set (Fig. 2), where the top 4 metabolites were selected as the feature subset for training and validation (Table 2). Interestingly, the meta-learners EVTREE, NSC, KNN, and KKNN consistently underperformed for predicting OS and PFS (Fig. 2). The meta-learners outperformed individual base learners for the maximum performance achieved across all feature subsets in the external validation set (Supplementary Fig. 3), at the cost of increased variance and slightly decreased average performance. Overall, high prediction accuracy was achieved during both internal cross-validation and external test set validation using SVM and BLR meta-learners for OS and PFS, respectively, demonstrating that tumor core biopsy derived metabolomic data is useful for survival prognosis even with a small sample size. For comparison to these results we evaluated the performance of a nomogram, a more traditional method commonly applied in clinical prognosis, using covariate clinical data as predictors (Supplementary Fig. 4). The nomogram significantly underperformed the metabolomic-based ensemble machine learning approach.
Guanine, choline and creatine were identified as significant between long and short OS groups (Fig. 3) and were all included in the top 7 metabolites selected by the EFS score. Increased relative abundances of these metabolites were associated with shorter OS. Guanine can arise from guanosine through the action of phosphate alpha-D-ribosyltransferase as part of the general purine metabolism pathway (reaction R02147 (KEGG)) (Supplementary Fig. 6). Guanine and guanosine are integral components of various cellular proteins involved in cancer progression (Wang et al., 2021), and abundance of guanosine nucleotides has been observed in cancer (Huang et al., 2021). Abnormal choline cellular metabolism leading to increased levels of choline-containing precursors is considered a hallmark of oncogenesis and tumor progression (Glunde et al., 2011). Malate, which is involved in both the TCA cycle and glycolysis/gluconeogenesis (Supplementary Fig. 6), was marginally increased in patients with shorter OS (Fig. 3). Urinary malate dehydrogenase (MDH) has been discovered as a biomarker able to differentiate lung cancer patients from control subjects and increases with worsening disease stage (Ma et al., 2021). The same study also found increased expression of MDH in lung cancer tissue compared to normal lung tissue in a small patient cohort. It is difficult to link malate levels to MDH activity since malate is also involved in glycolysis and gluconeogenesis towards the production of pyruvate, although it is possible that increased malate seen in patients with short OS could be partially due to MDH expression. Further study is needed to investigate the relationship between the metabolome and proteome in lung cancer patients.
Higher relative abundance of leucine, acetyl-L-carnitine, adenosine and malate were marginally associated with short OS (Fig. 3). These metabolites were also within the top 7 ranked metabolites selected by EFS score (Table 2). Leucine is a branched-chain amino acid (BCAA); along with isoleucine and valine; these essential amino acids are needed by tumors for protein synthesis and oxidation for energy purposes (Ananieva & Wilkinson, 2018). Adenosine is known to accumulate in tumors, promoting angiogenesis and tumor growth and dampening the immune response (Spychala, 2000). Adenosine is also involved with production of adenine through the action of phosphate alpha-D-ribosyltransferase (reaction R01561 (KEGG)) (Supplementary Fig. 6). Leucine and tryptophan were selected within the top 3 most important metabolites for predicting short vs. long PFS by EFS score (Table 2), and increased relative abundance was associated with shorter PFS (Fig. 3). Significant metabolic pathways associated with PFS include valine, leucine, and isoleucine degradation/biosynthesis, and tryptophan metabolism (Supplementary Table 2). Tryptophan, an essential amino acid, has been recognized as an important compound in lung cancer (Li & Zhao, 2021) and is known to be involved with immune system suppression (Mellor & Munn, 2004; Munn et al., 2005; Pilotte et al., 2012). Although decreased tryptophan in blood circulation is characteristic of lung cancer compared to healthy controls, it has not been able to differentiate between early and advanced stage of lung cancer (Ren et al., 2011). Interestingly, our study shows that increased tryptophan levels were associated with shorter PFS (Fig. 3).Tryptophan, along with other essential amino acids, is obtained exclusively from the diet, which may represent a confounding variable when measuring its abundance. In contrast, decreased relative abundance of glutamine was marginally significantly associated with shorter PFS (Fig. 3). Glutamine is a major carbon, nitrogen, and energy substrate in tumors (Jiang et al., 2019), known to be uptaken at higher rates by cancer cells (Gottfried et al., 2012); lower abundance may indicate enhanced tumor cell proliferative activity along with immune system suppression (Sikalidis, 2015), both of which can lead to more aggressive cancer.
