
Abstract
Introduction
The initial decades of the twenty-first century have witnessed striking technical advances that have made it possible to detect, identify and quantitatively measure large numbers of plasma or tissue metabolites. In parallel, similar advances have taken place in our ability to sequence DNA and RNA. Those advances have moved us beyond studies of single metabolites and single genetic polymorphisms to the study of hundreds or thousands of metabolites and millions of genomic variants in a single cell or subject. It is now possible to merge and integrate large data sets generated by the use of different “-omics” techniques to increase our understanding of the molecular basis for variation in disease risk and/or drug response phenotypes.
Objectives
This “Brief Review” will outline some of the challenges and opportunities associated with studies in which metabolomic data have been merged with genomics in an attempt to gain novel insight into mechanisms associated with variation in drug response phenotypes, with an emphasis on the application of a pharmacometabolomics-informed pharmacogenomic research strategy and with selected examples of the application of that strategy.
Methods
Studies that used pharmacometabolomics to inform and guide pharmacogenomics were reviewed. Clinical studies that were used as the basis for pharmacometabolomics-informed pharmacogenomic studies, published in five independent manuscripts, are described briefly.
Results
Within these five manuscripts, both pharmacokinetic and pharmacodynamic metabolomics approaches were used. Candidate gene and genome-wide approaches that were used in concert with these metabolomic data identified novel metabolite-gene relationships that were associated with drug response phenotypes in these pharmacometabolomics-informed pharmacogenomics studies.
Conclusion
This “Brief Review” outlines the emerging discipline of pharmacometabolomics-informed pharmacogenomics in which metabolic profiles are associated with both clinical phenotypes and genetic variants to identify novel genetic variants associated with drug response phenotypes based on metabolic profiles.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
1.1 Pharmacometabolomics
The concept that metabolomic profiles (metabotypes) might help make it possible to predict drug response phenotypes and to understand their underlying mechanisms has evolved rapidly over the past decade (Yap et al. 2006; Kaddurah-Daouk et al. 2008; Gavaghan McKee et al. 2006). Metabotype variation reflects variation resulting from both environmental and genomic factors. As a result, metabolomics may provide a more comprehensive view of overall molecular variation that contributes to individual differences in response to drug therapy than would genomics alone (Ellero-Simatos et al. 2014). The technical evolution that has occurred in our ability to accurately measure large numbers of metabolites in plasma or tissues in order to profile individual molecular variation has been reviewed elsewhere (Kaddurah-Daouk et al. 2008; Kaddurah-Daouk and Weinshilboum 2014; Wishart 2016). It is clear that metabolomics is only one of a series of measures of biological variation that might contribute to inter-individual differences in drug response. However, merging metabolomic data with other “-omics” information has the potential to provide novel insights into mechanisms underlying individual differences in disease risk or drug response phenotypes.
1.2 Pharmaco-omics
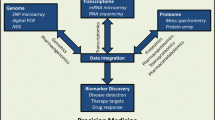
A major focus of medical research is on the identification of molecular mechanisms that contribute to disease pathophysiology to make it possible to understand the pathways involved and to identify novel therapeutic targets. However, many of those studies have often focused on only a specific piece of the larger puzzle, e.g., a single gene, mRNA transcript, protein or metabolite based on our current understanding of a particular disease or therapeutic target pathway. Technical advances that have occurred in recent decades have made it possible to move to broader studies of large scale “-omics” data including genomics, transcriptomics, proteomics and metabolomics (Fig. 1). These -omics approaches each individually moves us toward broader, less-biased studies that have the potential to reveal novel mechanisms underlying disease pathophysiology and response to drug therapy. However, even though the application of this type of approach represents a significant advance, most studies have continued to focus on the use of only a single -omic technique without evaluating the potential contribution of different -omics. Combined -omics approaches might provide novel insights that individual -omics may not identify. For example, pharmacogenetics, with a focus on single genes and their impact on drug response, has evolved into pharmacogenomics with the application of genome-wide studies of drug clinical phenotypes (Wang et al. 2011). However, the addition of other -omics data, that is, the evolution of pharmacogenomics to become “pharmaco-omics” is now possible and might be required, especially for studies of the drug therapy of complex diseases with multiple different underlying pathophysiologic mechanisms.

