
Abstract
Introduction
Chromatography with mass spectrometry (MS) is a technique of choice for metabolomic analysis of plant extracts. Single dimension gas chromatography (1DGC) with MS leads to poorly resolved metabolites of complex Eucalyptus spp. leaf oil secondary metabolites and consequently limited metabolic coverage of secondary compounds. Multidimensional chromatography with high resolution MS can contribute to advances in this field.
Objectives
Deeper insight into metabolite composition and variation for Eucalyptus spp. leaf oils through systematic untargeted metabolic profiling using comprehensive two-dimensional GC (GC × GC) with high resolution time-of-flight MS (accTOFMS), using generalised processes for metabolite identification.
Methods
GC × GC separation used cryogenic modulation, with standard length polar first dimension and short fast analysis non-polar 2D columns. Compound tentative identification incorporated 1D and 2D retention information, retention indices, mass spectrum matching, and accurate mass MS data. Global metabolic profiles were interpreted through 2D contour plots and chemometric analysis.
Results
Strategies for metabolite screening and identification using GC × GC-accTOFMS were proposed. Considerably more components are detected and recognised than for 1DGC. Structured 2D molecular composition chromatographic patterns aid identification. ca. 400 metabolites were detected, 183 compounds were identified or tentatively identified, representing between 50.8–90.0% of the total ion count, comprising various chemical families. PCA revealed discriminating metabolites, allowing chemotaxonomic classification of species.
Conclusion
Expansion of metabolic coverage by using GC × GC-accTOFMS, and detailed 2D metabolic fingerprints of E. polybractea, E. citriodora, E. radiata and E. globulus leaf oils were established. This high resolution analytical platform, and identification strategy can be adapted to metabolic analysis of other plant extracts.
Graphical abstract
Phytoconstituents of four Australian eucalypt leaf oils were profiled using high resolution GC × GC-accurate mass TOFMS. Two-dimensional plots illustrated significant expansion of metabolic coverage. PCA discriminated metabolites of the eucalypts.

Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
The genus Eucalyptus, an Australian native, comprising more than 700 species belonging to the Myrtaceae family, is one of the world’s most important and widely cultivated hardwood trees (Cossalter and Pye-Smith 2003; Hantao et al. 2013). The aromatic volatile oil steam-distilled from its foliage [referred to as Eucalyptus oil (EO)], is among the world’s top traded essential oils in terms of volume (Batish et al. 2008). The study of EO has attracted much attention; this penetrating oil reportedly exhibits anti-microbial, anti-bacterial, anti-septic, fungicidal and nematicidal activities (Pandey et al. 2000; Ramezani et al. 2002; Sartorelli et al. 2007; Cermelli et al. 2008; Mulyaningsih et al. 2010; Tyagi and Malik 2011). EO is comprised of two major types, the cineole-rich/medicinal-type and citronellal-rich/lemon-scented-type (the latter produced mainly by E. citriodora). The cineole-type EO is primarily used in medicinal/pharmaceutical industries, while the lemon scented oil is mainly for perfumery, cosmetics and functional food (flavouring agent) purposes (Goodger et al. 2007; Singh et al. 2012).
A recent upsurge in interest arises from the discovery of potent insecticidal and herbicidal/allelopathic activity of the volatile oil which may be derived from its foliage, suggesting future alternative bio-insecticides, crop protectants or bio-herbicides. Numerous studies have demonstrated that EO retards growth of vegetation and provides defence against phytopathogenic fungi, herbivors and harmful insects (Batish et al. 2008; Verdeguer et al. 2009; Zhang and Fu 2009). The extent of effects obtained from different species of eucalypt differ markedly, attributed to variability in metabolite composition. Different degrees of allelopathic potential of eucalypt have been highlighted; even though some Eucalyptus species (E. radiata; E. globulus) have 1,8-cineole as the major component, they display noticeable differences in activities (May and Ash 1990; Kohli et al. 1998). These discoveries prompted investigations to correlate these observations with metabolites present in EO. Trace components apparently play a critical role in mediating these activities, possibly by synergistic effects with other components (Hummelbrunner and Isman 2001; Nerio et al. 2010). Detailed profiling using advanced comprehensive approaches, expanding the metabolic coverage, would be valuable in identifying the full suite of metabolites, and aid exploration of possible synergistic relationships between chemical components (including minor constituents) and bio-activity.
Extensive chemical diversity associated with high variability in intrinsic physicochemical properties of plant secondary products, make separation, detection and identification of phytochemicals challenging (Wolfender et al. 2015). Whilst gas chromatography (GC) combined with mass spectrometry (MS) is pre-eminent for global profiling of plant secondary metabolites, the inability of GC–MS to distinguish between components of similar structure and/or mass spectra is a major limitation, especially where GC resolution is a limiting factor. Given the functional importance of isomers in biology, this is an important constraint (Fernie et al. 2004; Rathahao-Pari et al. 2016). Targeted analysis using specific capabilities in MS (triple quadrupole analysis with selected reaction monitoring, or multiple reaction monitoring) may elucidate known analytes of interest in the presence of complex matrix interferences, such that improved GC separation is less critical. However, this is not suited to untargeted profiling of plant metabolites, since they invariably focus on known target compounds. Therefore, utilisation of multiple separation dimensions is important for comprehensive analysis of a complex plant metabolome. Comprehensive two-dimensional GC (GC × GC), offering high resolution and increased peak capacity has considerable potential as a tool for metabolomics (Dettmer et al. 2013; Chin and Marriott 2014; Wong et al. 2014). Semi-automated data processing for metabolomics applications have been proposed (Koek et al. 2011). More usual, is the use of GC–MS as a part of the strategy of metabolite identification using multiple instrumental techniques (Lopez-Sanchez et al. 2015).
