
Abstract
The use of glucocorticoids as growth promoters for meat-producing animals is strictly regulated within the European Union. However, in the past few years, a higher frequency of non-compliant bovine urine samples for prednisolone has been noticed, which could not be directly related to fraudulent use of prednisolone. As such, questions have risen about the origin of this compound. Unfortunately, at present, no decisive strategy has been established to discriminate between endogenous and exogenous prednisolone. In this study, an untargeted metabolomics strategy, based on Orbitrap and QqTOF mass spectrometry, was deployed to reveal urinary biomarkers, which are indicative for the exogenous administration of the synthetic glucocorticoid prednisolone. For this purpose, prednisolone was administered intramuscularly and per os to 12 bovines and a total of 2700 urine samples were collected before, during and after treatment. Multivariate statistical data analysis (i.e. OPLS-DA) revealed four differentiating metabolites that allowed discrimination between urine samples collected before and during prednisolone administration. None of these compounds were present in urine containing endogenous prednisolone, of which the formation was induced by the administration of a synthetic analogue of adrenocorticotropic hormone. Only one metabolite was retained as a highly suitable biomarker during growth-promoting and therapeutic prednisolone treatment, with 93.4 % sensitivity and 96.3 % specificity. Besides, this compound could be detected up to 4 days after a single therapeutic per os prednisolone administration. Based on accurate mass, isotope pattern, and MS/MS spectra, this compound was putatively annotated and is suggested as an actionable biomarker for exogenous prednisolone administration.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
1 Introduction
Natural glucocorticoids (cortisol and cortisone) are involved in various physiological processes, closely related to immune activity (inflammation). As such, their anti-inflammatory properties have led to the development of synthetic analogs, which proved even more potent. Prolonged exposure to synthetic glucocorticoids, like prednisolone, results in growth-promoting side effects. Therefore, synthetic glucocorticoids may be fraudulently administered to meat-producing animals (Hardy et al. 2012; Leporati et al. 2013). In order to protect consumers against potential harmful residues, present in animal derived food products, the therapeutic use of synthetic glucocorticoids in livestock has been strictly regulated in the European Union (CD 96/23/EC). Maximum residue limits (MRLs) have been introduced for betamethasone, dexamethasone, methylprednisolone and prednisolone in various edible tissues of animal origin (CD 37/2010). Moreover, the use of synthetic glucocorticoids is completely prohibited for the sole purpose of increasing the body weight of bovines.
In light of the national control plans within the European Union, urine analyses are of critical importance in monitoring illegal administration of glucocorticoids. Recently, the European Commission reported in the Commission Staff Working Document ‘Implementation of national residue monitoring plans in the member states’ (2009–2012) an increasing occurrence of prednisolone residues (3.12–179.72 μg L−1) in bovine urine samples without any direct evidence for illegal administration. A number of hypotheses have been suggested to explain this specific finding, i.e. prednisolone could be generated by physiologic metabolic processes under influence of stress, which resulted in higher cortisol levels (during transport and slaughtering) (Pompa et al. 2011; Ferranti et al. 2011; Vincenti et al. 2012) or by faecal microbial contamination of urine (Arioli et al. 2010). The latter hypothesis arose from the close structural relationship of prednisolone to cortisol, only differing by one double bond on ring A at the C1–C2 position, which has recently been evidenced by de Rijke et al. (2014). Indeed, in vitro incubation experiments of cortisol with bovine liver enzymes showed a significant decrease of cortisol together with formation of prednisolone within 6 h. To take into account potential other origins for prednisolone at concentration levels below 5 μg L−1, European Reference Laboratories suggested a threshold level for prednisolone in bovine urine of 5 μg L−1 (ADVIES07-2013; de Rijke et al. 2014).
At this time, a method for direct discrimination between endogenous and exogenous prednisolone has not yet been established. One powerful and promising strategy could be the use of isotope ratio mass spectrometry (IRMS). This has already been applied to differentiate synthetic testosterone and estradiol from natural hormones in urine, by considering their inherent and different 13C/12C ratios (de la Torre et al. 2001; Buisson et al. 2005; Bulska et al. 2014). However, some significant disadvantages such as low sensitivity and labour-intensive clean-up are associated with this technique (Blokland et al. 2012). A second promising approach would be an untargeted metabolomics strategy, aiming at the identification of potential biomarkers that allow discrimination between endogenous formation and exogenous administration of prednisolone. This biomarker could, after evaluation, be implemented in the frame of national control plans as screening method (EURL 2014). The use of full-scan methods, preferably by means of high resolution mass spectrometry (HRMS), such as time-of-flight (TOF) (Wicklund et al. 2008), Fourier Transform Ion Cyclotron Resonance (Marshall 2000) or Fourier Transform Orbitrap MS (Pinel et al. 2011; Van Meulebroek et al. 2014), is highly encouraged for such biomarker investigations. Indeed, by screening biological samples with full-scan HRMS, a virtually unlimited number of compounds can be analyzed simultaneously and retrospective post-acquisition evaluation of data can reveal unidentified and/or unknown metabolites (Van Meulebroek et al. 2014). The identification of these metabolites is currently seen as the major bottleneck in the interpretation of metabolomics experiments (Neumann and Böcker 2010). In this regard, MS/MS or fragmentation data may enclose valuable identification potential. To obtain these types of data (i.e. both accurate molecular mass and fragmentation pattern) within a single analytical run, hybrid HRMS instruments like Q-Orbitrap-MS and Quadrupole Time-of-Flight-MS are most designated (Neumann and Böcker 2010; Quintás et al. 2012; Jia et al. 2014; Díaz et al. 2014). For further identity confirmation, it is recommended to validate the structure candidates by nuclear magnetic resonance spectroscopy (NMR) (Tardieu et al. 2010; Soininen et al. 2014) or by matching the retention time and mass spectra with those of authentic reference standards (Sumner et al. 2007), although these are of course not readily available for every compound (Scalbert et al. 2009).
The aim of this work was to reveal potential biomarkers for exogenous prednisolone administration in bovine urine by using a metabolomic fingerprinting approach. To this extent, an in vivo study was conducted with 12 adult cows that subsequently were subjected to a growth-promoting treatment (low dosage long-term) and a therapeutic treatment (high dosage short-term) with prednisolone. Collected urine samples were analyzed by both full-scan UHPLC-Orbitrap-MS and UHPLC-QqTOF-MS to acquire the specific metabolic fingerprints corresponding to the different prednisolone treatments. Next, multivariate analysis by means of orthogonal-partial least squares-discriminant analysis (OPLS-DA) was employed to search for differentiating metabolites linked to exogenous prednisolone administration. After metabolite discovery, it is necessary to evaluate the classification performance of the newly defined compounds (Parikh and Philbrook 2014). The performance of the newly obtained metabolites was defined by sensitivity and specificity (Adams et al. 1993; McLoughlin et al. 2000). In addition, the urinary excretion kinetics of the revealed biomarkers were monitored after a single therapeutic per os dose of prednisolone (Verhagen et al. 2004). Insights in these metabolites’ kinetics could indeed be of special interest to extend the detection period of illegal prednisolone abuse, i.e. when the detection limit of prednisolone is more rapidly reached than that of the biomarker(s).
