
Abstract
We aimed to evaluate the effect of caffeine on viability, apoptosis, migration, redox profile and modulatory effect of the purinergic system of cutaneous melanoma cells. The melanoma cells SK-MEL-28 and non-tumoural CCD-1059sk cells were treated for 24 h with different concentrations of caffeine. Cell viability was evaluated by a biochemical assay and fluorescence microscopy, and flow cytometry assessed apoptosis induction. A wound-healing assay assessed cell migration. The redox profile was evaluated by the levels of markers of reactive oxygen species (ROS), nitric oxide (NOx), total thiols (PSH) and non-protein thiols (NPSH). RT-qPCR and flow cytometry assessed the expression of CD39 and CD73. ATPase/ADPase and AMPase enzyme activities were evaluated by hydrolysis of ATP, ADP and AMP nucleotides. A bioluminescent assay assessed extracellular ATP levels. Caffeine significantly reduced melanoma cell viability and migration and did not affect non-tumoural cells. Caffeine increased ROS levels and improved PSH levels in melanoma cells. Furthermore, caffeine reduced CD39 and CD73 expression, decreased ATP, ADP and AMP nucleotide hydrolysis and increased extracellular ATP levels. We have shown that caffeine reduces metastatic cutaneous melanoma cell viability and migration, induces ROS generation and improves PSH levels. In an unprecedented manner, we also showed that caffeine reduces the expression of CD39 and CD73 and, consequently, ATPase/ADPase/AMPase hydrolytic activity of ectonucleotidases, thus displacing the CD39/CD73 axis and increasing extracellular ATP levels. Therefore, caffeine may be an interesting compound for clinical trials with the CD39/CD73 axis as a therapeutic target.
Graphical Abstract

Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Cutaneous melanoma (CM) is one of the most aggressive skin cancer types due to its highly metastatic nature and low survival rate [1]. The incidence of this disease is increasing. In 2020, 324,635 new cases and 57,043 deaths occurred worldwide; considering these rates, it can be predicted that by 2040 more than 500,000 new cases will emerge [2, 3]. CM has been strongly linked to harm caused by ultraviolet (UV) radiation due to excessive sunbathing [4].
Currently, immune system checkpoint inhibitors such as pembrolizumab and nivolumab (inhibitors of programmed cell death protein 1 [PD-1]) and ipilimumab (inhibitor of cytotoxic T lymphocyte antigen 4 [CTLA-4]) have been used in the management of many cancers and are used as a first-line strategy to treat patients with a prognosis of unresectable or metastatic melanoma [5, 6]. In addition, inhibitor molecules that directly target the signalling pathways responsible for controlling cell growth and division have emerged and are referred to as ‘targeted therapies’, such as V-Raf murine sarcoma viral oncogene homolog B (BRAF) and mitogen-activated protein kinase (MEK) inhibitors, which can target neoplasms with these mutated genes. However, only 20 to 30% of patients administered targeted therapies showed a complete and durable response, and biomarkers still need to be defined to predict who will benefit from these drugs [7]. Furthermore, the correct application of targeted therapy seems to be primordial in melanoma since findings from the literature showed that treatment duration was strongly associated with the risk of cancer progression [8].
The purinergic system is a key signalling pathway recently linked to CM since it regulates cell proliferation, angiogenesis and apoptosis. Orchestration between components is regulated by ectonucleotidases in the tumour microenvironment (TME), with adenosine triphosphate (ATP), adenosine diphosphate (ADP), adenosine monophosphate (AMP) nucleotides and adenosine (Ado) nucleoside being important molecules, in addition to the expression of P1 and P2 receptors [9,10,11]. Manica et al. [12] showed that Ecto-NTPDase (CD39) and Ecto-5′-nucleotidase (CD73) are enzymes involved in the pathophysiology of CM and could be an important target for cancer therapies as these enzymes act in the enzymatic axis and control the hydrolysis of the major purinergic signalling molecule, ATP.
In this context, several bioactive compounds have been studied to be applied as adjuvant therapies, given their cost-effectiveness, such as rosmarinic acid [13] and caffeic acid [14]. Another interesting substance is the coffee-derived methylxanthine, caffeine (1,3,7-trimetilxantina), which is found in coffee grains such as Coffea arabica, tea leaves of Camellia sinensis and cacao powder [15]; this substance is a promissory antineoplastic natural compound. Studies have shown that caffeine can mitigate neoplastic cell growth, migration, invasion and progression [16, 17] and act as an antagonist of A2A receptors, exhibiting antineoplastic effects [18]. In addition, caffeine seems to be involved in redox balance [19].
There are several pieces of literature relating the actions of caffeine on the CD39/CD73 axis in metastatic CM cells, as well as the implications of blocking these pathways on the purinergic system. In this study, we aimed to evaluate in vitro the antineoplastic effects of caffeine on metastatic CM cells. We hypothesise that caffeine may have antitumor potential against metastatic CM cells by modulating the expression of components involved in purinergic signalling.