Various reports describe chemotyping of EO of dissimilar species using GC–MS (Boland et al. 1991; Bendaoud et al. 2009; Cheng et al. 2009), though previous work did not address the complexity of metabolites in the oils, nor limitations of 1DGC as a result of limited peak capacity of the 1D separation, particularly where low abundance components suffer overlap with major components. As research shifts toward comprehensive “-omics” approaches for measurement of plant metabolomes, deep insights of biochemical regulation within plants, improved detection, and global compound identification is critical. Here, GC × GC with high resolution quadrupole time-of-flight MS (GC × GC-accTOFMS) is described for untargeted metabolic profiling of leaf oils of E. polybractea, E. citriodora, E. radiata, and E. globulus. Metabolite screening strategies and identification are proposed, based on a detailed study regarding metabolite diversity among eucalypt generated in a single comprehensive high resolution analytical approach. This study provides valuable information for studies of biological variation of secondary metabolites in eucalypts, their associated bioactive activities (allelopathic, insecticidal, etc.) and factors that trigger metabolite formation. Although GC × GC fully resolves many more compounds than 1DGC, its ability to provide a commensurate increase in metabolite coverage was of interest.
2 Materials and methods
2.1 Materials
2.1.1 Chemicals and reagents
Myrcene, α-pinene, β-pinene, carvone, methyl eugenol, 1,8-cineole, 1-octen-3-ol, cuminaldehyde, terpinolene, linalool oxide, p-cymene, camphor, car-3-ene, bornyl acetate, sabinene, cis-ocimene, α-terpinyl acetate, γ-terpinene, menthone, cinnamyl acetate, p-anisaldehyde, fenchone and estragole were provided by Australian Botanical Products (Hallam, Australia). Limonene, terpinen-4-ol, α-terpineol, linalyl acetate and linalool standards were provided by FGB Natural Products Pty Ltd (Oakleigh South, Australia). HPLC grade dichloromethane and hexane were purchased from Merck (Darmstadt, Germany). A series of n-alkanes (C7–C30) was purchased from Sigma–Aldrich (St. Louis, MO).
2.1.2 Eucalyptus leaf oil samples
All leaf oil samples were sampled from four plantation areas: the Inglewood region of Australia (E. polybractea), South Africa (E. radiata), and Yunnan (E. citriodora and E. globulus). They were provided by FGB Natural Products Pty Ltd. All collected oil samples were steam distilled from the foliage of the plant. Yields ranged from ca. 0.9 to 1.1% of wet material weight. Samples were stored refrigerated (4 °C), and were diluted in dichloromethane (1.0, 0.5 and 0.1% v/v) prior to injecting into the GC.
2.2 Methods
2.2.1 GC × GC-accQTOFMS system
Separations were conducted on an Agilent 7890 A GC coupled with a 7200 series quadrupole time-of-flight mass spectrometer (QTOFMS; Agilent Technologies, Mulgrave, Australia), retrofitted with an Everest model longitudinally modulated cryogenic system (LMCS, Chromatography Concepts Ltd, Doncaster, Australia). Chromatographic separation was performed using a first dimension (1D) SUPELCOWAX®10 column of dimension 30 m × 0.25 mm I.D. × 0.25 µm film thickness (d f); (Supelco, Bellefonte, PA), with a Rxi®-5Sil MS second dimension (2D) column (1 m × 0.1 mm I.D. × 0.1 μm d f ; Restek Corp, Bellefonte, PA) connected by a deactivated Press-Tight connector (Restek) through the LMCS. Modulation was performed at 0 °C with modulation period (P M) of 5 s. Helium was used as carrier gas (99.999% purity) at a constant flow rate of 1.2 mL min−1. The chromatographic conditions were: oven temperature program, 40 °C (hold 2 min), increased at 3 °C min−1 to 240 °C (hold 20 min); injector temperature, 230 °C; injection volume, 1 μL and using split ratios of 100:1 or 50:1. The outlet of the 2D column was connected to the MS source via deactivated fused silica (0.45 m × 0.10 mm I.D.). The QTOFMS was operated in total transfer ion mode through the quadrupole sector, so the MS functioned as an accurate mass (acc)TOFMS instrument. The ion source T was 280 °C, transfer line T was 250 °C, with ionisation of 70 eV. A mass range of 45–400 Da was used, with TOF mass resolution of 2 GHz extended dynamic range. GC-accTOFMS experiments were conducted using the same column configuration and instrument conditions, except the cryogenic modulation process was not performed. Agilent MassHunter software was used for modulation control, data acquisition and processing. A schematic of the system configuration is illustrated in Fig. S1 (Supporting Information).
2.3 Data handling
Data acquisition and processing were performed using Agilent MassHunter ver. B.06.00 (Agilent Technologies). NIST (National Institute of Standards and Technology) 11 MS spectrum library was used for spectrum searching and identification. Retention indices (RI) were calculated using the Van den Dool and Kratz equation, relative to C7–C30 n-alkanes (note that t R is determined as the total retention time on both the 1D and 2D columns) effectively corresponding to the polar SUPELCOWAX®10 column. Replicate analyses, and of the same sample at different concentration indicated acceptable reproducibility of retention time and index values. Metabolite identification levels were classified according to minimum reporting standards guidelines defined by the Metabolomics Standards Initiative (Sumner et al. 2007). Contour plots were generated by exporting MassHunter data in CSV file format, followed by data conversion to 2D matrix (according to the P M and data acquisition rate) using in-house software (2D GC converter), with 2D plots generated using Transform software (ver. 3.3, Fortner Research, VA). Precise data acquisition rates (nominally 50 Hz) were assessed by calculating the number of data points in a given time period. Principal component analysis (PCA) was performed using Multibase (NumericalDynamics.com, Tokyo, Japan), and Microsoft Excel Version 14.0.7140.5002 (Microsoft Corporation, Remond, WA).