2 Experimental
2.1 Animals
In this study, a diverse group of cattle was compiled in order to include the potential influence of biological variation on the specific biomarker identification. More specifically, twelve healthy cows of a mixed breed, varying age (2–6 years) and with a body weight between 360 and 570 kg were housed for 8 months under controlled experimental conditions in the animal facilities of the Centre d’Economie Rurale (CER) (Marloie, Belgium). The animals were fed a commercially available diet, commonly applied in zootechnical practice, with ad libitum access to water and hay. During the entire study, animals were kept in three separate groups (4 animals per group) and all housed in a half covered pen. Prior to the in vivo study, an initial acclimatization period of 14 days was considered. In order to consider animal growth during the in vivo study, the animals were weighted at the start and at the end of the experiment, but no significant differences were observed. The in vivo study was approved by CER’s Ethical Committee (CE/Sante/ET/004).
2.2 Experimental protocol
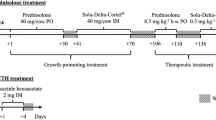
After the acclimatization period, which served as control, all animals underwent the same sequential prednisolone treatments: a growth-promoting treatment (long-term, 40 mg/cow/day) followed by a therapeutic treatment (short-term, 0.5 mg kg−1 b.w./day) (Fig. 1). The specific dosages were based on literature findings (Cannizzo et al. 2011; Ferranti et al. 2011; Vincenti et al. 2009) to ensure relevant levels of prednisolone and potential metabolites in urine.

Schematic representation of the in vivo study, indicating the various experimental sections with oral (PO) and intramuscular (IM) prednisolone administrations and their duration. This experimental protocol was specifically executed to each individual animal (n = 12)
The growth-promoting treatment started with 30 consecutive days of oral administration of 40 mg/day of prednisolone (Sigma-Aldrich St. Louis, USA) (Day + 1 till Day + 30) (PO1), followed by a washout period of 10 days (WO1). Next, intramuscular injections of 40 mg/day of Solu-Delta-Cortef® (prednisolone sodium succinate, Zoetis, Zaventem, België) were given for 30 consecutive days (Day + 41 till Day + 70) (IM1). Before the start of the therapeutic prednisolone treatment, a washout period of 35 days was considered (WO2).
During the therapeutic prednisolone treatment, a similar experimental set-up was implemented. First, the animals received 0.5 mg kg−1 b.w. of prednisolone per os for 5 days (Day + 106 till Day + 110) (PO2), which was followed by a washout period of 25 days (WO3). Next, intramuscular injections of 0.5 mg kg−1 b.w. of Solu-Delta-Cortef® were administered during 5 consecutive days (Day + 136 till Day + 140) (IM2). A final washout period of 32 days (Day + 141 till Day + 172) was respected (WO4).
During the periods of oral administration, one capsule containing the appropriate amount of prednisolone, using lactose as excipient, was given in the morning just after feeding, using a capsule launcher. The intramuscular injections were placed in the neck and were alternated every day from the left to the right side in order to minimize irritation.
2.3 Sample collection
Urine samples were collected in the morning, 5 min before prednisolone administration, by a veterinarian using a probe (to prevent faecal contamination), immediately portioned into 15 mL tubes, and stored in the dark at −80 °C until analysis (De Clercq et al. 2013). As for the sampling rate, during the acclimatization period, urine samples were collected daily. During the therapeutic and growth-promoting prednisolone treatments and washout periods, samples were collected every 5 days.
2.4 Reagents
Standards of prednisolone, prednisone, cortisone, cortisol, 20β-dihydrocortisone and methylprednisolone were purchased from Sigma-Aldrich (St. Louis, USA). Standards of 20α-dihydroprednisolone and 20β-dihydroprednisolone were from Steraloids (Rhode Island, USA). Internal standards were cortisol-d4 (Sigma-Aldrich) and prednisolone-d8 (TRC, Canada). Primary stock solutions were prepared in ethanol at a concentration of 200 μg mL−1 and stored in dark glass bottles at −20 °C. Working solutions were made in ethanol at a range of 0.1–10 μg mL−1.
Reagents were of analytical grade when used for extraction purposes and obtained from VWR International (Merck, Darmstadt, Germany). For UHPLC-HRMS applications, reagents were of LC–MS Optima grade and obtained from Fisher Scientific (Loughborough, UK). Ultrapure water was obtained by usage of a purified-water system.
2.5 Sample preparation
A detailed description of the analytical procedure for extraction and purification of urine samples has been described by De Clercq et al. (2013). In brief, a 5 mL aliquot of urine was spiked with internal standards (cortisol-d4 and prednisolone-d8) to obtain final concentration levels of 10 μg L−1. Next, a twofold liquid–liquid extraction with pure tert-butyl methylether was performed. The organic phases were collected, pooled and dried under a gentle stream of nitrogen at a temperature of 50 °C. The residue was dissolved in 100 μL solvent, reflecting the initial mobile phase conditions, and transferred to a vial for UHPLC-HRMS analysis.
2.6 Analytical detection
2.6.1 UHPLC-Orbitrap-MS
Untargeted analysis of urine samples was performed by UHPLC-Orbitrap mass spectrometry, according to the validated method of De Clercq et al. (2013). Chromatographic separation was thereby achieved on an Accela UHPLC system (Thermo Fisher Scientific, San José, USA), equipped with a Nucleodur Isis C18 column (1.8 μm, 100 mm × 2 mm, Macherey–Nagel, Düren, Germany). The binary solvent system consisted of 0.1 % aqueous formic acid (A) and 0.1 % formic acid in acetonitrile (B). The applied gradient program and other chromatographic parameters are presented in the supplementary table. High-resolution mass spectrometric analysis was performed on an Exactive™ single-stage Orbitrap mass spectrometer (Thermo Fisher Scientific, San José, USA), equipped with a heated electrospray ionization probe (HESI-II), operating in the polarity switching mode. Instrument control and data processing were carried out by Xcalibur 2.1 software (Thermo Fisher Scientific, San José, USA).
2.6.2 UHPLC-QqTime-of-Flight
For structure elucidation of the differentiating metabolites, urine samples were also analysed by UHPLC-QqTOF-MS. This hybrid system enables full-scan HRMS analysis in combination with HRMRM like fragmentation (MRMHR). The TripleTOF® 4600 mass analyser (SCIEX, California, USA) was coupled to a UHPLC UltraLC 100-XL system (SCIEX), consisting of an Eksigent pumping system, autosampler and degasser. The parameters of the chromatographic separation, using a Nucleodur C18 Isis column (1.8 μm, 100 mm × 2 mm, Macherey–Nagel), are summarized in the supplementary table.
The TripleTOF® 4600 mass analyser was equipped with a DuoSpray™ source, comprising an electrospray (TIS) as well as an atmospheric pressure chemical ionization (APCI) inlet. Ionization of the compounds was achieved using the electrospray inlet (TurboIonSpray® TIS) and operated separately in the positive and negative ion mode. For every 10 samples, automated calibration was performed using an external calibrant delivery system (CDS), which infuses calibration solution prior to sample analysis.