Material and methods
Chemicals and reagents
All chemicals and reagents were analytical grade from Sigma-Aldrich (Sigma-Aldrich, USA) and Merck (Germany). The caffeine (1,3,7-trimethylxanthine) was obtained from Sigma-Aldrich (CAS number: 58-08-2, EC number: 200-362-1, PubChem Substance ID: 24900939, anhydrous, purity ≥ 99%, molecular weight 194.19 g/mol). The cells SK-MEL-28 and CCD-1059sk were purchased from the Cell Bank of Rio de Janeiro (BCRJ), Brazil. Cell culture medium, plates and flasks used for culture procedures were obtained from Gibco™ Thermo Fisher Scientific (Grand Island, USA) and Invitrogen Life Technologies (Carlsbad, USA). Master Mix for qPCR was purchased from Promega Corporation®, and primers were synthesised by Thermo Fisher Scientific (Grand Island, USA). Anti-human CD39 and CD73 antibodies were purchased from BD Pharmingen™, and the readers were made by flow cytometry (BD ACCURI C6). For fluorescence assays, a fluorescence microscope was used (Nikon® Eclipse TS2-FL).
Cell culture and caffeine exposure
We treated a tumoural and a non-tumoural cell lineage under the same experimental conditions to verify the cytotoxic effects of caffeine and its antineoplastic potential. Thus, both SK-MEL-28 and CCD-1059sk were independently grown in flasks containing Dulbecco’s Modified Eagle’s Medium (DMEM) (Gibco™) with penicillin/streptomycin (1%) supplemented with 10% foetal calf serum. The cell cultures were maintained under appropriate conditions (5% carbon dioxide (CO2) at 37 °C). We used the medium to dissolve caffeine and obtain the different treatment concentrations (0.5 mM, 1.0 mM, 2.0 mM, 4.0 mM and 6.0 mM) based on a previous study [17]. The negative control group (CT) cells received only the culture medium, and the neoplastic cells were treated with the antineoplastic dacarbazine (dac) at a concentration of 1 mg/ml. All cells were exposed for 24 h to respective treatments.
Peripheral blood mononuclear cells (PBMCs) were collected by venipuncture from five healthy subjects. Cells were cultured under the same conditions as the cancer cells using Roswell Park Memorial Institute (RPMI) medium with penicillin/streptomycin (1%) supplemented with 10% foetal calf serum. This work was approved by the Human Ethics Committee of the Federal University of Fronteira Sul (UFFS, Campus Chapecó, SC, Brazil) under protocol number 822.782. Blood collection occurred after all donors had given written consent to participate in the study.
Cell viability assay
An assay using 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) as a substrate was used to evaluate the cytotoxicity of caffeine on SK-MEL-28 and CCD-059sk cells, following a study carried out by Mosmann [20]. Briefly, both cell lines were independently seeded in 96-well plates, in 4 replicates, at densities of 1 × 105 cells/well and treated with different concentrations of caffeine. After the exposure times, the MTT (Sigma-Aldrich, USA) (5 mg/ml in PBS) was added, and the plates were incubated at 37 °C for 2 h. The supernatant was discarded, and 200 μl of dimethylsulfoxide (DMSO) was added to dissolve the formazan crystals formed by the viable cells’ reduction of the MTT salt. The absorbance was measured at 570 nm using a Multiskan GO 96-well plate reader (Thermo Fisher Scientific).
Fluorescence microscopy assay
In addition to the MTT assay, we also used fluorescence microscopy to assess tumour cell viability. For this purpose, approximately 1 × 105 cells/well were seeded in 96-well plates, washed twice with PBS and stained with acridine orange (AO) to check for morphological changes as described by McGahon et al. [21]. AO is taken up by viable cells and stains double-stranded (ds) and single-stranded (ss) nucleic acids. When AO diffuses into cells, it emits green fluorescence after excitation at 480–490 nm in viable cells. For tetramethylrhodamine ethyl ester (TMRE) staining, the cells received the same preparation approach as AO staining and were incubated for 30 min with 100 μl of a solution containing TMRE (20 nM). The fluorescence photographs were taken under excitation at 550 nm [22] in triplicates at ×10 magnification and adjusted linearly for brightness and contrast with the software Fotos (Windows®).
Cell apoptosis assay
For the apoptosis assay, cells exposed to different concentrations of caffeine for 24 h were harvested, and the cell pellets were washed twice with cold PBS. An Annexin V-FITC and propidium iodide (PI) apoptosis detection kit (BD Biosciences) was used according to the manufacturer’s instructions. In brief, cells were resuspended at 1 × 106 cells/ml in 100 μl binding buffer. Annexin V-FITC (5 μl) and 5 μl PI (20 μg/ml) were added to the cell suspension and then incubated for 15 min at room temperature in the dark and mixed with 400 μl binding buffer. The analyses were done on a BD Accuri™ C6 Plus, with readings taken for every 20,000 events.
Cell migration assay
To verify neoplastic cell migration, we performed the experimental protocol proposed by Justus et al. [23]. Thus, we seeded 1 × 106 cells/well in a monolayer in 6-well plates. After reaching 100% confluence, we made a wound with a sterile tip and photographed it with optical microscopy (×4 magnification). Then, we exposed the cells to the respective treatment. After 24 h, we discarded the treatment, washed the cells with saline and proceeded with the image acquisition. Five different points on the wound were measured using ImageJ software to perform the statistical analyses.
Assessment of redox profile
All experiments to assess the redox profile of cells after caffeine treatment were performed independently three times and in three replicates.
Reactive oxygen species (ROS)
A commercial fluorometric intracellular ROS kit (Sigma® Life Science, Germany) was used to determine intracellular ROS levels according to the manufacturer’s protocol. In the presence of ROS, 2’,7’-dichlorofluorescein diacetate (H2DCF-DA), a cell-permeable non-fluorescent substance, is oxidised into a fluorescent molecule, 2',7'-dichlorofluorescein (DCF). The colourimetric product was measured at an excitation wavelength of 490 nm and emission of 520 nm (Thermo Scientific™ Varioskan™ LUX).