3 Results
Metabolic profiles of leaf oils from E. polybractea (EP), E. citriodora (EC), E. radiata (ER) and E. globulus (EG) were analysed by GC × GC-accTOFMS in order to classify differences in expression of their metabolites.
3.1 Assessment of GC × GC chromatographic parameters and operating conditions
Separation performance of GC × GC is dependent on the right column combination and also the selectivity of the stationary phases. Prior experience with similarly complex samples of agarwood (Aquilaria malaccensis) extracts allowed choice of a suitable column combination here (i.e. a polar × non-polar set) to effect appropriate separation within the 2D space (Wong et al. 2015). To obtain acceptable GC × GC separation and support metabolite identification and coverage, both P M and modulation temperature (T M) were investigated by using the EC leaf oil. The effect of T M over the range − 20–20 °C was investigated; too low T M, may lead to incomplete re-mobilisation of high boiling point compounds; at too high T M the more volatile components might not be trapped effectively, causing some breakthrough. T M = 0 °C was chosen as a compromise. Similarly, a larger P M will reduce wrap-around, but results in reduced 1D resolution (i.e. insufficient modulation events performed across a peak), potentially causing overlapping of some peaks already separated in 1D. P M = 5 s was subsequently chosen. To validate the accuracy of linear temperature programmed retention indices (RI) using GC × GC-accTOFMS, experimental RI (RI cal) for a mixture of monoterpenes were determined and compared with reference RI data (RI ref; Supporting Information Table S1); a good linear correlation (R2 = 0.9994) between RI cal and RI ref was obtained. Adequate reproducibility of the GC × GC experiment, and reliable index calculation are two prerequisites for metabolite identification.
3.2 Metabolic profiling of eucalypt leaf oils using GC × GC-accTOFMS
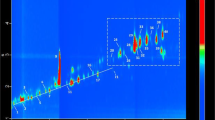
GC–accTOFMS analysis of EP leaf oil (Fig. 1A) shows a reasonably complex chromatogram, with many peak overlaps apparent or suspected (due to lack of sufficient peak separation). Therefore GC-accTOFMS is unable to adequately resolve and identify the large proportion of metabolites present within the oil. Hence, higher resolution GC × GC-accTOFMS utilising 2D separation in real-time with high resolution MS detection was conducted. Coupling the two columns with different separation mechanisms (here, polar 1D and non-polar 2D phases) and cryogenic modulation, with accurate mass MS detection, results in enhanced metabolite profiling with respect to the total coverage of measured metabolites. The gain in metabolic coverage can be readily observed. 1DGC analysis (Fig. 1A) enables the detection of ca. 90 compounds in EP leaf oil, while GC × GC analysis indicated detection of ca. 302 compounds (Fig. 1C), corresponding to a threefold increase in detected components.

GC × GC-accTOFMS analysis of E. polybractea leaf oil. Ai 1D GC‒accTOFMS analysis, Aii expansion of rectangle region in (Ai), Bi linear presentation of the GC result using cryogenic modulation with P M = 5 s, Bii expansion of rectangle region in (Bi), C 2D contour plot of data shown in (B)
Identification of phytochemicals by conventional GC–MS is often based on comparison of both detected compound spectra with those recorded in MS libraries, plus retention indices. There is a distinct possibility of rejecting low abundance or co-eluting compounds, which consequently have lower threshold MS match scores. This is a universal concern for almost all metabolic analysis tasks using chromatography with MS platforms. As GC × GC provides increased peak separation, and cryogenic focusing gives better peak sensitivity (i.e. higher S/N ratios; contrast Fig. 1B with Fig. 1A), an increased number of compounds with high match scores is expected. Hence, GC × GC analysis provides improved component peak assignment in contrast to 1DGC, especially for trace metabolites. The likelihood of having more than one matching library entry with similar score for a detected compound is a major constraint to identification; the highest match score cannot be guaranteed as correct, particularly for isomers. High resolution TOFMS provides increased confidence for components with different molecular formulae, especially those comprising heteroatomic species; (Table 1; Supporting Information Table S2), as indicated for mass spectra for two tentatively identified compounds (level 2 identifications (L2); Supporting Information Fig. S2), m-cumenol (alkyl phenol) and phytol (diterpenic alcohol), with mass accuracy values <12 ppm for corresponding ion formulae. However, not all molecular ions will necessarily be observed with electron ionisation (Fig. S2) due to complete fragmentation of the molecular ion. Hence, molecular ions alone are insufficient for identification. Base peak ions may provide an advantage to discriminate against molecules having similar exact masses. Both α-caryophyllene and γ-selinene have the same exact masses (204.1878 Da) but have base peak ions of 93.07043 ([C7H9]+) and 189.16393 ([C14H21]+), respectively. Use of retention index (RI) values (RI cal vs. RI ref) as a filter adds another degree of identification certainty, and may reduce the number of possible compounds generated from the NIST library search. However, RI for the 2D column (2 RI; non-polar phase) is not used for the current study, due to the lower accuracy threshold for solutes having higher retentions (von Mühlen and Marriott 2011; Jiang et al. 2015).