The mass spectrometer was operated in the information-dependent acquisition (IDA) mode, combining full-scan TOF-MS survey and MS/MS experiments. During the full-scan survey, spectral data about all ions, present within the selected m/z scan range of 150–650 Da, were acquired for each scan. In addition, for every scan the fragmentation profiles of the top five abundant ions were generated by MS/MS. However, ions that occurred seven consecutive times within the top five of most abundant ions were excluded for MS/MS experiments for 1 s. More details can be found in the supplementary table. Instrument control was carried out by Analyst® TF 1.6 software (SCIEX, California, USA).
2.6.3 1H-NMR
The NMR spectra of the revealed metabolite biomarker were measured on an Avance II Bruker spectrometer operating at a 1H frequency of 700 MHz and equipped with a 1-mm 1H/13C/15N TXI-z probe to allow maximum sensitivity, taking into account the limited amount of available sample. The dried sample (<10 μg) was dissolved in ±10 µL MeOD-d4 (99.96 % D) in order to minimize potential signal interferences from the solvent. All spectra were referenced to the residual solvent-signals at 3.31 (5) ppm for the 1H frequency.
2.7 Chemometric data analysis
In this study, the general workflow of data acquisition and analysis can be organized into multiple steps. A first step relates to the extraction of urine samples. Due to the large number of collected urine samples, sample preparation and analysis were performed in subsequent batches of four animals. The first batch considered of the urine samples of animals 1, 5, 7 and 12. The second batch contained the urine samples of animals 2, 4, 6 and 11, and the last batch included urine of animals 3, 8, 9 and 10. Each batch was first analyzed on the Exactive™ and later on reanalyzed on the TripleTOF® 4600. Samples were analysed in a random order during each batch. As such, both full-scan data for metabolomic fingerprinting and MS/MS fragmentation patterns for identification were acquired. Instrumental stability (quality control measure) during mass spectrometric analyses was verified by considering standard injections. These injections were run at the beginning and repeated every 25 samples. This mixture consisted of cortisol, cortisone, dihydrocortisone, prednisolone, prednisone, methylprednisolone, 20α-dihydroprednisolone and 20β-dihydroprednisolone. The following average coefficients of variance (n = 162) were obtained for the various glucocorticoids: 6.39 % for cortisol, 5.51 % for cortisone, 6.79 % for dihydrocortisone, 3.09 % for prednisolone, 4.35 % for prednisone, 8.93 % for methylprednisolone, 7.61 % for 20α-dihydroprednisolone and 6.94 % for 20β-dihydroprednisolone. Based on the manuscript of Shah et al. (2000), the acceptance criterion to ensure system stability was set at 15 %. Since all absolute and average CVs were well below 15 %, appropriate stability during analysis was concluded. In addition, relative retention time changes during analysis were <1 % (i.e. <0.04 min).
The second step involved data pre-processing (peak list generation), thereby using either Sieve™ 2.1 (Thermo Fisher Scientific, San José, USA) or MarkerView™ 1.2.1.1 (SCIEX, California, USA) software, to respectively process Exactive™ (raw.files) and TripleTOF® 4600 (wiff.files) data files. Taking into account that both full-scan data sets enclosed information about both positively and/or negatively charged ions, it was decided to handle both ionization modes separately during peak list generation (Van Meulebroek et al. 2014; Hjerpsted et al. 2014). Applied parameter values included an m/z-range of 150–650 Da, a m/z width of 5 ppm, a retention time window ranging from 1 to 9.5 min, a peak intensity threshold of 100,000 arbitrary units, a maximum peak width of 0.5 min and a maximum number of 10,000 frames. Furthermore, corrections for inherent chromatographic variability along samples were made during the peak alignment process. This strategy rendered a list of features (detected ions) that were characterized by their m/z-value, retention time and signal abundance. The signal intensities of these ions were for each sample normalized by the average signal intensity of the two deuterium labeled internal standards i.e. cortisol-d4 and prednisolone-d8, which were supplemented prior to extraction. Moreover, since urine is a matrix subjected to potential dilution effects, a secondary normalization was implemented. As suggested by Jacob et al. (2014), normalization by means of specific gravity (Pocket Refractometer™, Atago, Tokyo) based on the Levine–Fahy equation (Levine and Fahy 1945) (correction factor ranged from 1.003 to 1.0044) was implemented.
In the final step, multivariate statistical analysis was performed by means of SIMCA™ 13 software (Umetrics, Malmö, Sweden) in order to reveal significant differences between the metabolic fingerprints, associated with the various treatments. For this particular purpose, OPLS-DA was implemented, for revealing metabolite ions with discriminating power towards the samples’ class membership. Prior to this OPLS-DA modelling, data were log-transformed and Pareto scaled (1/√SD, where SD is the standard deviation) to induce normality and to standardize the range of independent X-variables, respectively (van den Berg et al. 2006). Within the applied modelling strategy, a qualitative relationship between the X-matrix (detected metabolite ions) and the dependent Y-variable (prednisolone treatment samples ‘Treatment’ or control urine samples ‘Control’) was established. In this regard, the Y-variable was defined as a qualitative variable, representing the class membership (Theodoridis et al. 2012; Wold et al. 2001; Matthew and William 2003). The advantage of OPLS compared to conventional PLS relates to the applied model rotation whereby class separation is found in the first predictive component (correlated variation) and variation that is not correlated with class separation is found in the orthogonal components (orthogonal variation) (Trygg and Wold 2002). Model-validity was verified by performing a sevenfold internal cross validation-analysis of variance (CV-ANOVA, p value <0.01) (Jung et al. 2011), permutation testing, and considering three model characteristics (R2(X), R2(Y) and Q2(Y)). The first model characteristic corresponds to the predictive and orthogonal variation in X that is explained by the model, the second characteristic defines the total sum of variation in Y that is explained by the model and the third refers to the goodness of prediction, calculated by full cross-validation (Eriksson et al. 2007; Jung et al. 2011). Response permutation testing was performed to estimate the significance of the generated models, whereby the order of elements in the Y-vector was randomly permutated 100 times (Eriksson et al. 2007).
2.8 Preliminary biomarker validation
After discovery of differentiating metabolites, it is necessary to evaluate the performance and usefulness of the defined compounds (Koulman et al. 2009) whereby a distinction should be made between analytical method validation and clinical qualification. Validation is defined as the process of assessing the biomarker and its measurement performance characteristics, and determining the range of conditions under which the biomarker will give reproducible and accurate data (Lee et al. 2006; Wagner 2002). While clinical qualification is the evidentiary process of linking a biomarker with biological processes and clinical endpoints (Wagner 2002). Both processes are intertwined since the aim is to link the biomarker with its intended use (Chau et al. 2008). In this study, the selectivity, bio-analytical performance, and urinary excretion kinetics of the proposed marker molecules were included as preliminary validation. Statistical analysis was performed using SPSS™ statistics 21.0.