Nitric oxide (NOx) detection
We used a modified Griess method to detect nitrate or nitrite in samples with adaptations [24,25,26]. First, the Griess reagent was prepared by adding 2% sulphanilamide and 0.2% N-1-naftil-etilendiamina-diclorhidrato (NED) to 5% of orthophosphoric acid (H2PO4). Subsequently, approximately 100 μl of the samples was mixed with 100 μl of the Griess reagent and incubated at 37 °C for 20 min. The absorbance of the purple-azo-dye product was measured at 540 nm. We used a calibration curve prepared with sodium nitrate to determine the NOx amount in samples. The results were expressed in μM.
Levels of total thiol (PSH) and non-protein thiol (NPSH)
Both thiol contents were determined according to Elmann [27] with adjustments. For the total thiol assay, 30 μl of supernatant in a 96-well plate was added to 200 μl of potassium phosphate buffer (PPB) (1 M, pH 6.8) and 20 μl of 5,5′-dithiobis (2-nitrobenzoic acid) (DTNB) with immediate reading. For the non-protein thiol, the samples were previously deproteinised to 10% with an equal sample volume of trichloroacetic acid (TCA), and the remaining supernatant was used. Then, 40 μl of supernatant was mixed with 260 μl of PPB and 15 μl with read immediately. All reads were measured at 412 nm. The results were expressed in μM having a cysteine standard curve as the parameter.
Purinergic system assays
CD39 and CD73 gene expression
Total RNA from melanoma cells was extracted with TRIzol® according to the manufacturer’s instructions. RNA was quantified using a Thermo Scientific™ Varioskan™ LUX, and the purity of the RNA was assessed using a 260/280 nm absorbance ratio. RNA was treated with 1 U/DNase amplification grade (Invitrogen) for 30 min at 37 °C. Double-stranded complementary DNA (cDNA) was synthesised from 200 ng of total RNA with random hexamer primers using a high-capacity cDNA reverse transcription kit (Thermo Scientific, USA) according to the manufacturer’s instructions. Molecular assays were performed using 5 μl cDNA sample, 10 μl 2X SYBR® Green PCR Master Mix (Thermo Scientific, USA), 2 μl forward primer (500 nM), 2 μl reverse primer (500 nM) and 1 μl ultrapure water DNAse- and RNAse-free to a final volume of 20 μl. Thermal cycling to amplify each transcript was as follows: initial denaturation at 95 °C for 5 min, 60 cycles of denaturation at 95 °C for 15 s, and annealing/extension at 60 °C for 60 s. Melting curve analyses were performed to verify product identity. Samples were run in duplicate, and results were expressed relative to GAPDH levels. Data were then normalised to a calibrator sample using the ΔΔCq method. We used the following sequences of oligos (5′–3′): GAPDH (F): CTCCTCACAGTTGCCATGTA; GAPDH (R): GTTGAGCACAGGGTACTTTATTG; CD39 (F): GCCCTGGTCTTCAGTGTATTAG; CD39 (R): CTGGCATAACCTACCTACTCTTTC; CD73 (F): GTGCCTTTGATGAGTCAGGTAG; CD73 (R): TTCCTTTCTCTCGTGTCCTTTG.
Detection of CD39 and CD73 protein expression by flow cytometry
After growing under caffeine exposure for 24 h, SK-MEL-28 cells were counted at 1 × 106 cells/ml using a haemocytometer. The cell suspension was washed in duplicate with PBS supplemented with 10% FBS and centrifuged for 5 min at 400g. Then, following the manufacturer’s recommendations, the cell pellets were resuspended in a buffer solution and incubated for 30 min with purified antibodies (1:10 dilution) PE mouse anti-human CD39 (clone A1 (RUO), catalogue number: 567157) and PE mouse anti-human CD73 (clone AD2 (RUO), catalogue number: 550257) (BD Pharmingen™). Each experimental procedure was done separately and incubated accordingly with antibody targets (CD39 or CD73). The same number of cells was incubated without antibodies as a non-stained control. After the elapsed times, all samples were washed with PBS, and 10,000 events were immediately acquired by flow cytometry (BD ACCURI C6). Subsequently, the acquired results were analysed using the FlowJo V10 software and expressed as a percentage (%) of CD39 or as a percentage (%) of CD73-positive cells in relation to the control in caffeine untreated cells (CT). All experiments were performed independently three times and in three replicates.
Assessment of ATPase/ADPase/AMPase activities of ectonucleotidases
The enzymatic activities of ATPase/ADPase/AMPase were performed according to Pilla et al. [28] and Lunkes et al. [29]. A reaction system for ATPase/ADPase was prepared containing 5 mM CaCl2, 100 mM NaCl, 5 mM KCl, 6 mM glucose and 50 mM Tris-HCl buffer, pH 7.4. The reaction system was the same for AMPase, with the exception of 10 mM MgCl2 instead of CaCl2. Briefly, after protein normalisation in saline (0.9% NaCl), 20 μl of cell samples was added to the reaction mixture of each enzyme and pre-incubated at 37 °C for 10 min. The enzymatic reaction was initiated by adding the specific substrates for each enzyme, ATP and ADP for ATPase/ADPase and AMP for AMPase and incubated at 37 °C for 70 min. Finally, reactions were stopped by the addition of 150 μl of 10% TCA. From the total well content, 30 μl was transferred to a new plate, 300 μl of malachite green as a colourimetric reagent was added and the released inorganic phosphate was determined at 630 nm. Control wells were performed to correct for non-enzymatic hydrolysis. A standard curve was prepared with KH2PO4, and the results were expressed as nmol of Pi released/min/mg of protein. All experiments were performed independently three times and in three replicates.