Pharmacometabolomics-informed Pharmacogenomics. Genomics contributes to the variation observed at the transcriptomic level, proteomic level and metabolomics level, resulting in a clinical phenotype. In addition, the environment can influence all of the -omics leading to a clinical phenotype. The interaction of metabolomics with clinical outcome measures and genomic variants (as indicated by the double-ended arrows) could reveal novel pathways associated with clinical phenotypes, which can then be validated by functional genomic studies and by investigating the interaction of those genetic variants with clinical outcomes. This may be particularly helpful for heterogeneous disorders in which traditional approaches have been disappointing
1.3 Pharmacometabolomics meets pharmacogenomics
Prior to completion of the Human Genome Project (Lander et al. 2001), it was hoped that genomic variants might provide insight into much of the inter-individual variation in disease risk and response to drug therapy. It quickly became clear that genomic variants alone could explain only a portion of that variation. In addition, it has also been difficult to apply genomics to complex disease in which similar phenotypes might arise from a variety of different pathophysiologic processes. Of the -omics disciplines depicted graphically in Figure 1, metabolomics and genomics might be particularly useful when applied in concert. The genome is consistent across different cell and tissue types within an individual—although epigenomics and the regulation of genomic expression is highly cell- and tissue-dependent—and since metabolomics is the “-omic ” data type that appears to be most closely related to clinical phenotypes, the integration of these two might be particularly useful to study multi-genic, complex diseases. Furthermore, although the application of genomic techniques such as genome wide association studies (GWAS) has been highly successful (Ingle et al. 2010; Ingle et al. 2013), especially in situations in which the phenotype is clearly defined, it has been less successful when applied to complex and less well defined phenotypes such as those for many psychiatric diseases. It is for that reason that the application of a research strategy that begins with metabolomics and allows the metabolomic data to “guide” genomics, e.g., pharmacometabolomics-informed pharmacogenomics, one type of pharmaco-omics study, might be particularly useful in selected situations.
2 Pharmacometabolomics-informed pharmacogenomics
There are two main subgroups of metabolites that can be used for pharmacometabolomics-informed pharmacogenomic studies: exogenous metabolites (those produced by biotransformation of the pharmacological agent, i.e., the drug) and endogenous metabolites. In other words, these two subgroups result from pharmacokinetic (PK) and pharmacodynamic (PD) variation, respectively, that might contribute to individual differences in drug response. The following brief examples will serve to illustrate both exogenous and endogenous pharmacometabolomics-informed pharmacogenomic studies.
2.1 Antidepressant pharmacometabolomics-informed pharmacogenomics
Samples from the Mayo Clinic NIH-Pharmacogenomics Research Network-Antidepressant Pharmacogenomics Medication Study (PGRN-AMPS) have been used to perform a series of pharmacometabolomics-informed pharmacogenomic analyses that have included both exogenous and endogenous metabolomics. This study involved nearly 800 patients with major depressive disorder (MDD) who were treated with citalopram or escitalopram–highly selective serotonin reuptake inhibitors (SSRIs). Clinical depression symptoms were assessed with both the Hamilton rating scale for depression (HAMD) and the quick inventory of depressive symptomatology-clinician rated (QIDS-C16) at baseline and after 4 and 8 weeks of SSRI treatment. Blood samples were collected at baseline and after 4 and 8 weeks of SSRI treatment for genomic and metabolomic analyses. The data from this study have been used to analyze both parent drug and drug metabolite concentrations and to assay endogenous metabolite variations associated with genomic single nucleotide polymorphisms (SNPs). A GWAS for association with clinical response phenotypes (e.g. remission and response) for the initial 529 patients enrolled in the study has also been published (Ji et al. 2013). All of these GWAS included adjustment for appropriate covariates such as age, sex, drug and prescribed dose.
2.1.1 Citalopram/escitalopram drug and drug metabolite studies
One of the metabolomic analyses conducted for the PGRN-AMPS study involved a GWAS for blood drug and drug metabolite concentrations assayed on an LC MS/MS quantitative platform (Ji et al. 2014). GWA studies of escitalopram and S-didesmethylcitalopram concentrations identified associations with SNPs across CYP2C19 (p = 4.1E−09) and CYP2D6 (p = 2.0E−16), respectively—the two enzymes that had previously been reported to metabolize escitalopram to form S-didesmethylcitalopram. In addition, after correcting for the CYP2C19 signal, the ratio of these two metabolites identified SNPs near TRIML1 (p = 7.08E−09), a gene not previously associated with drug metabolism. Therefore, this study identified variants across genes already known to be involved in the metabolism of citalopram/escitalopram as well as a novel gene not previously known to play a role in drug biotransformation. Of perhaps more importance were the data for endogenous metabolites, which are described subsequently.