2.9 Identification of biomarkers
After evaluation of the relevant biomarkers, various identification steps were undertaken, thereby using the TripleTOF® 4600 full-scan MS and MS/MS spectra. In first instance, an in silico based strategy was applied. Elemental compositions were thereby determined using Formula Finder, a tool within MasterView™ (SCIEX), and based on precursor mass, fragment masses and isotopic pattern. The elements that were allowed for formula prediction were restricted to the basic elements of natural metabolites, i.e. hydrogen (H), carbon (C), oxygen (O), nitrogen (N) and sulphur (S). To this extent, a mass deviation window of 5 ppm was allowed. Subsequently, chemical formulas were screened towards an in-house database, which comprises the elemental composition of 1693 steroidal compounds (based on the 11th catalogue edition of steroids from Steraloids inc.). Next, structural elucidation was pursued by applying a combinatorial-based prediction strategy, thereby using the MetFrag software tool (Wolf et al. 2010). Within this approach, the acquired fragmentation spectrum of an ion is matched towards theoretically predicted fragments of candidate structures from public compound libraries (ChemSpider and PubChem), which allows to rank candidate chemical structures. A second identification strategy was based on compound purification (preparative chromatography) and subsequent 1H-NMR analysis.
3 Results and discussion
3.1 Peak list generation
Peak list generation using Orbitrap-MS data resulted in a metabolic fingerprint, which enclosed 9952 positively charged and 9494 negatively charged ions. However, by excluding the 13C containing ion species, the fingerprint was reduced to only 6637 positively and 5626 negatively charged monoisotopic ions. The same strategy was applied to the QqTOF-MS data and yielded a metabolic fingerprint, which comprised 5085 positive and 8036 negative monoisotopic ion species.
3.2 Predictive modeling
The acquired data matrices were normalized and reorganized into a control group (Control, n = 120) and a treatment group (Treatment, n = 216), which comprised all urine samples that were either collected prior to or during prednisolone treatment. It should hereby be noted that all types of prednisolone administration (per os vs. intramuscular) and all different treatment strategies (growth-promoting vs. therapeutic, long- vs. short-term) were combined into the same ‘Treatment’ group. This was to guarantee that differentiating metabolites were relevant for exogenous prednisolone treatment across administration routes. The multivariate statistical analysis, which aimed to reveal such metabolites, will first be discussed for the data set that originated from the Orbitrap mass spectrometric analysis.
OPLS-DA models were separately constructed for the negatively and positively charged ions (Supplementary Fig. 1) and were each time evaluated through various validation strategies, i.e. CV-ANOVA (p < 0.01), permutation testing, and three model characteristics (R2(X), R2(Y), and Q2(Y)). With respect to the latter strategy, the following parameter values were obtained for the constructed OPLS-DA models (based on either the positive or negative ions, respectively): 0.408, 0.993 and 0.894, and 0.598, 0.977 and 0.782. In this context, good model predictability was assumed if R2(Y) is ≥0.5 (Hawkins et al. 2003; Jung et al. 2011). As such, based on the various validation strategies, overall good model quality was concluded. Next, to reveal the significance of particular ions to discriminate between the control and treatment class, an S-plot was constructed (Fig. 2a). Hereby, the x-axis indicates the contribution (covariance p1) of a particular ion towards class separation whereas the y-axis refers to the correlation (p(corr)) between samples and thus reliability of results (Wicklund et al. 2008; Xue et al. 2012). In this regard, ions with cut-off values of |p1| ≥ 0.05 and |p(corr)| ≥ 0.05 were considered as differentiating metabolites (Xue et al. 2012). The S-plot results were integrated with the VIP-scores, which indicate the relative importance of the ions to discriminate between classes (Dietmair et al. 2012). In this study, only ions with a VIP Score >3 were considered as potentially relevant. As such, 19 positively charged ions and 11 negatively charged ions were eventually retained and considered as differentiating metabolites, allowing to discriminate between endogenous and exogenous prednisolone. In order to determine which of these compounds could serve as an actual biomarker for xenobiotic prednisolone treatment, additional criteria were taken into consideration, i.e. the chromatographic performance (peak shape As ≤ 1.5) (Snyder et al. 1997), signal-to-noise ratio (≥3) (CD 2002/657/EC) and points over the peak (≥15) (Churchwell et al. 2005). Based on these criteria, 3 positively ionized metabolites were retained and all other metabolite ions excluded. These retained metabolite ions were more strongly detected in the samples that were collected from prednisolone treated animals (Table 1).

Loading S-plot representing the leading contribution of positive (A 1 , B 1 ) and negative (A 2 , B 2 ) ionized metabolite ions, respectively determined with Sieve™ (A) and MarkerView™ (B). Ions with VIP ≥ 3 and |p1| ≥ 0.05 and a |p(corr)| ≥ 0.05 are in black. Depending on the position in the S-plot and the associated p- and p(corr)-values, an ion was significant important towards ‘Treatment’ (upper, right part of the plot) or towards ‘Control’ (lower, left part of the plot)
The same multivariate strategy was applied to the dataset obtained from QqTOF mass analysis. Quality of the established OPLS-DA models was evaluated as described before, considering permutation testing, CV-ANOVA, and three model characteristics. For the constructed models, values for R2(X), R2(Y) and Q2(Y) were 0.371, 0.973 and 0.860 (for the positive ions) and 0.204, 0.877 and 0.665 (for the negative ions). The latter R2(X) values are rather low, which is explained by the fact that all prednisolone treatment data were combined, resulting in a higher variation. As such, overall good model quality was concluded. Interpretation of the data by means of S-plot and VIP-scores, thereby using the previously proposed criteria, yielded 16 positively and 10 negatively ion metabolites (Fig. 2b). A further exclusion of ions was established by applying the additional peak performance criteria, listed above, whereby only two positively charged ions were retained (Table 1).
Independently of the dataset used for generation of the peak lists, the multivariate data analysis assigned in both cases the same ion (i.e. metabolite biomarker) with the highest correlation to the exogenous prednisolone treatment. This particular ion (further referred to as metabolite 1) was characterized by an m/z-value of 283.1693 Da and a retention time ranging from 1.37 to 1.41 min. In total, four differentiating metabolites were retained.
3.3 Preliminary validation of suggested marker molecules
In literature, numerous metabolites have been claimed as biomarkers for discriminating exogenous treatment with e.g. growth promoters as opposed to natural prevalence. For example, 19-noretiochlanolone (Scarth et al. 2010) and 5α-estrane-3β,17α-diol (Pinel et al. 2010) have been revealed as biomarkers of nortestosterone treatment in porcines and bovines, respectively. However, the differentiating metabolites are in general rarely validated due to the absence of consistent validation guidelines, in essence needed to assign a metabolite the biomarker label (Koulman et al. 2009). Indeed, although various statistical methods and their limitations are described, no rigorous procedures or criteria are available to evaluate and validate biomarkers, required to endorse their widespread acceptance (Hunter et al. 2010; Puntmann 2009; Regal et al. 2013; Gika et al. 2014; Behrens et al. 2014). Upon further inclusion of data from multiple independent validation experiments, the proposal of a threshold as was earlier described by Pinel et al. (2015) and Kaabia et al. (2013) would be recommended. However, setting a threshold based on our preliminary validation data was considered to early. It is generally agreed that a cross-validation experiment, which includes the independent validation of the biomarker by replicating the experiment at different sites, gives high inter-observer and intra-observer reproducibility (Puntmann 2009; Koulman et al. 2009; Pinel et al. 2015). This was unfortunately not possible in light of the experimental complexity of this work. Therefore, a preliminary validation strategy was followed in this study comprising bio-analytical assessment, selectivity evaluation and evaluating urinary excretion kinetics (Hunter et al. 2010; Puntmann 2009; Regal et al. 2013; Gika et al. 2014; Behrens et al. 2014).