Extracellular ATP determination
To determine extracellular ATP levels, we used a Molecular Probes® ATP determination kit (Invitrogen™), a bioluminescence assay that contains recombinant firefly luciferase and its substrate D-luciferin. The assay is based on the requirement of luciferase for ATP in light production with a maximum emission of ~560 nm at pH 7.8 [30]. We combined the reaction components to make a standard reaction solution and adjusted the volumes to specific requirements with 1.25 μg/ml firefly luciferase, 50 μM D-luciferin and 1 mM DTT in 1x reaction buffer. Afterwards, we mixed 10 μl of the supernatant with 90 μl of the reaction solution and incubated for 15 min. Then, we measured the luminescence. An ATP standard curve was prepared at concentrations from 1 nM to 1 μM. All experiments were performed independently three times and in three replicates.
Protein determination
The method of Bradford [31] was employed for protein determination using bovine serum albumin as a standard and the protein samples were adjusted according to each assay in mg/ml.
Statistical analysis
All measurements were performed statistically using an analysis of variance followed by a post hoc test using GraphPad Prism 9 software. All data were expressed as mean ± standard deviation. Differences between groups in relation to the variables studied were evaluated by one-way ANOVA, followed by a post hoc Dunnett’s multiple comparison test. Differences in the probability of rejecting the null hypothesis as < 5% (P < 0.05) were considered statistically significant. Statistical significance was defined for P-values of *P < 0.05, **P < 0.01, ***P < 0.001 and ****P < 0.0001.
Results
Caffeine reduces viability of metastatic melanoma cells and induces apoptosis
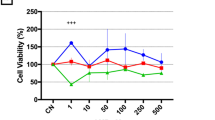
We verified the effect of caffeine on the viability of melanoma cells and non-tumour cells, as shown in Fig. 1A–C. Caffeine reduced the viability of melanoma cells after 24 h of treatment at concentrations of 0.5 mM, 2.0 mM, 4.0 mM, 6.0 mM (P < 0.0001) and significantly decreased viability at 1.0 mM (P < 0.001) compared to control, showing it is cytotoxic to tumour cells. All concentrations of caffeine had a higher effect than dacarbazine (P < 0.05) used as a positive antineoplastic control (Fig. 1A). In addition, caffeine induced apoptosis in melanoma cells at concentrations of 0.5 mM (P < 0.0001), 4.0 mM (P < 0.0001) and 6.0 mM (P<0.0001) (Fig. 1B). On the other hand, caffeine had no cytotoxic effect on the viability of the non-tumoural cell models (CCD-1059sk and PBMCs) (Fig. 1C and D). A fluorescent microscopy assay reinforced the cytotoxic effect of caffeine since there was a reduction in the fluorescence intensity percentage of cells stained with AO at concentrations of 4.0 mM, 6.0 mM (P<0.01) and 2.0 mM (P<0.05) compared to control (Fig. 2A). More pronounced results were found with TMRE staining, with reduction in fluorescence intensity percentage at 0.5 mM and 1.0 mM (P<0.05), 2.0 mM and 4.0 mM (P<0.01) and 6.0 mM (P < 0.001) in relation to control (Fig. 2A). These sets of results corroborate the effects of caffeine on the viability of melanoma cells using an MTT and fluorescence microscopy assay.

Cell viability and apoptosis of SK-MEL-28 cells and cell viability of CCD-1059sk. An MTT assay indicated that all concentrations of caffeine treatments substantially reduced the viability of SK-MEL-28 cells (A) while having no effect on the non-tumoural cells CCD-1059sk (C). The treatment with caffeine induced apoptosis at concentrations of 0.5 mM, 1.0 mM, 4.0 mM and 6.0 mM (B). All experiments were performed independently three times and in three replicates. Data are presented as mean ± SD. Statistical analysis: one-way ANOVA followed by a post hoc Dunnett’s multiple comparisons test. Values with P < 0.05 were considered statistically significant. *(P < 0.05); **(P < 0.01); ***(P < 0.001); ****(P < 0.0001). CT, control; dac, dacarbazine

Microscope fluorescence and migration of SK-MEL-28 cells. A Microscope fluorescence assay stained with AO showed that caffeine reduced cell viability at 2.0 mM, 4.0 mM and 6.0 mM and at all tested caffeine concentrations when stained with tetramethylrhodamine ethyl ester (TMRE). B For cell migration, caffeine affected the reduction in wound-healing closure for all concentrations tested. All experiments were performed independently three times and in three replicates. Data are presented as mean ± SD. Statistical analysis: one-way ANOVA followed by a post hoc Dunnett’s multiple comparisons test. Values with P < 0.05 were considered statistically significant. *(P < 0.05); **(P < 0.01); ***(P < 0.001); ****(P < 0.0001). CT, control; dac, dacarbazine
Caffeine inhibits migration of metastatic CM cells
As shown in Fig. 2B, caffeine treatment reduced the SK-MEL-28 cell migration at all concentrations. The treatment concentrations of 1.0 mM, 2.0 mM, 4.0 mM and 6.0 mM (P < 0.0001) exhibit more potential than 0.5 mM (P < 0.001) compared to control. As expected, dacarbazine (1 mg/ml) also reduced cell migration (P < 0.0001). These results showed that doses higher than 0.5 mM of caffeine could inhibit melanoma cell migration, similar to dacarbazine, a common chemotherapy drug used in melanoma therapy.