2.1.2 Citalopram/escitalopram and endogenous metabolites
2.1.2.1 Glycine pharmacometabolomics-informed pharmacogenomics
In addition to studying drug and drug metabolite concentrations and their associations with SNPs, plasma samples from a subgroup of the patients enrolled in the PGRN-AMPS study were used to perform metabolomic analyses. The first metabolomic study analyzed 20 escitalopram remitters and 20 non-remitters using a semi-quantitative gas chromatography—mass spectrometry (GC-MS) platform that measured 251 metabolites, 97 of which were identified. The pathway most highly associated with percent change in QIDS-C16 was the “nitrogen metabolism pathway”, with glycine demonstrating the most significant association. Candidate glycine metabolism pathway genes were then selected for tag SNP genotyping in the entire PGRN-AMPS patient population available at that time—512 MDD patients who had been treated with SSRIs. SNPs across the GLDC gene were associated with a number of measures of clinical outcomes and the SNP with the lowest p-value (rs10975641) was also associated with SSRI response in a replication cohort from a separate MDD treatment study. Furthermore, functional genomic studies involving electrophoretic mobility shift assays for this SNP demonstrated altered protein binding in central nervous system-derived cell lines but not in other cell lines. This series of findings began with a metabolomic analysis of samples from just 40 MDD patients at the extremes of SSRI response, followed by a tag SNP “candidate pathway” genomic study (Ji et al. 2011).
2.1.2.2 Serotonin pharmacometabolomics-informed pharmacogenomics
As a follow-up to the initial SSRI pharmacometabolomics-informed pharmacogenomics study that implicated glycine in SSRI response, plasma samples from 290 of the PGRN-AMPS patients were assayed using a quantitative, targeted liquid chromatography electrochemical coulometric array (LCECA) metabolomics platform. This analysis identified and quantified metabolites primarily from the tryptophan, tyrosine and tocopherol pathways, metabolites that included serotonin—a metabolite that is effectively the target for SSRIs which block the reuptake of serotonin by the monoamine transporter encoded by the SLC6A4 gene (Jacobsen et al. 2014; Zhong et al. 2009). Of the 31 metabolites identified by use of this platform, plasma serotonin was the metabolite most highly associated with SSRI clinical outcomes. Therefore, serotonin concentration at baseline and the change in serotonin concentration after 4 or 8 weeks of SSRI treatment were used as phenotypes for GWA studies. Although this study was performed with only a relatively small group of MDD patients treated with SSRIs, two major SNP signals were identified in the serotonin GWA studies—TSPAN5 and ERICH3, both of which were associated with plasma serotonin and were shown by functional genomic studies to alter serotonin concentrations in cell culture media (Fig. 2) (Gupta et al. 2016).