Structured 2D chromatographic behaviour in the contour plot aids classification of structurally related compounds into chemical groupings based on clustering of their elution in the GC × GC plane, based on chemical composition. This can support characterisation of a compound, by relating 2D chromatographic position to possible chemical families and/or structures. Thus, compounds with match scores ≥80% and mass accuracy within ±12 ppm (but without supporting RI ref data) that are located outside the expected corresponding family group cluster, will be rejected. The generalised procedural steps employed for compound identification (Fig. 2; taking the peak with 1 t R of 1183.2 s and 2 t R of 3.2 s, for example), commenced with (1): the mass spectrum for the component at a given 1 t R and 2 t R; (2): the library match, revealing 6 compounds with relatively high match factor; (3): consideration of mass precision/accuracy (ppm) of molecular and base ions; (4): calculation of retention index, as isobaric compounds (e.g. chemical subclasses of mono- or sesquiterpenes) could not be differentiated solely by accurate mass and/or fragmentation patterns; and finally (5): consideration of 2D structure relationships for chemical class position.

Workflow diagram of the process employed for compound identification in GC × GC-accTOFMS analysis. A peak at 1D t R = 1183.2 s and 2D t R = 3.2 s is used as an example to illustrate the overall selection and identification process
Detected compounds are identified based on comparison of mass spectra to standard compounds where available, or using the NIST 11 MS spectrum database, and by comparison of GC retention indices with values reported in the literature for a poly(ethylene glycol) phase column. Higher MS match score values correspond to better MS correlation. Mass accuracies were determined for the base ion masses of the respective components. A match score ≥80%, with consistent RI cal (within ±20 of RI ref) values and mass accuracy values (within ±12 ppm), were employed as criteria for the tentative identification (L2) of compounds. For compounds that do not have available RI ref values, m/z ion mass fragmentation patterns with corresponding relative ion abundances and ion formulae were determined to aid tentative identification. A total of ca. 400 compounds (estimated from the number of peak contours in the 2D plots, Fig. 3; Table S2) were detected; of these 172 were tentatively identified (L2), 11 were positively identified (via co-injection of standards; level 1 identification, L1), while the remainder remain unidentified. Among these 183 components (representing 88.7% (EP), 50.8% (EC), 90.0% (ER) and 82.2% (EG) of the total ion counts of the respective samples), there are 91 monoterpenoids, 52 sesquiterpenoids, 1 diterpenoid and 39 other components. These results indicate that GC × GC-accTOFMS provides much higher metabolic coverage for the characterisation of phytochemicals compared with 1DGC, explained as mainly due to the dual separation experiment (1D and 2D) leading to better resolution of components in 2D space.

2D contour plots of the analysed eucalypt leaf oils. Ai E. polybractea, Aii expansion of braced region in (Ai), Bi E. citriodora, Bii expansion of braced region in (Bi), Ci E. radiata, Cii expansion of braced region in (Ci) and Di E. globulus, Dii expansion of braced region in (Di). a monoterpenic hydrocarbons, b sesquiterpenic hydrocarbons, c oxygenated monoterpenes, d oxygenated sesquiterpenes. The numbering of compounds is given in Table S2
Contour plots (Fig. 3) demonstrate that terpenic compounds are organised mainly into four major structural clusters in the 2D separation space: monoterpenic/sesquiterpenic hydrocarbons are located at a lower 1D retention and 2D higher retention; their oxygenated analogues locate at a higher 1D retention and lower 2D retention. The low polarity 2D column phase leads to reduced 2D retention for more polar oxygenated species. Results show that earlier 1D retention terpene or sesquiterpene components in GC × GC analysis have low-to-moderate polarity; they elute later on 2D. Later 1D eluting components will be more polar, and are primarily oxygenated. Many of these show extensive 1D co-elution, usually with 2–4 overlapping compounds for every ‘peak’ on the 1D column (Fig. 3). These results clearly demonstrate that the complexity of EO is much greater than that which might be suspected from 1DGC analysis. With GC × GC, minor constituents which otherwise overlap larger components in 1D may still be well characterised and quantified due to 2D separation. From the chromatographic retention patterns, their chemical nature may be indicated.
Supporting Information (Fig. S3) presents various selected mass spectra for closely eluting compounds (showing 1 t R differences of ≤0.8 s) in Table S2 (indicated as bold entries), which in most cases have quite distinct spectra, and therefore identities. These selected examples are only a subset of such closely eluting compounds that confound the analysis, but serve to illustrate that often quite dissimilar spectra are obtained, and offer independent library identities. It is important to note that the separation window in 2D (5 s) is sufficient to allow the resolution of suspected overlapping components, due to the zone compression effect providing narrow modulated peaks (w b of < 450 ms for peaks with counts ≤106). This supports the case that the better resolution of GC × GC does indeed lead to a greater peak density in the chromatographic space.