In addition to analyzing metabolites between short vs. long survival groups, we report relative abundance of significant metabolites between event versus non-event groups (Supplementary Fig. 5). Interestingly, increased levels of several metabolites which were associated with short survival (guanine, choline, leucine, acetyl-L-carnitine, adenosine) (Fig. 3A) or short progression (tryptophan, glutamine) (Fig. 3B) were not associated with events (OS or PFS) (Supplementary Fig. 5). Leucine, which was increased for long PFS (Fig. 3), was marginally significantly associated with progression at lower levels (Supplementary Fig. 5). Discrepancies like these may occur because events and length of time to events or censorship are not necessarily correlated. These results confirm the value of analyzing survival time length in addition to event occurrence, separately. Glycine, serine and threonine metabolism was the top and only significant metabolic pathway for OS (Supplementary Table 2). Although glycine was not detected in our patient samples, metabolite hits relevant to this pathway include choline, creatine (Supplementary Fig. 6) and pyruvate. Glycine is associated with tumorigenesis and is an integral component of glutathione, and, therefore, required for cellular redox balance (Amelio et al., 2014; Jain et al., 2012). Glycine uptake and catabolism is known to promote rapid cancer cell proliferation (Jain et al., 2012).
The results of this study demonstrate that short vs. long term survival of NSCLC patients can be predicted in an external validation cohort using an ensemble machine learning approach that analyzes metabolomic data extracted from patient tumor core biopsies, with the ultimate goal to help clinical decision-making. The approach resulted in higher accuracy predictions than traditional clinical methods such as nomograms based on patient covariate data (staging, age, and sex). A small sample size and potential confounding factors (differences in biopsy collection date relative to date of diagnosis, smoking status, and type and duration of treatment) are potential limitations. Nevertheless, previous work has highlighted the predictive capability of metabolomic data in spite of such potential confounding factors, showing that adjusting for smoking status, cancer stage and age at diagnosis, and time between sample collection and diagnosis had minimal significant effect on metabolite hazard ratios associated with prostate cancer-specific mortality (Huang et al., 2019). The results imply some degree of overfitting due to slightly decreased classification performance on the external validation set (Fig. 2). However, adequate predictive performance of the proposed ensemble machine learning approach to analyze tumor core biopsy metabolomic data is shown with AUROC > 0.9 on internal validation data and AUROC > 0.8 on external validation data. Future work will evaluate the proposed approach with larger sample sizes and in the context of prospective evaluation, including prediction of event vs. censorship (death or progression).
Data availability
Datasets used are in (Metabolomics Workbench (MetWB), RRID:SCR_013794, www.metabolomicsworkbench.org) (Study ST001527).
Abbreviations
- BGAM:
-
Boosted generalized additive model
- BGLM:
-
Boosted general linear model
- BLR:
-
Boosted logistic regression
- EVTREE:
-
Tree models from genetic algorithms
- KNN:
-
K-nearest neighbors
- MANN:
-
Model averaged neural network
- MLP:
-
Multi-layer perceptron
- NB:
-
Naïve Bayes
- NNET:
-
Neural network (single layer)
- NNFE:
-
Neural network with feature extraction
- NSC:
-
Nearest shrunken centroids
- ORFSVM:
-
Oblique random forest with SVM as splitting model
- RF:
-
Random forest
- RLR:
-
Regularized logistic regression
- RRF:
-
Regularized random forest
- SPLS:
-
Sparse partial least squares
- SVM:
-
Support vector machines
- SVMPoly:
-
Support vector machine with polynomial kernel
- WKNN:
-
Weighted k-nearest neighbors
References
Agrawal, A., Misra, S., Narayanan, R., Polepeddi, L., & Choudhary, A. (2012). Lung cancer survival prediction using ensemble data mining on seer data. Scientific Programming, 20, 920245.
Amelio, I., Cutruzzola, F., Antonov, A., Agostini, M., & Melino, G. (2014). Serine and glycine metabolism in cancer. Trends in Biochemical Sciences, 39, 191–198.
Ananieva, E. A., & Wilkinson, A. C. (2018). Branched-chain amino acid metabolism in cancer. Current Opinion in Clinical Nutrition and Metabolic Care, 21, 64–70.
Bamji-Stocke, S., van Berkel, V., Miller, D. M., & Frieboes, H. B. (2018). A review of metabolism-associated biomarkers in lung cancer diagnosis and treatment. Metabolomics, 14, 81.
Chong, J., Soufan, O., Li, C., Caraus, I., Li, S., Bourque, G., Wishart, D. S., & Xia, J. (2018). MetaboAnalyst 4.0: Towards more transparent and integrative metabolomics analysis. Nucleic Acids Research, 46, W486–W494.
Collino, S., Martin, F. P., & Rezzi, S. (2013). Clinical metabolomics paves the way towards future healthcare strategies. British Journal of Clinical Pharmacology, 75, 619–629.