Identification of novel genetic variants associated with serotonin concentrations in major depressive disorder (MDD) patients treated with selective serotonin reuptake inhibitors (SSRIs). Plasma serotonin concentrations were associated with the most clinical outcomes for MDD patients treated with SSRIs and, therefore, serotonin concentration was used as a phenotype for GWA studies. a GWAS of baseline plasma serotonin identified two SNP signals associated with serotonin concentration: TSPAN5 (rs11947402, p = 7.84E−09) and ERICH3 (rs11580409, p = 9.28E−08). These two signals were also identified after SSRI treatment for 4 and 8 weeks. Variant TSPAN5 genotype was associated with higher baseline and larger decreases in plasma serotonin concentrations, whereas variant ERICH3 genotype was associated with lower baseline and smaller decreases in plasma serotonin concentrations after SSRI treatment. b Variant (V) TSPAN5 genotype and wildtype (WT) ERICH3 genotypes were associated with higher serotonin concentrations and could explain 18.8 % of the variation observed in this population
2.2 Aspirin pharmacometabolomics-informed pharmacogenomics
The Heredity and Phenotype Intervention (HAPI) Heart Study (Mitchell et al. 2008) was designed to study the molecular basis for variation in aspirin response at both genomic and metabolomic levels and also to integrate these two -omics data sets. Although variation in aspirin response is highly heritable, few genetic variants had been associated with individual variation in aspirin response. This aspirin response study was performed with subjects from the Old Order Amish population—a highly homogeneous population (Cross 1976). Specifically, serum from 165 HAPI Heart Study participants who had been exposed to aspirin was assayed using a semi-quantitative untargeted GC-MS platform. The identified metabolites were then associated with response to aspirin which was measured using ex vivo assays of collagen-stimulated platelet aggregation. Not only were purine pathway metabolites altered after treatment with aspirin, but purine metabolites were also associated with differences in response between 40 “good” and 36 “poor” responders. Therefore, associations between aspirin response and purine pathway gene SNPs were investigated, and SNPs across the adenosine kinase (ADK) gene were associated with aspirin response (p = 3.4E−04). The SNP with the lowest p value was replicated in 341 participants from the Pharmacogenomics of Antiplatelet Intervention study (p = 0.002). The authors also tested the association of concentrations of purine metabolites with the top SNP and found that the SNP was associated with concentrations of a series of purine metabolites both before and after aspirin intervention. Therefore, by utilizing both genomic and metabolomic analyses in concert, this study was able to identify a novel genetic locus that may play a role in individual variation in response to aspirin (Yerges-Armstrong et al. 2013; Lewis et al. 2013).
3 Discussion
Healthcare professionals have always attempted to “tailor” their treatments to individual patients. However, they often lacked adequate objective data to truly “individualize” drug therapy. The current era of Precision Medicine promises to make it possible to utilize a combination of different -omics data to identify underlying molecular pathways and variation in the genes encoding proteins in those pathways that can help explain individual phenotypes. Therefore, pharmacometabolomics-informed pharmacogenomics—a term coined in 2012 (Ji et al. 2011)—represents one strategy at the interface of metabolomics and genomics that will clearly be an important aspect of the movement to Precision Medicine. While this “Brief Review” has focused on pharmacometabolomics-informed pharmacogenomics, it should be emphasized that there have also been metabolomics-informed genomic studies designed to identify SNPs associated with inter-individual metabotype variation that is independent of pharmacological response. In other words, metabolomics-informed genomic studies do not have to focus on metabolite variations after pharmacological treatment but may also be used to investigate genetic variation(s) that may impact metabolomic variation in the population or that are related to disease pathophysiology (Illig et al. 2010; Gieger et al. 2008; Suhre et al. 2011). It will be important to compare the results of those studies with the results of pharmacometabolomic-informed pharmacogenomic studies to identify genetic variants that contribute to metabolomic variation independent of drug response.
While approaches that use multiple -omics provide methods to investigate heterogeneous and complex diseases, there are challenges that must be addressed when utilizing large -omic datasets. Metabolomics can be assayed using a variety of different platforms that will identify anywhere from a handful of metabolites to hundreds of known metabolites. Increasing numbers of metabolites can result in a dimensionality issue in which there are many more metabolites than subjects or samples so it may be necessary to implement further filters to select individual metabolites or sets of metabolites from the full list of identified metabolites. As proposed here, multiple -omics approaches can effectively used as hypothesis-generating steps to be tested with functional validation and replication within independent samples of the population. As in any study, a larger sample size will provide more power for statistical analyses or models to be used but cost may be a limiting factor. It is important to keep these challenges (cost, dimensionality and sample size) in mind when selecting a platform for metabolomics analyses.
We have briefly outlined several studies that have applied a pharmacometabolomics-informed pharmacogenomic research strategy to help identify metabolomic profiles associated with variation in pharmacological response-followed by the identification of SNPs/genes that are associated with metabolomic profiles. It should be emphasized that each of the studies elected to integrate three types of data for their subjects (clinical outcomes, SNP genotypes, and metabolomics) using slightly different approaches, but each serves to illustrate the potential value of the addition of metabolomic data to the “traditional” genotype-phenotype association approach. It is already clear that one -omic measure, i.e., genomics, transcriptomics, proteomics or metabolomics alone may not be sufficient to explain a clinical phenotype nor can it provide adequate information to individualize drug therapy. We are moving progressively toward the use of multiple -omics in concert to sub-classify phenotypes and variation in response to medications. Future studies should attempt to use pharmacometabolomics together with pharmacogenomics to enhance our understanding of biochemical pathways that play a role in individual variation in drug response phenotypes. This approach may have the potential to take us beyond current disease phenotypes by using drugs as probes to help identify genetic and metabolomic variants that may contribute to individual differences in response to pharmacological agents.