The detailed analysis of eucalypt leaf oils allowed the positive/tentative identification of 120 secondary compounds in EP, 58 in EC, 76 in ER and 100 in EG. Table S2 summarises an extended list of all identified secondary metabolites and relative abundance based on total ion counts; a considerable variation (both qualitative and quantitative) between the species can be observed. An overall metabolic composition of the analysed eucalypt leaf oils is depicted in Fig. 4. Monoterpenic compounds predominated in all species, both in relative amount (45.0–83.7%) and in the number of compounds detected. 1,8-Cineole was the major compound in EP, ER and EG, accounting for ≥39.8% of the total ion count; citronellal was the principal constituent for EC (29.3%). Whilst oxygenated monoterpenes dominated the profiles, other abundant compounds detected were the hydrocarbon monoterpenes α-pinene (0.5–3.2%), limonene (0.3–8.4%), p-cymene (0.4–5.5%), and the monoterpenic alcohols terpinen-4-ol (0.03–3.6%) and α-terpineol (0.1–5.9%). Notable differences between the four species appeared in the sesquiterpenic profiles. A large number of sesquiterpenes were detected (54 compounds), but they comprised <16% of the total oil composition. Both qualitative and quantitative differences were observed in the sesquiterpene profiles: alloaromadendrene (0.8%) and globulol (0.4%) were the main compounds in EP, whereas β-caryophyllene (0.4%) and bicyclogermacrene (1.3%) were the principal sesquiterpenes of ER. Alloaromadendrene (1.8%), globulol (2.6%), viridiflorol (1.1%), α-eudesmol (1.8%) and β-eudesmol (2.9%), were the main compounds of these fractions in EG leaf oil. A small amount (0.09%) of oxygenated diterpenes was found in EG.

Relative metabolic compositions (%) of the analysed eucalypt leaf oils. EP E. polybractea, EC E. citriodora, ER E. radiata, EG E. globulus. MO monoterpenic oxide, MH monoterpenic hydrocarbon, MA monoterpenic aldehyde, OM monoterpenic alcohol, OS oxygenated sesquiterpene, SH sesquiterpenic hydrocarbon, MK monoterpenic ketone, MAC monoterpenic acetate, OT other subgroups of metabolites (refer to Table S2), UN not defined (unidentified)
4 Discussion
Leaf oils of different Eucalyptus spp. could be differentiated by the presence or absence of certain metabolites, and this can be supported by visual discrimination and retention time comparison to identify discriminators for each species with reference to the obtained 2D contour profiles (Fig. 3), where each observed contour spot denotes a specific secondary compound. Specifically, PCA was performed on the basis of the selected 27 identified compounds (approximately based on largest relative abundance) in respective species to objectively identify differences between metabolic profiles of the analysed eucalypts. The obtained loading plot and score plot (Fig. 5) represented 85.7% of the metabolic composition. The scores clustered in four groups according to the eucalypt species. The main explanatory variables (based on the loading plot) were citronellal, isopregol, isopulegol, citronellic acid and β-caryophyllene for EC; trans-pinocarveol, globulol, β-eudesmol, and α-eudesmol for EG. However, the significance of this is not yet apparent, and further studies covering additional uniform sample sets of each species, covering different geographical origins are warranted to increase confidence in class attributes and chemical resemblance to different chemotypes.

Principal component analysis of 27 selected metabolites (based on relative abundance) in analysed eucalypt leaf oils, A loading plot and B score plot. Percentage of variance explained by each principal component is indicated in parenthesis. EP E. polybractea, EC E. citriodora, ER E. radiata, EG E. globulus. a α-pinene, b β-pinene, c sabinene, d α-phellandrene, e limonene, f 1,8-cineole; g γ-terpinene, h cis-β-ocimene, i p-cymene, j p-cymenene, k citronellal, l isopregol, m isopulegol, n β-caryophyllene, o terpinen-4-ol, p alloaromadendrene, q trans-pinocarveol, r citronellol acetate, s cryptone t α-terpineol, u terpinyl acetate, v 3,7-dimethyl-2-octen-1-ol, w caryophyllene oxide, x globulol, y α-eudesmol, z β-eudesmol, z* citronellic acid
The obtained metabolic compositions might shed light on different biological activities of eucalypt leaf oils. Albeit bioactivities of plant extracts are generally attributed to particular compounds (mainly major constituents), a synergistic phenomenon among corresponding mixtures have been shown to result in a higher bioactivity compared to the isolated individual component (Nerio et al. 2010). A number of reports have indicated that minor compounds may act additively or synergistically to achieve the desired biological effects, in line with evolutionary hypotheses of plant defence where a single phytochemical alone might not be the basis for mediating deterrence (Becerra et al. 2009; Agrawal 2011). For instance, some studies have highlighted that 1,8-cineole (a major constituent) might not be the principal chemical responsible for the allelopathic suppression of weeds (Angelini et al. 2003; Verdeguer et al. 2009). These indicate that minor constituents also contribute to the allelopathic and insect repellent activities, and reflect the importance of compositional complexity in conferring bioactivity to natural terpenoid mixtures.
In the current study, phenethyl propionate was found to be present only in the EP leaf oil. This compound is well known for its herbicidal activity and has been patented as a formulation in herbicide (Dayan et al. 2009). Citral, another phytotoxic metabolite that displayed contact herbicidal activity (Bessette 2000), was found to be present in both ER and EG. Interestingly, both EP and EG oils were found to contain thymol and carvacrol—phytochemicals that show broad-spectrum insecticidal activities. β-caryophyllene (a strong repellent against Aedes aegypti) was found within the leaf oils of all the analysed eucalypt samples (Gillij et al. 2008). Notably, the presence of caryophyllene oxide in some of the eucalypts (EP, EC and EG) indicated possible repellent activity against Attini ants (leaf-cutting ant—a major pest affecting agricultural and forestry production) (Boulogne et al. 2012). Eugenol (a fast acting contact insecticidal compound) was found to be present only within EC (Dayan et al. 2009). Additionally, phytol is present in EG leaf oil; this diterpene alcohol has been reported to have high repellent activity against Anopheles gambiae (Odalo et al. 2005). Considering some reported studies on the discrepancy of activities exerted by leaf oils of different eucalypt, it may be suggested that these minor secondary compounds can account for some of the differences in their overall activities (Inouye et al. 2001; Vilela et al. 2009). However, the perception of plant metabolite interactions is presumably far more complex than what might be expected.