Doppalapudi, S., Qiu, R. G., & Badr, Y. (2021). Lung cancer survival period prediction and understanding: Deep learning approaches. International Journal of Medical Informatics, 148, 104371.
Glunde, K., Bhujwalla, Z. M., & Ronen, S. M. (2011). Choline metabolism in malignant transformation. Nature Reviews Cancer, 11, 835–848.
Gottfried, E., Kreutz, M., & Mackensen, A. (2012). Tumor metabolism as modulator of immune response and tumor progression. Seminars in Cancer Biology, 22, 335–341.
Howlader NNA, Krapcho M, Miller D, Brest A, Yu M, Ruhl J, Tatalovich Z, Mariotto A, Lewis DR, Chen HS, Feuer EJ, Cronin KA (EDS). based on November 2020 SEER data submission, posted to the SEER web site, April 2021. SEER Cancer Statistics Review, 1975–2018, National Cancer Institute, Bethesda
Huang, F., Huffman, K. E., Wang, Z., Wang, X., Li, K., Cai, F., Yang, C., Cai, L., Shih, T. S., Zacharias, L. G., Chung, A., Yang, Q., Chalishazar, M. D., Ireland, A. S., Stewart, C. A., Cargill, K., Girard, L., Liu, Y., Ni, M., … Deberardinis, R. J. (2021). Guanosine triphosphate links MYC-dependent metabolic and ribosome programs in small-cell lung cancer. Journal of Clinical Investigation, 79, 1629.
Huang, J., Weinstein, S. J., Moore, S. C., Derkach, A., Hua, X., Mondul, A. M., Sampson, J. N., & Albanes, D. (2019). Pre-diagnostic serum metabolomic profiling of prostate cancer survival. Journals of Gerontology. Series a, Biological Sciences and Medical Sciences, 74, 853–859.
Jain, M., Nilsson, R., Sharma, S., Madhusudhan, N., Kitami, T., Souza, A. L., Kafri, R., Kirschner, M. W., Clish, C. B., & Mootha, V. K. (2012). Metabolite profiling identifies a key role for glycine in rapid cancer cell proliferation. Science, 336, 1040–1044.
Jiang, J., Srivastava, S., & Zhang, J. (2019). Starve cancer cells of glutamine: Break the spell or make a hungry monster? Cancers (basel), 11, 804.
Kim, H., Goo, J. M., Lee, K. H., Kim, Y. T., & Park, C. M. (2020). Preoperative CT-based deep learning model for predicting disease-free survival in patients with lung adenocarcinomas. Radiology, 296, 216–224.
Lai, Y. H., Chen, W. N., Hsu, T. C., Lin, C., Tsao, Y., & Wu, S. (2020). Overall survival prediction of non-small cell lung cancer by integrating microarray and clinical data with deep learning. Science and Reports, 10, 4679.
Li, C., & Zhao, H. (2021). Tryptophan and its metabolites in lung cancer: basic functions and clinical significance. Frontiers in Oncology, 11, 707277.
Liang, W., Zhang, L., Jiang, G., Wang, Q., Liu, L., Liu, D., Wang, Z., Zhu, Z., Deng, Q., Xiong, X., Shao, W., Shi, X., & He, J. (2015). Development and validation of a nomogram for predicting survival in patients with resected non-small-cell lung cancer. Journal of Clinical Oncology, 33, 861–869.
Lim, W., Ridge, C. A., Nicholson, A. G., & Mirsadraee, S. (2018). The 8(th) lung cancer TNM classification and clinical staging system: Review of the changes and clinical implications. Quantitative Imaging in Medicine and Surgery, 8, 709–718.
Ma, Y. C., Tian, P. F., Chen, Z. P., Yue, D. S., Liu, C. C., Li, C. G., Chen, C., Zhang, H., Liu, H. L., Zhang, Z. F., Chen, L., Zhang, B., & Wang, C. L. (2021). Urinary malate dehydrogenase 2 is a new biomarker for early detection of non-small-cell lung cancer. Cancer Science, 112, 2349–2360.
McShane, L. M., Altman, D. G., Sauerbrei, W., Taube, S. E., Gion, M., Clark, G. M., Statistics Subcommittee of the, N. C. I. E. W. G. O. C. D. (2005). REporting recommendations for tumor MARKer prognostic studies (REMARK). Nature Clinical Practice. Urology, 2(416), 422.
Mellor, A. L., & Munn, D. H. (2004). IDO expression by dendritic cells: Tolerance and tryptophan catabolism. Nature Reviews Immunology, 4, 762–774.
Miller, H. A., Rai, S. N., Yin, X., Zhang, X., Chesney, J. A., van Berkel, V. H., & Frieboes, H. B. (2022). Lung cancer metabolomic data from tumor core biopsies enables risk-score calculation for progression-free and overall survival. Metabolomics, 18, 31.