3.3.1 Bioanalytical assessment
Sensitivity and specificity of the four revealed differentiating metabolites are considered of critical importance in gauging their validity as markers for exogenous (per os and intramuscular) administration of prednisolone (Weiss et al. 1985; McLoughlin et al. 2000; Matabosch et al. 2013). Hereby, sensitivity indicates the true positive rate, i.e. the biomarker’s presence during treatment, whereas specificity relates to the true negative rate, i.e. the biomarker’s absence during control. In this study, sensitivity was determined by assessing the presence of each marker metabolite in the urine samples, obtained during the different prednisolone treatments (Treatment) (Table 2), whereby only metabolites 1 and 4 were assigned a total sensitivity of >90 %. Specificity was determined in the urine samples that were collected during the acclimatization period (Control) and wash-out periods (WO) between the different prednisolone treatments. The first time point of the wash-out periods was excluded due to the unknown elimination kinetics of the differentiating metabolites. Metabolites 1, 2, and 3 were effectively absent in more than 90 % of the urine samples, collected outside the prednisolone treatment periods, which indicates a low chance of false positive findings (Matsui 2006).
The sensitivity and specificity of each ion were subsequently visualized using receiver-operating characteristic (ROC) curves and its summary index, i.e. the area under curve (AUC) (Table 2). Normally, the AUC ranges from 0.5 (the area under the diagonal line, representing the extent of class discrimination based on random chance) to 1 (perfect discrimination) (Pepe et al. 2004). In this study, the AUC obtained for metabolite 1 indicates perfect discrimination, whereas for the other metabolites excellent discrimination could be achieved (Parikh and Philbrook 2014).
The odds ratio (Grund and Sabin 2010) was determined to quantify how strong the presence of each biomarker was correlated with prednisolone treatment. In this context, a high odds ratio indicates a strong correlation. The odds ratio of metabolite 1 was much higher than the odds ratios of the other metabolites (Table 2), which evidenced the strong correlation between this biomarker and prednisolone treatment (Grund and Sabin 2010).
In conclusion, based on the outlined parameters, metabolite 1 is attributed the highest potential to serve as a reliable biomarker for exogenous prednisolone administration.
3.3.2 Selectivity
To verify that newly defined differentiating metabolites for prednisolone treatment were not present in urine that contained endogenous prednisolone, 12 animals (cfr. Sect 2.1) were intramuscularly injected with 2 mg tetracosactide hexaacetate (Utrecht University, Faculty of Veterinary Medicine), a synthetic analogue of adrenocorticotropic hormone (ACTH), corresponding to 200 I.U. of ACTH (Pompa et al. 2011). After 4 h, prednisolone was detected in all urine samples, thereby reaching concentration levels that were similar to those obtained during growth-promoting treatments. However, none of the four defined differentiating metabolites were present in these samples, endorsing the ability of these particular metabolites to distinguish between exogenously administered (per os and intramuscular) and endogenously synthesized prednisolone (Bertocchi et al. 2013; Ferranti et al. 2013).
3.3.3 Urinary excretion kinetics
By achieving proper insights into the urinary kinetic profiles, the actionable sensitivity in terms of detection window and screening capacities of the proposed biomarkers could be confirmed (Verhagen et al. 2004; Goniewicz et al. 2009; Pinel et al. 2015). More specifically, an additional in vivo experiment was performed in which a single cow (milking cow, 3.5 years, 550 kg body weight) received a single dose of 0.5 mg prednisolone per kg−1 b.w. and per os. Urine was collected at different time points during the first 32 h (4 h–6 h 30 min–10 h–21 h–24 h–26 h 30 min–29 h 30 min–31 h 15 min) after administration and alterations in the peak area of the marker metabolites and prednisolone were monitored. Additional urine samples were collected 4 and 7 days after prednisolone administration (Fig. 3). During this experiment, all metabolites were detected, whereby metabolite 1 was five times more abundant (in terms of peak area) in comparison with the other metabolites. Moreover, metabolite 1 was still present at detectable concentration levels (S/N > 3) at the moment that prednisolone was no longer detected in the collected urine samples, i.e. until 4 days after treatment. This specifically endorses the suitability of metabolite 1 as an actionable biomarker for exogenous exogenous prednisolone administration. However, metabolites 2, 3, and 4 may fulfill a supporting role in determining the origin of prednisolone in non-compliant bovine urine samples.

Urinary metabolite excretion profile after administration of a single dose of 0.5 mg/kg body weight prednisolone per os (indicated with arrow) to one milking cow, with Metabolite 1 (a); Metabolite 2 (b); Metabolite 3 (c); Metabolite 4 (d) and Prednisolone (e). Results are expressed as peak area
3.4 Tentative identification of relevant biomarkers
The elemental composition of the four revealed biomarkers was determined by means of FormulaFinder (MasterView™), thereby integrating the information about the precursor accurate mass, fragment masses and isotopic patterns. Together with the maximally allowed mass deviation of 5 ppm, only one candidate chemical formula (Table 3) was suggested for each biomarker and thus used for subsequent structural assessment. For this particular purpose, the experimental MS/MS spectra from each metabolite ion were matched towards the in silico predicted fragmentation pattern from candidate chemical structures, corresponding to the determined chemical formula. These candidate structures were retrieved from publically available databases (i.e. Metlin, PubChem and ChemSpider) and our in-house database. The respective use of MetFrag and MasterView™ software allowed to retain a ‘best fitting’ candidate structure (Table 3) to the degree of matching fragmentation profiles.
As metabolite 1 was proposed as the most potent biomarker for exogenous prednisolone administration, additional efforts were made to reach the highest level of identification (Sumner et al. 2007). Since no authentic reference standard was available for the candidate structure, 1H-NMR analysis was attempted to confirm the metabolite’s identity. For this reason, all urine samples collected during prednisolone treatments were pooled and preparative HPLC was performed in order to obtain a relatively pure fraction of the compound. Unfortunately, 1H-NMR analysis was not able to reveal the compound’s structure, which was mainly due to the available low absolute quantities (<10 μg) that could be collected for this compound.
For now, all revealed biomarkers were putatively annotated (Table 3), thereby reaching the second highest level of identification, as defined by Sumner et al. (2007) since a standard for identification at the highest level of confidence is lacking. Besides, the physiological activity and as a result the link to prednisolone administration of the newly defined compounds could not be demonstrated yet, because no literature about these compounds was found. The only retrieved background information comprised the identity of the compounds in online databases Chemspider and/or PubChem.