Ecologically, the biosynthesised array of secondary metabolites may serve to confound the capacity for natural competitors or herbivores to evolve resistance to all of the secondary compounds (i.e. plant signaling in response to environmental stimuli), and hence slow the rate of breakthrough of the plant’s defence. Resolving the mechanisms involved in the interactions (additive, synergistic or even antagonistic) between metabolites resulting in the overall observed activities will require further detailed investigation. It is not the aim of this study to draw conclusions on the interrelation of designated metabolites to the overall bioactivities of eucalypt leaf oils. Nevertheless, it is shown that a more comprehensive approach allows improved expression of the “metabolic pool” of these biosynthesised metabolites in a single analysis with well-resolved components. Tentative identification of some phytochemicals (octene-1,2-diol, undecatrien-3-ol, sulcatone, phenethyl pivalate, 1,8-terpin, jasmone, gleenol, etc.) that have not been previously identified in some of the analysed eucalypts, raises some pertinent questions on their formation mechanism and ecological significance. It is demonstrated that the study of such samples offers a rich tapestry with which to work, largely due to the number of secondary metabolites tentatively proposed or unidentified. Future studies that focus on unambiguous identification of detected secondary compounds by methods such as sequential heart-cut multidimensional GC (GC–GC), preparative GC–GC in combination with nuclear magnetic resonance spectroscopy or X-ray crystallography (if the targeted compound is crystalline) would be valuable, supported by a more complete reference standard set (Eyres et al. 2008).
Being confined to a small number of samples of each species, the findings presented above are exploratory in nature and interpretation of putative biochemical variations between Eucalyptus spp. should be drawn with care, as their relevance and consistency needs to be further evaluated. Importantly, the presented metabolic diversity of different eucalypt leaf extracts can serve to provide a basis for plant scientists, biochemists and microbiologists to explore molecular mechanisms behind the microevolutionary events of secondary metabolism and eucalypt adaptations to environmental demands, potentially providing insights or clues to their natural biological activities (Brakhage et al. 2009). It is thus worthwhile to explore the metabolic compositions of EOs to a deeper extent, as described herein, which could serve as clues in searching for important biosynthetic pathways, and might serve to unravel the response (i.e. signaling metabolites) of eucalypts towards environmental stimuli, extending the knowledge on the physiological role of the produced secondary metabolites which confer biological advantages for the producer. Additionally, the identified diverse array of secondary metabolites might serve as an important extension to the recent reported genome database of E. grandis, correlating with some of the identified terpene synthase genes responsible for mediating the synthesis of these specialised metabolites (Myburg et al. 2014).
5 Concluding remarks
Here, we report detailed untargeted metabolic profiling of secondary metabolites in E. polybractea, E. citriodora, E. radiata and E. globulus leaf oils using high resolution GC × GC-accTOFMS analysis. This extends the metabolic coverage when compared with conventional 1DGC, allowing deeper characterisation of the metabolic composition of eucalypt leaf oils. Terpenic profiles presented in 2D contour plots allow visual discrimination of the metabolic composition among interspecies of Eucalyptus. This can be extended to chemotaxonomical applications such as metabolite fingerprinting and more complete characterisation of leaf oils of different Eucalyptus spp. (i.e. for defining chemotypes—visually recognised 2D patterns), provided that more samples of the same species and of different provenances are analysed to increase confidence. A schematic of the metabolite identification procedures based on mass spectra, mass accuracy, retention indices, and solute retentions in the 2D chromatographic space were proposed. Specifically, PCA allows classification of different eucalypt leaf oils into their corresponding species, characterised according to their metabolic composition and providing information on discriminating metabolites in each species. This high resolution platform together with proposed identification procedures can be applied to untargeted profiling of other plant derived extracts, allowing significant gain in metabolite coverages. This raises the possibility of future work to identify and decode the function of terpene synthase genes responsible for mediating biosynthesis of these specialised metabolites.
References
Agrawal, A. A. (2011). Current trends in the evolutionary ecology of plant defence. Functional Ecology, 25(2), 420–432.
Angelini, L. G., Carpanese, G., Cioni, P. L., Morelli, I., Macchia, M., & Flamini, G. (2003). Essential oils from Mediterranean Lamiaceae as weed germination inhibitors. Journal of Agriculture and Food Chemistry, 51(21), 6158–6164.
Βatish, D. Ρ., Singh, Η. P., Κohlia, Ρ. Κ., & Κaura, S. (2008). Eucalyptus essential oil as a natural pesticide. Forest Ecology and Management, 256(12), 2166–2174.
Becerra, J. X., Noge, K., & Venable, D. L. (2009). Macroevolutionary chemical escalation in an ancient plant–herbivore arms race. Proceedings of the National Academy of Sciences of the United States of America, 106(43), 18062–18066.
Bendaoud, H., Bouajila, J., Rhouma, A., Savagnac, A., & Romdhane, M. (2009). GC/MS analysis and antimicrobial and antioxidant activities of essential oil of Eucalyptus radiata. Journal of the Science of Food and Agriculture, 89(8), 1292–1297.