Miller, H. A., Yin, X., Smith, S. A., Hu, X., Zhang, X., Yan, J., Miller, D. M., van Berkel, V. H., & Frieboes, H. B. (2021). Evaluation of disease staging and chemotherapeutic response in non-small cell lung cancer from patient tumor-derived metabolomic data. Lung Cancer, 156, 20–30.
Munn, D. H., Sharma, M. D., Baban, B., Harding, H. P., Zhang, Y., Ron, D., & Mellor, A. L. (2005). GCN2 kinase in T cells mediates proliferative arrest and anergy induction in response to indoleamine 2,3-dioxygenase. Immunity, 22, 633–642.
Pan, H., Shi, X., Xiao, D., He, J., Zhang, Y., Liang, W., Zhao, Z., Guo, Z., Zou, X., Zhang, J., & He, J. (2017). Nomogram prediction for the survival of the patients with small cell lung cancer. Journal of Thoracic Disease, 9, 507–518.
Pilotte, L., Larrieu, P., Stroobant, V., Colau, D., Dolusic, E., Frederick, R., de Plaen, E., Uyttenhove, C., Wouters, J., Masereel, B., & van den Eynde, B. J. (2012). Reversal of tumoral immune resistance by inhibition of tryptophan 2,3-dioxygenase. Proc Natl Acad Sci U S A, 109, 2497–2502.
Randall, R. L., & Cable, M. G. (2016). Nominal nomograms and marginal margins: What is the law of the line? The Lancet Oncology, 17, 554–556.
Ren, Y. P., Tang, A. G., Zhou, Q. X., & Xiang, Z. Y. (2011). Clinical significance of simultaneous determination of serum tryptophan and tyrosine in patients with lung cancer. Journal of Clinical Laboratory Analysis, 25, 246–250.
Riley, R. D., Sauerbrei, W., & Altman, D. G. (2009). Prognostic markers in cancer: The evolution of evidence from single studies to meta-analysis, and beyond. British Journal of Cancer, 100, 1219–1229.
Shahrjooihaghighi, A., Frigui, H., Zhang, X., Wei, X., Shi, B., & Trabelsi, A. (2017). An ensemble feature selection method for biomarker discovery. Proc IEEE Int Symp Signal Proc Inf Tech, 2017, 416–421.
She, Y., Jin, Z., Wu, J., Deng, J., Zhang, L., Su, H., Jiang, G., Liu, H., Xie, D., Cao, N., Ren, Y., & Chen, C. (2020). Development and validation of a deep learning model for non-small cell lung cancer survival. JAMA Network Open, 3, e205842.
Sikalidis, A. K. (2015). Amino acids and immune response: A role for cysteine, glutamine, phenylalanine, tryptophan and arginine in T-cell function and cancer? Pathology Oncology Research, 21, 9–17.
Spychala, J. (2000). Tumor-promoting functions of adenosine. Pharmacology & Therapeutics, 87, 161–173.
Wang, J., Wu, Y., Li, Y., Wang, Y., Shen, F., Zhou, J., & Chen, Y. (2021). Guanosine monophosphate synthase upregulation mediates cervical cancer progression by inhibiting the apoptosis of cervical cancer cells via the Stat3/P53 pathway. International Journal of Oncology. https://doi.org/10.3892/ijo.2021.5183
Xu, Y., Hosny, A., Zeleznik, R., Parmar, C., Coroller, T., Franco, I., Mak, R. H., & Aerts, H. (2019). Deep learning predicts lung cancer treatment response from serial medical imaging. Clinical Cancer Research, 25, 3266–3275.
Zhang, J., Fan, J., Yin, R., Geng, L., Zhu, M., Shen, W., Wang, Y., Cheng, Y., Li, Z., Dai, J., Jin, G., Hu, Z., Ma, H., Xu, L., & Shen, H. (2019). A nomogram to predict overall survival of patients with early stage non-small cell lung cancer. Journal of Thoracic Disease, 11, 5407–5416.
Zhang, Z., & Kattan, M. W. (2017). Drawing nomograms with R: Applications to categorical outcome and survival data. Ann Transl Med, 5, 211.
Funding
VvB and HBF acknowledge partial support by National Cancer Institute grant R15CA203605 (Frieboes).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of interest
The authors declare that they have no competing interests.
Informed consent
Not applicable.
Research involving human participants and/or animals
This study did not involve human participants or animals.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Miller, H.A., van Berkel, V.H. & Frieboes, H.B. Lung cancer survival prediction and biomarker identification with an ensemble machine learning analysis of tumor core biopsy metabolomic data. Metabolomics 18, 57 (2022). https://doi.org/10.1007/s11306-022-01918-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11306-022-01918-3