4 Conclusion
The aim of this study was to identify biomarkers, which have the ability to discriminate between endogenous formation and exogenous administration of the synthetic glucocorticoid prednisolone. A strategy of metabolic fingerprinting was performed to assess potential metabolite perturbations in the urine of cows, treated with prednisolone. Four metabolite ions were found to emerge during prednisolone treatment. The selectivity of the markers was proved, since none of these compounds were present in urine containing endogenous prednisolone, of which the formation was induced by ACTH administration. Besides, biological relevance of these ions was determined by means of sensitivity and specificity. This showed that only one metabolite was highly suitable as biomarker during growth-promoting and therapeutic prednisolone treatment, reflected by 93.4 % sensitivity and 96.3 % specificity. The urinary excretion profiles of the four metabolites were considered as an additional criterion. The most potent compound could be detected up to 4 days after a single per os prednisolone administration. The identity and qualification as fit-for-purpose of the proposed biomarker ‘Metabolite 1’ needs to be further explored through independent data sets that cover a larger population, different ages, sex, origin of feeding, etc. Moreover, further validation of the proposed biomarker to confirm its specificity for prednisolone treatment as opposed to treatment with other glucocorticoids or growth promoters should be performed.
References
Adams, J. E., Bodor, G. S., Dávila-Román, V. G., et al. (1993). Cardiac troponin I. A marker with high specificity for cardiac injury. Circulation, 88(1), 101–106.
Arioli, F., Fidani, M., Casati, A., Fracchiolla, M. L., & Pompa, G. (2010). Investigation on possible transformations of cortisol, cortisone and cortisol glucuronide in bovine faecal matter using liquid chromatography-mass spectrometry. Steroids, 75(4), 350–354.
Behrens, T., Bonberg, N., Casjens, S., Pesch, B., & Brüning, T. (2014). A practical guide to epidemiological practice and standards in the identification and validation of diagnostic markers using a bladder cancer example. Biochimica et Biophysica Acta, 1844, 145–155.
Bertocchi, L., Dusi, G., Ghidelli, V., et al. (2013). Investigation on the origin of prednisolone in urine and adrenal glands of cows. Food Additives & Contaminants. Part A, Chemistry, Analysis, Control, Exposure & Risk Assessment, 30(6), 1055–1062.
Blokland, M. H., Van Tricht, E. F., Van Rossum, H. J., Sterk, S. S., & Nielen, M. W. F. (2012). Endogenous steroid profiling by gas chromatography-tandem mass spectrometry and multivariate statistics for the detection of natural hormone abuse in cattle. Food Additives and Contaminants. Part A, Chemistry, Analysis, Control, Exposure and Risk Assessment, 29(7), 1030–1045.
Buisson, C., Hebestreit, M., Weigert, A. P., et al. (2005). Application of stable carbon isotope analysis to the detection of 17beta-estradiol administration to cattle. Journal of Chromatography A, 1093(1–2), 69–80.
Bulska, E., Gorczyca, D., Zalewska, I., Pokrywka, A., & Kwiatkowska, D. (2014). Analytical approach for the determination of steroid profile of humans by gas chromatography isotope ratio mass spectrometry aimed at distinguishing between endogenous en exogenous steroids. Journal of Pharmaceutical and Biomedical Analysis,. doi:10.1016/j.jpba.2014.11.017.
Cannizzo, F. T., Capra, P., Divari, S., Ciccotelli, V., Biolatti, B., & Vincenti, M. (2011). Effects of low-dose dexamethasone and prednisolone long term administration in beef calf: Chemical and morphological investigation. Analytica Chimica Acta, 700, 95–104.
Chau, C. H., Rixe, O., McLeod, H., & Figg, W. D. (2008). Validation of analytical methods for biomarkers employed in drug development. Clinical Cancer Research, 14(19), 5967–5976.
Churchwell, M. I., Twaddle, N. C., Meeker, L. R., & Doerge, (2005). Improving LC-MS sensitivity through increases in chromatographic performance: comparisons of UPLC-ES/MS/MS to HPLC-ES/MS/MS. Journal of Chromatography B. Analytical Technologies in the Biomedical and Life Sciences, 825(2), 134–143.
Commission Decision 2002/657/EC (2002). Official Journal of the European Community L221:8. Retrieved February 12, 2015 from http://eur-lex.europa.eu/LexUriServ/LexUriServ.do?uri=OJ:L:2002:221:0008:0036:EN:PDF.
Commission Decision 96/23/EC (1996). Official Journal of the European Community, L125, 10-32. Retrieved February 12, 2015 from http://eur-lex.europa.eu/legal-content/NL/TXT/PDF/?uri=CELEX:31996L0023&from=EN.
Commission Regulation 37/2010 (2010). Official Journal of the European Community, No. L15/1, 11, 26, 47 and 56. Retrieved February 12, 2015 from http://ec.europa.eu/health/files/eudralex/vol-5/reg_2010_37/reg_2010_37_en.pdf.
Commission staff working document on the implementation of national residue monitoring plans in the member states in 2009 (2009). Brussel, Belgium. Retrieved February 12, 2015 from http://ec.europa.eu/food/food/chemicalsafety/residues/workdoc_2009_en.pdf.
Commission staff working document on the implementation of national residue monitoring plans in the member states in 2010, Brussel, Belgium. . Retrieved February 12, 2015 from http://ec.europa.eu/food/food/chemicalsafety/residues/workdoc_2010_en.pdf.
Commission staff working document on the implementation of national residue monitoring plans in the member states in 2011, Brussel, Belgium. . Retrieved February 12, 2015 from http://ec.europa.eu/food/food/chemicalsafety/residues/docs/workdoc_2011_en.pdf.
Commission staff working document on the implementation of national residue monitoring plans in the member states in 2012, Brussel, Belgium. . Retrieved February 12, 2015 from http://ec.europa.eu/food/food/chemicalsafety/residues/docs/workdoc_2012_en.pdf.
De Clercq, N., Vanden Bussche, J., Croubels, S., Delahaut, P., & Vanhaecke, L. (2013). A validated analytical method to study the long-term stability of natural and synthetic glucocorticoids in livestock urine using ultra-high performance liquid chromatography coupled to Orbitra-high resolution mass spectrometry. Journal of Chromatography A, 1301, 111–121.
de la Torre, X., González, J. C., Pichini, S., Pascual, J. A., & Segura, J. (2001). 13C/12C isotope ratio MS analysis of testosterone, in chemicals and pharmaceutical preparations. Journal of Pharmaceutical and Biomedical Analysis, 24(4), 645–650.
de Rijke, E., Zoontjes, P. W., Samson, D., Oostra, S., Sterk, S. S., & van Ginkel, L. A. (2014). Investigation of the presence of prednisolone in bovine urine. Food Additives and Contaminants. Part A, Chemistry, Analysis, Control, Exposure and Risk Assessments, 31(4), 605–613.
Díaz, R., Pozo, O. J., Sancho, J. V., & Hernández, F. (2014). Metabolomic approaches for orange origin discrimination by ultra-high performance liquid chromatography coupled to quadrupole time-of-flight mass spectrometry. Food Chemistry, 157, 84–93.
Dietmair, S., Hodson, M. P., Quek, L. E., Timmins, N. E., Gray, P., & Nielsen, L. K. (2012). A multi-omics analysis of recombinant protein production in Hek293 cells. PLoS One, 7(8), 1–15.