Bessette, S. M. 2000. Essential oils and their components as herbicides. WO 2000051436.
Boland, D. J., Brophy, J. J., & House, A. P. N. (1991). Eucalyptus leaf oils, use, chemistry, distillation and marketing (pp. 11–155). Melbourne: Inkata Press.
Boulogne, I., Petit, P., Ozier-Lafontaine, H., Desfontaines, L., & Loranger-Merciris, G. (2012). Insecticidal and antifungal chemicals produced by plants: a review. Environmental Chemistry Letters, 10(4), 325–347.
Brakhage, A., Gierl, A., Hartmann, T., & Strack, D. (2009). Evolution of metabolic diversity. Phytochemistry, 70(15–16), 1619–1620.
Cermelli, C., Fabio, A., Fabio, G., & Quaglio, P. (2008). Effect of eucalyptus essential oil on respiratory bacteria and viruses. Current Microbiology, 56(1), 89–92.
Cheng, S.-S., Huang, C.-G., Chen, Y.-J., Yu, J.-J., Chen, W.-J., & Chang, S.-T. (2009). Chemical composition and larvicidal activities of leaf essential oils from two eucalyptus species. Bioresource Technology, 100(1), 452–456.
Chin, S.-T., & Marriott, P. J. (2014). Multidimensional gas chromatography beyond simple volatiles separation. Chemical Communications, 50(64), 8819–8833.
Cossalter, C., & Pye-Smith, C. (2003). Fast-wood forestry: myths and realities (pp. 13–20). Indonesia: Center for International Forestry Research.
Dayan, F. E., Cantrell, C. L., & Duke, S. O. (2009). Natural products in crop protection. Bioorganic & Medicinal Chemistry , 17(12), 4022–4034.
Dettmer, K., Almstetter, M. F., Wachsmuth, C. J., & Oefner, P. J. (2013). Comprehensive two-dimensional gas chromatography for metabolomics. In W. Weckwerth & G. Kahl (Eds.), The handbook of plant metabolomics (pp. 77–91). Weinheim: Wiley-VCH Verlag GmbH & Co.
Eyres, G. T., Urban, S., Morrison, P. D., Dufour, J.-P., & Marriott, P. J. (2008). Method for small-molecule discovery based on microscale-preparative multidimensional gas chromatography isolation with nuclear magnetic resonance spectroscopy. Analytical Chemistry, 80(16), 6293–6299.
Fernie, A. R., Trethewey, R. N., Krotzky, A. J., & Willmitzer, L. (2004). Metabolite profiling: from diagnostics to systems biology. Nature Reviews Molecular Cell Biology, 5, 763–769.
Gillij, Y. G., Gleiser, R. M., & Zygadlo, J. A. (2008). Mosquito repellent activity of essential oils of aromatic plants growing in Argentina. Bioresource Technology, 99(7), 2507–2515.
Goodger, J. Q. D., Connelly, C. A., & Woodrow, I. E. (2007). Examination of the consistency of plant traits driving oil yield and quality in short-rotation coppice cultivation of Eucalyptus polybractea. Forest Ecology and Management, 250(3), 196–205.
Hantao, L. W., Aleme, H. G., Passador, M. M., Furtado, E. L., de Lima Ribeiro, F. A., Poppi, R. J., & Augusto, F. (2013). Determination of disease biomarkers in Eucalyptus by comprehensive two-dimensional gas chromatography and multivariate data analysis. Journal of Chromatography A, 1279, 86–91.
Hummelbrunner, L. A., & Isman, M. B. (2001). Acute, sublethal, antifeedant, and synergistic effects of monoterpenoid essential oil compounds on the tobacco cutworm, Spodoptera litura (Lep., Noctuidae). Journal of Agriculture and Food Chemistry, 49(2), 715–720.
Inouye, S., Takizawa, T., & Yamaguchi, H. (2001). Antibacterial activity of essential oils and their major constituents against respiratory tract pathogens by gaseous contact. Journal of Antimicrobial Chemotherapy, 47(5), 565–573.
Jiang, M., Kulsing, C., Nolvachai, Y., & Marriott, P. J. (2015). Two-dimensional retention indices improve component identification in comprehensive two-dimensional gas chromatography of saffron. Analytical Chemistry, 87(11), 5753–5761.
Koek, M. M., van der Kloet, F. M., Kleemann, R., Kooistra, T., Verheij, E. R., & Hankemeier, T. (2011). Semi-automated non-target processing in GC × GC–MS metabolomics analysis: applicability for biomedical studies. Metabolomics, 7(1), 1–14.
Kohli, R. K., Batish, D. R., & Singh, H. P. (1998). Eucalypt oils for the control of Parthenium (Parthenium hysterophorus L.). Crop Protection, 17(2), 119–122.
Lopez-Sanchez, P., de Vos, R. C. H., Jonker, H. H., Mumm, R., Hall, R. D., Bialek, L., Leenman, R., Strassburg, K., Vreeken, R., Hankemeier, T., Schumm, S., & van Duynhoven, J. (2015). Comprehensive metabolomics to evaluate the impact of industrial processing on the phytochemical composition of vegetable purees. Food Chemistry, 168, 348–355.
May, F., & Ash, J. (1990). An assessment of the allelopathic potential of Eucalyptus. Australian Journal of Botany, 38(3), 245–254.