References
Cross, H. E. (1976). Population studies and the old order Amish. Nature, 262(5563), 17–20.
Ellero-Simatos, S., Lewis, J. P., Georgiades, A., Yerges-Armstrong, L. M., Beitelshees, A. L., Horenstein, R. B., et al. (2014). Pharmacometabolomics reveals that serotonin is implicated in aspirin response variability. CPT: Pharmacometrics and Systems Pharmacology, 3, e125. doi:10.1038/psp.2014.22.
Gavaghan McKee, C. L., Wilson, I. D., & Nicholson, J. K. (2006). Metabolic phenotyping of nude and normal (Alpk:ApfCD, C57BL10 J) mice. Journal of Proteome Research, 5(2), 378–384. doi:10.1021/pr050255h.
Gieger, C., Geistlinger, L., Altmaier, E., Hrabe de Angelis, M., Kronenberg, F., Meitinger, T., et al. (2008). Genetics meets metabolomics: A genome-wide association study of metabolite profiles in human serum. PLoS Genetics, 4(11), e1000282. doi:10.1371/journal.pgen.1000282.
Gupta, M., Neavin, D., Liu, D., Biernacka, J., Hall-Flavin, D., Bobo, W. V., et al. (2016). TSPAN5, ERICH3 and selective serotonin reuptake inhibitors in major depressive disorder: Pharmacometabolomics-informed pharmacogenomics. Molecular Psychiatry. doi:10.1038/mp.2016.6.
Illig, T., Gieger, C., Zhai, G., Romisch-Margl, W., Wang-Sattler, R., Prehn, C., et al. (2010). A genome-wide perspective of genetic variation in human metabolism. Nature Genetics, 42(2), 137–141. doi:10.1038/ng.507.
Ingle, J. N., Liu, M., Wickerham, D. L., Schaid, D. J., Wang, L., Mushiroda, T., et al. (2013). Selective estrogen receptor modulators and pharmacogenomic variation in ZNF423 regulation of BRCA1 expression: Individualized breast cancer prevention. Cancer Discovery, 3(7), 812–825. doi:10.1158/2159-8290.cd-13-0038.
Ingle, J. N., Schaid, D. J., Goss, P. E., Liu, M., Mushiroda, T., Chapman, J. A., et al. (2010). Genome-wide associations and functional genomic studies of musculoskeletal adverse events in women receiving aromatase inhibitors. Journal of Clinical Oncology, 28(31), 4674–4682. doi:10.1200/jco.2010.28.5064.
Jacobsen, J. P., Plenge, P., Sachs, B. D., Pehrson, A. L., Cajina, M., Du, Y., et al. (2014). The interaction of escitalopram and R-citalopram at the human serotonin transporter investigated in the mouse. Psychopharmacology (Berl), 231(23), 4527–4540. doi:10.1007/s00213-014-3595-1.
Ji, Y., Biernacka, J. M., Hebbring, S., Chai, Y., Jenkins, G. D., Batzler, A., et al. (2013). Pharmacogenomics of selective serotonin reuptake inhibitor treatment for major depressive disorder: Genome-wide associations and functional genomics. The pharmacogenomics Journal, 13(5), 456–463. doi:10.1038/tpj.2012.32.
Ji, Y., Hebbring, S., Zhu, H., Jenkins, G. D., Biernacka, J., Snyder, K., et al. (2011). Glycine and a glycine dehydrogenase (GLDC) SNP as citalopram/escitalopram response biomarkers in depression: Pharmacometabolomics-informed pharmacogenomics. Clinical Pharmacology and Therapeutics, 89(1), 97–104. doi:10.1038/clpt.2010.250.
Ji, Y., Schaid, D. J., Desta, Z., Kubo, M., Batzler, A. J., Snyder, K., et al. (2014). Citalopram and escitalopram plasma drug and metabolite concentrations: Genome-wide associations. British Journal of Clinical Pharmacology, 78(2), 373–383. doi:10.1111/bcp.12348.