Caffeine induces oxidative stress in metastatic CM cells
We found that caffeine increased ROS levels at a concentration of 6.0 mM (P < 0.0001) in melanoma cells (Fig. 3A). Dacarbazine caused an increase in NOx levels (P < 0.01) (Fig. 3B). On the other hand, concentrations of 2.0 mM, 4.0 mM and 6.0 mM caffeine also improved the PSH levels (P < 0.05) (Fig. 3C). For the other oxidative profile markers that were evaluated, we did not find any statistically significant differences when compared to control following caffeine exposure.

Oxidative stress marker levels in SK-MEL-28 cells. Caffeine increased ROS levels at 6.0 mM (A) and PSH levels at 2.0 mM, 4.0 mM and 6.0 mM (C). There was no statistical significance for NOx (B) and NPSH (D) levels. All experiments were performed independently three times and in three replicates. Data are presented as mean ± SD. Statistical analysis: one-way ANOVA followed by a post hoc Dunnett’s multiple comparisons test. Values with P < 0.05 were considered statistically significant. *(P < 0.05); **(P < 0.01); ***(P < 0.001); ****(P < 0.0001). CT, control; dac, dacarbazine
CD39 and CD73 expression is modulated by caffeine in metastatic melanoma cells
Firstly, we employed RT-qPCR to assess CD39 and CD73 gene expression, as shown in Fig. 4A and D. After 24 h of treatment, we found that caffeine reduced CD39 gene expression similarly at 0.5 mM and 2.0 mM (P < 0.05) and more significantly at concentrations of 4.0 mM (P < 0.01) and 6.0 mM (P < 0.0001) (Fig. 4A). We also found that caffeine reduced gene expression of CD73, with statistical significance at 2.0 mM and 4.0 mM (P < 0.05) and was more evident at 6.0 mM (P < 0.01) (Fig. 4D). Dacarbazine significantly increased gene expression of CD73 (P < 0.0001) (Fig. 4D). Secondly, we evaluated protein expression of CD39 and CD73 ectonucleotidases by flow cytometry, as shown in Fig. 4B, C, E, and F. Converging this data with the gene expression results, we confirmed that caffeine significantly reduced CD39 protein expression at 2.0 mM (P < 0.0001), 4.0 mM (P < 0.05) and 6.0 mM (P < 0.0001). Dacarbazine treatment was also able to reduce CD39 protein expression (P < 0.0001) (Fig. 4B and C). For CD73, we found that caffeine significantly reduced the expression of this protein at 0.5 mM, 2.0 mM and 4.0 mM (P < 0.0001). In addition, dacarbazine significantly increased the protein expression of CD73 (P < 0.0001) (Fig. 4E and F). These results show that important ectonucleotidases involved in neoplastic purinergic signalling can modulate gene and protein expression by caffeine.

CD39 and CD73 gene and protein expression. All tested concentrations of caffeine were able to reduce CD39 gene expression (A). In the same way, CD39 protein expression was reduced at 2.0 mM, 4.0 mM and 6.0 mM concentrations (B, C). For CD73, it is possible to notice that caffeine treatment reduced gene expression at 2.0 mM, 4.0 mM and 6.0 mM (D) and protein expression at 0.5 mM, 2.0 mM and 4.0 mM (E-F). All experiments were performed independently three times and in three replicates. Data are presented as mean ± SD. Statistical analysis: one-way ANOVA followed by a post hoc Dunnett’s multiple comparisons test. Values with P < 0.05 were considered statistically significant. *(P < 0.05); **(P < 0.01); ***(P < 0.001); ****(P < 0.0001). CT, control; dac, dacarbazine
Caffeine modulates enzymatic activities of ATPase/ADPase/AMPase ectonucleotidases and alters extracellular ATP levels in metastatic melanoma cells
To understand purinergic signalling beyond gene and protein expression, we evaluated ATPase/ADPase/AMPase activities by the hydrolysis of ATP, ADP, AMP and extracellular ATP (Fig. 5A–D). In our study, ATP hydrolysis was decreased at concentrations of 1.0 mM (P < 0.05), 2.0 mM (P < 0.0001), 4 mM (P < 0.0001) and 6.0 mM (P < 0.001) compared to control (Fig. 5A). ADP hydrolysis was increased at 0.5 mM (P < 0.001) and significantly decreased in the range of 1.0–6.0 mM (P < 0.0001) of caffeine treatment when compared to control (Fig. 5B). In addition, AMP hydrolysis at 1.0 mM, 2.0 mM, 4.0 mM and 6.0 mM concentrations of caffeine treatment was significantly decreased in relation to control (P < 0.0001) (Fig. 5C). Ultimately, we found that caffeine increased extracellular ATP levels at 2.0 mM, 4.0 mM and 6.0 mM concentrations with a similar significance (P < 0.0001) (Fig. 5D). These results confirm the action of caffeine in decreasing the hydrolytic activity of ectonucleotidases in melanoma cells.