Mulyaningsih, S., Sporer, F., Zimmermann, S., Reichling, J., & Wink, M. (2010). Synergistic properties of the terpenoids aromadendrene and 1,8-cineole from the essential oil of Eucalyptus globulus against antibiotic-susceptible and antibiotic-resistant pathogens. Phytomedicine, 17(13), 1061–1066.
Myburg, A. A., Grattapaglia, D., Tuskan, G. A., Hellsten, U., Hayes, R. D., Grimwood, J., et al. (2014). The genome of Eucalyptus grandis. Nature, 510, 356–362.
Nerio, L. S., Olivero-Verbel, J., & Stashenko, E. (2010). Repellent activity of essential oils: a review. Bioresource Technology, 101(1), 372–378.
Odalo, J. O., Omolo, M. O., Malebo, H., Angira, J., Njeru, P. M., Ndiege, I. O., & Hassanali, A. (2005). Repellency of essential oils of some plants from the Kenyan coast against Anopheles gambiae. Acta Tropica, 95(3), 210–218.
Pandey, R., Kalra, A., Tandon, S., Mehrotra, N., Singh, H. N., & Kumar, S. (2000). Essential oils as potent source of nematicidal compounds. Journal of Phytopathology, 148(7–8), 501–502.
Ramezani, H., Singh, H. P., Batish, D. R., & Kohli, R. K. (2002). Antifungal activity of the volatile oil of Eucalyptus citriodora. Fitoterapia, 73(3), 261–262.
Rathahao-Paris, E., Alves, S., Junot, C., & Tabet, J.-C. (2016). High resolution mass spectrometry for structural identification of metabolites in metabolomics. Metabolomics, 12(1), 1–15.
Sartorelli, P., Marquioreto, A. D., Amaral-Baroli, A., Lima, M. E. L., & Moreno, P. R. H. (2007). Chemical composition and antimicrobial activity of the essential oils from two species of. Eucalyptus. Phytotherapy Research, 21(3), 231–233.
Singh, H. P., Kaur, S., Negi, K., Kumari, S., Saini, V., Batish, D. R., & Kohli, R. K. (2012). Assessment of in vitro antioxidant activity of essential oil of Eucalyptus citriodora (lemon-scented Eucalypt; Myrtaceae) and its major constituents. LWT-Food Science and Technology, 48(2), 237–241.
Sumner, L. W., Amberg, A., Barrett, D., Beale, M. H., Beger, R., Daykin, C. A., et al. (2007). Proposed minimum reporting standards for chemical analysis. Metabolomics, 3(3), 211–221.
Tyagi, A. K., & Malik, A. (2011). Antimicrobial potential and chemical composition of Eucalyptus globulus oil in liquid and vapour phase against food spoilage microorganisms. Food Chemistry, 126(1), 228–235.
Verdeguer, M., Blázquez, M. A., & Boira, H. (2009). Phytotoxic effects of Lantana camara, Eucalyptus camaldulensis and Eriocephalus africanus essential oils in weeds of Mediterranean summer crops. Biochemical Systematics and Ecology, 37(4), 362–369.
Vilela, G. R., de Almeida, G. S., D’Arce, M. A. B. R., Moraes, M. H. D., Brito, J. O., da Silva M. F. D.G., Silva, S. C., de Stefano Piedade, S. M., Calori-Domingues, M. A., & da Gloria, E. M. (2009). Activity of essential oil and its major compound, 1,8-cineole, from Eucalyptus globulus Labill., against the storage fungi Aspergillus flavus Link and Aspergillus parasiticus Speare. Journal of Stored Products Research, 45(2), 108–111.
von Mühlen, C., & Marriott, P. J. (2011). Retention indices in comprehensive two-dimensional gas chromatography. Analytical and Bioanalytical Chemistry, 401(8), 2351–2360.
Wolfender, J.-L., Marti, G., Thomas, A., & Bertrand, S. (2015). Current approaches and challenges for the metabolite profiling of complex natural extracts. Journal of Chromatography A, 1382, 136–164.
Wong, Y. F., Chin, S.-T., Perlmutter, P., & Marriott, P. J. (2015). Evaluation of comprehensive two-dimensional gas chromatography with accurate mass time-of-flight mass spectrometry for the metabolic profiling of plant–fungus interaction in Aquilaria malaccensis. Journal of Chromatography A, 1387, 104–115.
Wong, Y. F., Hartmann, C., & Marriott, P. J. (2014). Multidimensional gas chromatography methods for bioanalytical research. Bioanalysis, 6(18), 2461–2479.
Zhang, C., & Fu, S. (2009). Allelopathic effects of eucalyptus and the establishment of mixed stands of eucalyptus and native species. Forest Ecology and Management, 258(7), 1391–1396.
Acknowledgements
YFW gratefully acknowledges the provision of a Monash Graduate Scholarship and Monash International Postgraduate Research Scholarship. PJM acknowledges the Australian Research Council for a Discovery Outstanding Researcher Award; DP130100217. The authors also thank FGB Natural Products Pty Ltd and Australian Botanical Products Pty Ltd for providing the EOs and standards used in this study.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no potential conflicts of interest.
Informed consent
Informed consent was not required for the studies reported.
Research involving human and animal rights
This research does not involve human participants, nor animal studies.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Wong, Y.F., Perlmutter, P. & Marriott, P.J. Untargeted metabolic profiling of Eucalyptus spp. leaf oils using comprehensive two-dimensional gas chromatography with high resolution mass spectrometry: Expanding the metabolic coverage. Metabolomics 13, 46 (2017). https://doi.org/10.1007/s11306-017-1173-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11306-017-1173-3