Kaddurah-Daouk, R., Kristal, B. S., & Weinshilboum, R. M. (2008). Metabolomics: A global biochemical approach to drug response and disease. Annual Review of Pharmacology and Toxicology, 48, 653–683. doi:10.1146/annurev.pharmtox.48.113006.094715.
Kaddurah-Daouk, R., & Weinshilboum, R. M. (2014). Pharmacometabolomics: Implications for clinical pharmacology and systems pharmacology. Clinical Pharmacology and Therapeutics, 95(2), 154–167. doi:10.1038/clpt.2013.217.
Lander, E. S., Linton, L. M., Birren, B., Nusbaum, C., Zody, M. C., Baldwin, J., et al. (2001). Initial sequencing and analysis of the human genome. Nature, 409(6822), 860–921. doi:10.1038/35057062.
Lewis, J. P., Yerges-Armstrong, L. M., Ellero-Simatos, S., Georgiades, A., Kaddurah-Daouk, R., & Hankemeier, T. (2013). Integration of pharmacometabolomic and pharmacogenomic approaches reveals novel insights into antiplatelet therapy. Clinical Pharmacology and Therapeutics, 94(5), 570–573. doi:10.1038/clpt.2013.153.
Mitchell, B. D., McArdle, P. F., Shen, H., Rampersaud, E., Pollin, T. I., Bielak, L. F., et al. (2008). The genetic response to short-term interventions affecting cardiovascular function: Rationale and design of the Heredity and Phenotype Intervention (HAPI) Heart Study. American Heart Journal, 155(5), 823–828. doi:10.1016/j.ahj.2008.01.019.
Suhre, K., Shin, S. Y., Petersen, A. K., Mohney, R. P., Meredith, D., Wagele, B., et al. (2011). Human metabolic individuality in biomedical and pharmaceutical research. Nature, 477(7362), 54–60. doi:10.1038/nature10354.
Wang, L., McLeod, H. L., & Weinshilboum, R. M. (2011). Genomics and drug response. New England Journal of Medicine, 364(12), 1144–1153. doi:10.1056/NEJMra1010600.
Wishart, D. S. (2016). Emerging applications of metabolomics in drug discovery and precision medicine. Nature Reviews Drug Discovery. doi:10.1038/nrd.2016.32.
Yap, I. K., Clayton, T. A., Tang, H., Everett, J. R., Hanton, G., Provost, J. P., et al. (2006). An integrated metabonomic approach to describe temporal metabolic disregulation induced in the rat by the model hepatotoxin allyl formate. Journal of Proteome Research, 5(10), 2675–2684. doi:10.1021/pr0601584.
Yerges-Armstrong, L. M., Ellero-Simatos, S., Georgiades, A., Zhu, H., Lewis, J. P., Horenstein, R. B., et al. (2013). Purine pathway implicated in mechanism of resistance to aspirin therapy: Pharmacometabolomics-informed pharmacogenomics. Clinical Pharmacology and Therapeutics, 94(4), 525–532. doi:10.1038/clpt.2013.119.
Zhong, H., Hansen, K. B., Boyle, N. J., Han, K., Muske, G., Huang, X., et al. (2009). An allosteric binding site at the human serotonin transporter mediates the inhibition of escitalopram by R-citalopram: Kinetic binding studies with the ALI/VFL-SI/TT mutant. Neuroscience Letters, 462(3), 207–212. doi:10.1016/j.neulet.2009.07.030.
Funding
This work was supported, in part, by U19 GM61388 (the Pharmacogenomics Research Network), RO1 GM28157 and by R24 GM078233 (The Metabolomics Research Network for Drug Response Phenotype) and by RC2 GM092729 (The Metabolomics Network for Drug Response Phenotype).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare they have no conflicts of interest.
Informed consent
All the patients included in the studies described in this review were reported to have provided informed consent for their participation in the original research.
Rights and permissions
About this article

Cite this article
Neavin, D., Kaddurah-Daouk, R. & Weinshilboum, R. Pharmacometabolomics informs pharmacogenomics. Metabolomics 12, 121 (2016). https://doi.org/10.1007/s11306-016-1066-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11306-016-1066-x