ATP and ADP hydrolysis measured ATPase/ADPase activity, while AMPase activity was measured by AMP hydrolysis. Extracellular ATP levels were assessed by a sensitive bioluminescent assay. In the range of 1.0–6.0 mM of caffeine, the treatment reduced ATP hydrolysis (A). ADP nucleotide hydrolysis (B) increased with 0.5 mM but decreased with 1.0–6.0 mM caffeine. AMP hydrolysis after 24 h of caffeine treatment (1.0–6.0 mM) was also reduced (C). Extracellular ATP levels were increased at 2.0 mM, 4.0 mM and 6.0 mM concentrations (D). All experiments were performed independently three times and in three replicates. Data are presented as mean ± SD. Statistical analysis: one-way ANOVA followed by a post hoc Dunnett’s multiple comparisons test. Values with P < 0.05 were considered statistically significant. *(P < 0.05); **(P < 0.01); ***(P < 0.001); ****(P < 0.0001). CT, control; dac, dacarbazine
Discussion
Stage III and IV CM still have a poor prognosis, despite the development of targeted drugs and immune checkpoint modulators (63 to 68% and 22 to 30% survival rates in 5 years, respectively) [32]. In this sense, studies carried out by Lentini et al. [33], Gude et al. [34], Wrześniok et al. [19] and Tabolacci et al. [35] using other melanoma cell lineages also found caffeine antineoplastic potential. Fagundes et al. [36] showed that low concentrations of this compound could potentiate the effects of dacarbazine. In this study, we found that caffeine has an antineoplastic effect on metastatic melanoma cell lineage SK-MEL-28, reducing viability and inhibiting migration. Our study highlights, for the first time, that caffeine modulates the CD39/CD73 axis in SK-MEL-28 cells.
To achieve these findings, we searched for a possible cell pathway to more robustly explain the caffeine antineoplastic effect on SK-MEL-28 cells. In this field, Eini et al. [18] showed that caffeine has the potential to modulate the purinergic system. This extracellular cell–cell signalling pathway is a ubiquitous system considered a therapeutic target to be used in the treatment of CM and plays a role in regulating numerous physiological and pathological processes in mammals [37]. Research showed that ectonucleotidases, specifically CD39 and CD73, have critical functioning in the purinergic cascade, regulating nucleotides and nucleosides in the TME of melanoma [38,39,40]. We evaluated the gene and protein expression of CD39 and CD73 and the hydrolysis of ATP, ADP and AMP after treatment of CM cells with caffeine. Interestingly, we found that caffeine strongly decreased the expression of CD39 and CD73 and, consequently, the enzyme activity in nucleotide breakdown. It is important to emphasise that this enzymatic axis is responsible for regulating the amount of extracellular ATP in the purinergic cascade [11].
High ATP levels in the TME would be more available to exert agonistic actions on P2X receptors since it is the preferred molecule agonist for these receptor types [41]. Amongst them, P2X7 receptors are overexpressed in neoplastic cell membranes [42]. In melanoma, a high concentration of ATP can lead to cell death by activating the P2X7 receptors that are involved in mTOR signalling [43]. Considering that our results showed that the purinergic cascade could be displaced towards an increase in ATP, we measured the extracellular ATP levels by a sensitive bioluminescent assay to verify these results. We found high levels of extracellular ATP at the same caffeine concentrations where CD39 and CD73 genic, protein and hydrolytic activity was decreased, confirming that caffeine modulates purinergic signalling in SK-MEL-28 melanoma cells, specifically on the CD39/CD73 axis. One of the main concerns in developing anticancer therapies is that in addition to blocking ATP hydrolysis for a complete response, it would be necessary to block adenosine receptors, mainly A2A [44]. In this context, Eini et al. [18] demonstrated that caffeine can antagonise adenosine receptors (e.g. P1 receptors) and promote an effective antitumour immune response, making caffeine a promising xanthine in treating metastatic melanoma.
We also evaluated cell viability using an MTT assay and parallel fluorescence microscopy staining with AO and TMRE. Using flow cytometry, we also verified the potential of caffeine to induce apoptosis in melanoma cells. We found that caffeine substantially decreased SK-MEL-28 cell viability at all tested concentrations compared to CT, a similar effect to dacarbazine. At concentrations of 0.5 mM, 4.0 mM and 6.0 mM of treatment, caffeine induced apoptosis. Both in AO and TMRE staining, caffeine reduced fluorescence intensity percentage, confirming the results found with the MTT assay. A study in vitro that used very similar substance concentrations to ours found that caffeine, by decreasing HDAC1 activity and/or increasing p300 activity, significantly decreased RT2 glioma cell viability after 48 h of treatment [45]. Cheng et al. [46] also showed that, via cathepsin B and MAPK signalling, treatment with caffeine reduced the viability of the glioma cell lineages U-87MG, LN229 and GBM8401. Using Mel1 and Mel3 melanoma cells, Tabolacci et al. [35] showed significantly reduced cell proliferation after 2.0 mM of caffeine treatment in a dose-dependent manner. In parallel, we also performed a cell viability assay to evaluate the cytotoxic effect of caffeine on non-neoplastic cells using CCD-1059sk as a model. However, in our study, caffeine had no cytotoxic effect on non-tumoural cells.
Another important parameter related to melanoma is metastasis, which is a complex biological process that involves cell migration and colonisation of adjacent sites [47]. For metastasis to occur, cells adhere to the extracellular matrix (ECM) or other cells, with the specific binding of cell adhesion molecules such as selectins, integrins and cadherins to ECM ligands or other cells [48]. Methods to study cell migration, such as wound-healing scratch assays, are very useful research tools [23]. Thus, we assessed the effects of caffeine on cell migration and found that all treatment concentrations could reduce the migration of SK-MEL-28 cells. In addition, at high concentrations, caffeine also modifies cell morphology. Furthermore, as expected, dacarbazine also inhibited cell migration. Wang et al. [17] found reductions in prostate cancer cell migration in a study that combined atorvastatin and caffeine. Another study confirmed that caffeine inhibits NCI-H23 cell migration, a mechanism involved in lung cancer metastasis [49].