Eriksson, L., Trygg, J., & Wold, S. (2007). CV-ANOVA for significance testing of PLS and OPLS® models. Journal of Chemometrics, 22(11–12), 594–600.
EURL (European Union Reference Laboratory). (2014). EURL Reflection paper: Natural growth promoting substances in biological samples. Presence—and formation—of hormones and other growth promoting substances in food producing animals. Current approaches for enforcement and research needs for full implementation in residue control. Rikilt Wageningen UR.
Ferranti, C., delli Quadri, F., Palleschi, L., et al. (2011). Studies on the presence of natural and synthetic corticosteroids in bovine urine. Steroids, 76(6), 616–625.
Ferranti, C., Famele, M., Palleschi, L., Bozzetta, E., Pezzolato, M., & Draisci, R. (2013). Excretion profile of corticosteroids in bovine urine compared with tissue residues after therapeutic and growth-promoting administration of dexamethasone. Steroids, 78(9), 803–812.
Gika, H. G., Theodoridis, G. A., Plumb, R. S., & Wilson, I. D. (2014). Current practice of liquid chromatography–mass spectrometry in metabolomics and metabonomics. Journal of Pharmaceutical and Biomedical Analysis, 87, 12–25.
Goniewicz, M. L., Havel, C. M., Peng, M. W., et al. (2009). Elimination kinetics of the tobacco-specific biomarker and lung carcinogen 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol. Cancer Epidemiology, Biomarker and Prevention, 18(12), 3421–3425.
Grund, B., & Sabin, C. (2010). Analysis of biomarker data: logs, odds ratio and ROC curves. Current Opinion in HIV and AIDS, 5(6), 473–479.
Hardy, R. S., Raza, K., & Cooper, M. S. (2012). Endogenous glucocorticoids in inflammation: Contributions of systemic and local responses. Swiss Medical Weekly, 142, 1–10.
Hawkins, D. M., Basak, S. C., & Mills, D. (2003). Assessing model fit by cross-validation. Journal of Chemical Information and Modeling, 43(2), 579–586.
Hjerpsted, J. B., Ritz, C., Schou, S. S., Tholstrup, T., & Dragsted, L. O. (2014). Effect of cheese and butter intake on metabolites in urine using an untargeted metabolomics approach. Metabolomics, 10(6), 1176–1185.
Hunter, D. J., Losina, E., Guermazi, A., Burstein, D., Lassere, M. N., & Kraus, V. (2010). A pathway and approach to biomarker validations and quantification of osteoarthritis clinical trails. Current Drug Targets, 11(5), 536–545.
Jacob, C. C., Pinel, G., Biancotto, G., & Le Bizec, B. (2014). Evaluation of specific gravity as normalization strategy for cattle urinary metabolome analysis. Metabolomics, 10, 627–637.
Jia, W., Ling, Y., Lin, Y. H., Chang, J., & Chu, X. (2014). Analysis of additives in diary products by liquid chromatography coupled to quadrupole-orbitrap mass spectrometry. Journal of Chromatography A, 1336, 67–75.
Jung, Y. J., Lee, H. S., Kang, D. G., et al. (2011). 1H-NMR-based metabolomics study of cerebral infarction. Stroke, 42(5), 1282–1288.
Kaabia, Z., Pinel, G., Popot, M. A., Chouribery, L., Plou, P., Bonnaire, Y., & Le Bizec, B. (2013). Monitoring the endogenous steroid profile disruption in urine and blood upon nandrolone administration: An efficient and innovative strategy to screen for nandrolone abuse in entire male horses. Drug Testing and Analysis, 6, 376–388.
Koulman, A., Lane, G. A., Harrison, S. J., & Volmer, D. A. (2009). From differentiating metabolites to biomarkers. Analytical and Bioanalytical Chemistry, 394(4), 663–670.
Lee, J. W., Devanarayan, V., Barrett, Y. C., et al. (2006). Fit-for-purpose method development and validation for successful biomarker measurement. Pharmaceutical Research, 23(2), 312–328.
Leporati, M., Capra, P., Cannizzo, F. T., Biolatti, B., Nebbia, C., & Vincenti, M. (2013). Determination of prednisolone metabolites in beef cattle. Food Additives and Contaminants. Part A, Chemistry, Analysis, Control, Exposure and Risk Assessment, 30(6), 1044–1054.
Levine, L., & Fahy, J. P. (1945). Evaluation of urinary lead determinations. The Journal of Industrial Hygiene and Toxicology, 27, 217–223.
Marshall, A. G. (2000). Milestones in Fourier transform ion cyclotron resonance mass spectrometry technique development. International Journal of Mass Spectrometry and Ion Processes, 200(1–3), 331–356.
Matabosch, X., Pozo, O. J., Monfort, N., et al. (2013). Urinary profile of methylprednisolone and its metabolites after oral and topical administrations. The Journal of Steroid Biochemistry and Molecular Biology, 138, 214–221.
Matsui, S. (2006). Reducing false positive findings in statistical analysis of pharmacogenomics biomarker studies using high-throughput technologies. Current Drug Safety, 1(2), 135–141.
Matthew, B., & William, R. (2003). Partial least squares for discrimination. Journal of Chemometrics, 17(3), 166–173.
McLoughlin, N., Yin, D., Maltby, L., Wood, R. M., & Yu, H. (2000). Evaluation of sensitivity and specificity of two crustacean biochemical biomarkers. Environmental Toxicology and Chemistry, 19(8), 2085–2092.
Neumann, S., & Böcker, S. (2010). Computational mass spectrometry for metabolomics: Identification of metabolites and small molecules. Analytical and Bioanalytical Chemistry, 398(7–8), 2779–2788.
Parikh, C. R., & Philbrook, H. T. (2014). Key concepts and limitations of statistical methods for evaluating biomarkers of kidney disease. Journal of the American Society of Nephrology, 25(8), 1621–1629.
Pepe, M. S., Janes, H., Longton, G., Leisenring, W., & Newcomb, P. (2004). Limitations of the odds ratio in gauging the performance of a diagnostic prognostic, or screening marker. American Journal of Epidemiology, 159(9), 882–890.
Pinel, G., Chereau, S., Cesbron, N., Monteau, F., & Le Bizec, B. (2015). LC-HRMS based metabolomics screening model to detect various β-agonists treatments in bovines. Metabolomics, 11, 403–411.
Pinel, G., Rambaud, L., Monteau, F., Elliot, C., & Le Bizec, B. (2010). Estranediols profiling in calves’ urine after 17β-nandrolone laureate ester administration. The Journal of Steroid Biochemistry and Molecular Biology, 121(3–5), 626–632.
Pinel, G., Weigel, S., Lommen, A., et al. (2011). Assessment of two complementary liquid chromatography coupled to high resolution mass spectrometry metabolomics strategies for the screening of anabolic steroid treatments in calves. Analytica Chimica Acta, 700(1–2), 144–154.
Pompa, G., Arioli, F., Casati, A., Fidani, M., Bertocchi, L., & Dusi, G. (2011). Investigation of the origin of prednisolone in cow urine. Steroids, 76(1–2), 104–110.