In the context of melanoma, dacarbazine is one of the primary drugs used for treatment [50]. Dacarbazine targets the overproduction of ROS, a mechanism involved in the cytotoxic effect of this antineoplastic [51, 52]. Similarly, this mechanism was also linked with caffeine cytotoxicity potential in CM cells, where other cell lineages treated with this substance showed an increase in lipid peroxidation and depletion in glutathione levels leading to DNA damage with induced cell death [19, 36, 53, 54]. For this reason, we evaluated the oxidative biomarkers ROS and NOx levels in SK-MEL-28 cells treated with caffeine. We found an increased ROS level at 6.0 mM of treatment. Although caffeine did not alter NOx levels, dacarbazine induced an increase in NOx. On the other hand, PSH levels increased at high concentrations of caffeine within 24 h of treatment. This suggests that an increase in PSH levels at the same concentrations of caffeine that increased ROS may be a metabolic regulation to neutralise the overproduction of reactive species. With this, caffeine’s possible mechanism of action in reducing the viability and migration of melanoma cells may be linked to oxidative stress at high concentrations.
Study limitations
One of the limitations of our study was that we did not use CD39 and/or CD73 inhibitors to verify the specific activity of these enzymes in the hydrolysis of nucleotides in the purinergic cascade. Furthermore, we did not test the possible antagonistic effects of caffeine on P2X7 and A2A receptors.
Conclusion
This study shows that caffeine reduces viability, induces apoptosis, inhibits migration, induces oxidative stress and modulates the purinergic system components in metastatic CM cell lineage SK-MEL-28. In an unprecedented manner, we also showed that caffeine downregulates CD39 and CD73 expression and the hydrolysis of ATP, ADP and AMP, resulting in a displacement of the CD39/CD73 axis towards an increase in extracellular ATP. Thus, caffeine may be an interesting and promising coffee-derived compound to be widely studied in melanoma research, as it modulates purinergic signalling by controlling the levels of extracellular ATP by modulating the CD39/CD73 axis and the antagonistic effects on P1 receptors. Finally, further studies that focus on understanding the potential effects of caffeine on P2X7 and A2A receptors are required.
Data availability
All data are available in the main text or supplementary materials.
References
Paddock LE, Lu SE, Bandera EV, Rhoads GG, Fine J, Paine S, Barnhill R, Berwick M (2016) Skin self-examination and long-term melanoma survival. Melanoma Research 26:401
Ferlay J, Soerjomataram I, Ervik M, Dikshit R, Eser S, Mathers C, Rebelo M, Parkin DM, Forman D, Bray F in GLOBOCAN 2020, Global Cancer Observatory: cancer today. International Agency for Research on Cancer, Lyon https://gco.iarc.fr/today. Accessed 02 October 2022
Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, Bray F (2021) Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA A Cancer J Clin 71:209
Carr S, Smith C, Wernberg J (2020) Epidemiology and risk factors of melanoma. Surgical Clinics of North Am 100:1
O’Day SJ, Hamid O, Urba WJ (2007) Targeting cytotoxic T-lymphocyte antigen-4 (CTLA-4): a novel strategy for the treatment of melanoma and other malignancies. Cancer 110:2614
Carlino MS, Larkin J, Long GV (2021) immune checkpoint inhibitors in melanoma. Lancet 398:1002
Flaherty KT et al (2012) Improved survival with MEK inhibition in BRAF-mutated melanoma. N Engl J Med 367:107
Lee J et al (2023) BRAF inhibitor cessation prior to disease progression in metastatic melanoma: long-term outcomes. European Journal of Cancer 179:87
Gao Z, Dong K, Zhang H (2014) The roles of CD73 in cancer. BioMed Res Int 2014:1
Ferrari D, Malavasi F, Antonioli L (2017) A purinergic trail for metastases. Trends Pharmacol Sci 38:277
Di Virgilio F, Sarti AC, Falzoni S, De Marchi E, Adinolfi E (2018) Extracellular ATP and P2 purinergic signalling in the tumour microenvironment. Nat Rev Cancer 18:601
Mânica A, da Silva Rosa B, Bonadiman AM, Cardoso A, Paiz C, Siepko JVG, de Souza M, Moreno A, Moreno MRC, Schetinger VM, Bagatini MD (2019) The signaling effects of ATP on melanoma-like skin cancer. Cell Signal 59:122
da Silva GB, Yamauchi MA, Zanini D, Bagatini MD (2022) Novel possibility for cutaneous melanoma treatment by means of rosmarinic acid action on purinergic signaling. Purinergic Signal 18:61
Pelinson LP et al (2019) Antiproliferative and apoptotic effects of caffeic acid on SK-Mel-28 human melanoma cancer cells. Mol Biol Rep 46:2085