Puntmann, V. O. (2009). How-to guide on biomarkers: Biomarker definitions, validation and applications with examples from cardiovascular disease. Postgraduate Medical Journal, 85(1008), 538–545.
Quintás, G., Portillo, N., García-Cañaveras, J. C., Castell, J. V., Ferrer, A., & Lahoz, A. (2012). Chemometric approaches to improve PLSDA model outcome for predicting human non-alcoholic fatty liver disease using UHPLC-MS as a metabolomic profiling tool. Metabolomics, 8(1), 86–98.
Regal, P., Seijas, A., Cepeda, A., & Fente, C. (2013). Structure elucidation and HPLC-MS/MS determination of a potential biomarker for estradiol administration in cattle. Analytical and Bioanalytical Chemistry, 405(29), 9537–9546.
Scalbert, A., Brennan, L., Fiehn, O., et al. (2009). Mass-spectrometry-based metabolomics: Limitations and recommendations for future progress with particular focus on nutrition research. Metabolomics, 5(4), 435–458.
Scarth, J., Clarcke, A., Hands, J., Tealt, P., Macarthur, R., & Kay, J. (2010). Validation of a quantitative multi-residue urinary assay for the detection of androgen, oestrogen and progestagen abuse in the bovine. Chromatographia, 71(3), 241–252.
Shah, V. P., Midha, K. K., & Findlay, J. W. A. (2000). Bioanalytical method validation-a revisit with a decade of progress. Pharmaceutical Research, 17(12), 1551–1557.
Snyder, L. R., Kirkland, J. J., & Glajch, J. L. (1997). Practical HPLC method development (2nd ed.). London: Wiley.
Soininen, T. H., Jukarainen, N., Auriola, S. O. K., Julkunen-Tiitto, R., Karjalainen, R., & Vepsäläinen, J. J. (2014). Quantitative metabolite profiling of edible onion species by NMR and HPLC-MS. Food Chemistry, 165, 499–505.
Sumner, L. W., Amberg, A., Barrett, D., et al. (2007). Proposed minimum reporting standards for chemical analysis Chemical Analysis Working Group (CAWG) Metabolomics Standards Initiative (MSI). Metabolomics, 3(3), 211–221.
Tardieu, A., De Man, W., & This, H. (2010). Using one-dimensional (1D) and two-dimensional (2D) quantitative proton (1H) nuclear magnetic resonance spectroscopy (q NMR) for the identification and quantification of taste compounds in raw onion (Allium cepa L.) bulbs and in aqueous solutions where onion tissues are soaked. Analytical and Bioanalytical Chemistry, 398(7–8), 3139–3153.
Theodoridis, G. A., Gika, H. G., Want, E. J., & Wilson, I. D. (2012). Liquid chromatography-mass spectrometry based global metabolite profiling: A review. Analytica Chimica Acta, 711, 7–16.
Trygg, J., & Wold, S. (2002). Orthogonal projections to latent structures (O-PLS). Journal of Chemometrics, 16(3), 119–128.
van den Berg, R. A., Hoefsloot, H. C. J., Westerhuis, J. A., Smilde, A. K., & van der Werf, M. J. (2006). Centering, scaling, and transformations: improving the biological information content of metabolomics data. BMC Genomics, 7, 142–156.
Van Meulebroek, L., Vanden Bussche, J., De Clercq, N., Steppe, K., & Vanhaecke, L. (2014). A metabolomics approach to unravel the regulating role of phytohormones towards carotenoid metabolism in tomato fruit. Metabolomics,. doi:10.1007/s1130601407289.
Verhagen, H., Coolen, S., Duchateau, G., Hamer, M., Kyle, J., & Rechner, A. (2004). Assessment of the efficacy of functional food ingredients—introducing the concept “kinetics of biomarkers”. Mutation Research, 551(1–2), 65–78.
Vincenti, M., Girolami, F., Capra, P., et al. (2009). Study of dexamethasone urinary excretion profile in cattle by LC-MS/MS: Comparison between therapeutic and growth-promoting administration. Journal of Agricultural and Food Chemistry, 57(4), 1299–1306.
Vincenti, M., Leporati, M., Capra, P., et al. (2012). A field survey on the presence of prednisolone and prednisone in urine samples from untreated cows. Food Additives and Contaminants. Part A, Chemistry, Analysis, Control, Exposure and Risk Assessment, 29(12), 1893–1900.
Wagner, J. A. (2002). Overview of biomarkers and surrogate endpoints in drug development. Disease Markers, 18(2), 41–46.
Weiss, S. H., Goedert, J. J., Sarngadharan, M. G., Bodner, A. J., Gallo, R. C., & Blattner, W. A. (1985). Screening test for HTLV-III (AIDS agent) antibodies. Specificity, sensitivity and applications. JAMA, 253(2), 221–225.
Wetenschappelijk Comité van het Federaal Agentschap voor de veiligheid van de voedselketen, (2013). Retrieved February 12, 2015 from http://www.afsca.be/wetenschappelijkcomite/adviezen/_documents/ADVIES07-2013_NL_DossierSciCom2012-07.pdf.
Wicklund, S., Johansson, E., Sjöström, L., et al. (2008). Visualization of GC/TOF-MS-based metabolomics data for identification of biochemically interesting compounds using OPLS class models. Analytical Chemistry, 80(1), 115–122.
Wold, S., Sjöström, M., & Eriksson, L. (2001). PLS-regression: A basic tool of chemometrics. Chemometrics and Intelligent Laboratory Systems, 58(2), 109–130.
Wolf, S., Schmidt, S., Müller-Hannemann, M., & Neumann, S. (2010). In silico fragmentation for computer assisted identification of metabolite mass spectra. BMC Bioinformatics, 11, 148–160.
Xue, S. Y., Li, Z. Y., Zhi, H. J., et al. (2012). Metabolic fingerprinting investigation of Tussilago farfara L. by GC-MS and multivariate data analysis. Biochemical Systematics and Ecology, 41, 6–12.
Acknowledgments
This study was funded by the Federal Public Service of Health, Food Chain Safety and Environment (RT 11/03). The authors wish to thank Mieke Naessens and Joke Goedgebuer for their technical contribution to this manuscript. We are also grateful towards SCIEX for their support.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
Nathalie De Clercq, Julie Vanden Bussche, Lieven Van Meulebroek, Siska Croubels, Philippe Delathaut, Dieter Buyst, José Martins, Jianru Stahl-Zeng and Lynn Vanhaecke declare that they have no conflict of interest.
Research involving human participants and/or animals
All applicable international, national, and/or institutional guidelines for the care and use of animals were followed.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Supplementary Fig. 1
Score plots for OPLS-DA models representing the discrimination between control (Blue) and treatment (Green), respectively determined with Sieve™ (A) and MarkerView™ (B) and for positive (A1,B1) and negative (A2, B2) ionized metabolite ions.. Supplementary material 2 (TIFF 6593 kb)
Rights and permissions
About this article

Cite this article
De Clercq, N., Bussche, J.V., Van Meulebroek, L. et al. Metabolic fingerprinting reveals a novel candidate biomarker for prednisolone treatment in cattle. Metabolomics 12, 1 (2016). https://doi.org/10.1007/s11306-015-0887-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11306-015-0887-3