Ashihara H, Sano H, Crozier A (2008) Caffeine and related purine alkaloids: biosynthesis, catabolism, function and genetic engineering. Phytochemistry 69:841
Miwa S, Sugimoto N, Yamamoto N, Shirai T, Nishida H, Hayashi K, Kimura H, Takeuchi A, Igarashi K, Yachie A, Tsuchiya H (2012) Caffeine induces apoptosis of osteosarcoma cells by inhibiting AKT/mTOR/S6K, NF-κB and MAPK pathways. Anticancer Res 32:3643
Wang Z, Zhang L, Wan Z, He Y, Huang H, Xiang H, Wu X, Zhang K, Liu Y, Goodin S, Du Z, Zheng X (2020) Atorvastatin and caffeine in combination regulates apoptosis, migration, invasion and tumorspheres of prostate cancer cells. Pathol Oncol Res 26:209
Eini H, Frishman V, Yulzari R, Kachko L, Lewis EC, Chaimovitz C, Douvdevani A (2015) Caffeine promotes anti-tumor immune response during tumor initiation: involvement of the adenosine A2A receptor. Biochem Pharmacol 98:110
Wrześniok D, Rzepka Z, Respondek M, Beberok A, Rok J, Szczepanik K, Buszman E (2018) Caffeine modulates growth and vitality of human melanotic COLO829 and amelanotic C32 melanoma cells: preliminary findings. Food Chem Toxicol 120:566
Mosmann T (1983) Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods 65:55
McGahon AJ, Martin SJ, Bissonnette RP, Mahboubi A, Shi Y, Mogil RJ, Nishioka WK, Green DR (1995) In: Schwartz LM, Osborne BA (eds) Chapter 9 the end of the (cell) line: methods for the study of apoptosis in vitro in methods in cell biology. Academic Press, pp 153–185
Joshi DC, Bakowska JC (2011) Determination of mitochondrial membrane potential and reactive oxygen species in live rat cortical neurons. JoVE 2704
Justus CR, Leffler N, Ruiz-Echevarria M, Yang LV (2014) In vitro cell migration and invasion assays. JoVE 51046
Romitelli F, Santini SA, Chierici E, Pitocco D, Tavazzi B, Amorini AM, Lazzarino G, Di Stasio E (2007) Comparison of nitrite/nitrate concentration in human plasma and serum samples measured by the enzymatic batch Griess assay, ion-pairing HPLC and ion-trap GC–MS: the importance of a correct removal of proteins in the Griess assay. J Chromatograph B 851:257
Zahedi Asl S, Ghasemi A, Azizi F (2008) Serum nitric oxide metabolites in subjects with metabolic syndrome. Clin Biochem 41:1342
Tatsch E, Bochi GV, da Pereira SR, Kober H, Agertt VA, de Campos MMA, Gomes P, Duarte MMMF, Moresco RN (2011) A simple and inexpensive automated technique for measurement of serum nitrite/nitrate. Clin Biochem 44:348
Ellman GL (1959) Tissue sulfhydryl groups. Arch Biochem Biophys 82:70
Pilla C, Emanuelli T, Frassetto SS, Battastini AMO, Dias RD, Sarkis JJF (1996) ATP diphosphohydrolase activity (apyrase, EC 3.6.1.5) in human blood platelets. Platelets 7:225
Lunkes GI, Lunkes D, Stefanello F, Morsch A, Morsch VM, Mazzanti CM, Schetinger MRC (2003) Enzymes that hydrolyze adenine nucleotides in diabetes and associated pathologies. Thrombosis Res 109:189
Karamohamed S, Guidotti G (2001) Bioluminometric method for real-time detection of ATPase activity. BioTechniques 31:420
Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72:248
Miller R, Walker S, Shui I, Brandtmüller A, Cadwell K, Scherrer E (2020) Epidemiology and survival outcomes in stages II and III cutaneous melanoma: a systematic review. Melanoma Manag 7:MMT39
Lentini A, Kleinman HK, Mattioli P, Autuori-Pezzoli V, Nicoli L, Pietrini A, Abbruzzese A, Cardinali M, Beninati S (1998) Inhibition of melanoma pulmonary metastasis by methylxanthines due to decreased invasion and proliferation. Melanoma Res 8:131
Gude RP, Menon LG, Rao SG (2001) Effect of caffeine, a xanthine derivative, in the inhibition of experimental lung metastasis induced by B16F10 melanoma cells. J Exp Clin Cancer Res 20:287
Tabolacci M, Cordella S, Rossi M, Bonaccio A, Eramo C, Mischiati S, Beninati L, Iacoviello AF, Facchiano F (2021) Targeting melanoma-initiating cells by caffeine: in silico and in vitro approaches. Molecules 26:3619
Fagundes TR, Madeira TB, Melo GP, Bordini HP, Marinello PC, Nixdorf SL, Cecchini AL, Luiz RC (2022) Caffeine improves the cytotoxic effect of dacarbazine on B16F10 murine melanoma cells. Bioorganic Chem 120:105576
Burnstock G (1997) The past, present and future of purine nucleotides as signalling molecules. Neuropharmacology 36:1127
Young A, Ngiow SF, Barkauskas DS, Sult E, Hay C, Blake SJ, Huang Q, Liu J, Takeda K, Teng MWL, Sachsenmeier K, Smyth MJ (2016) Co-inhibition of CD73 and A2AR adenosine signaling improves anti-tumor immune responses. Cancer Cell 30:391
Soleimani A, Farshchi HK, Mirzavi F, Zamani P, Ghaderi A, Amini Y, Khorrami S, Mashayekhi K, Jaafari MR (2020) The therapeutic potential of targeting CD73 and CD73-derived adenosine in melanoma. Biochimie 176:21
Bagatini MD, Bertolin K, Bridi A, Pelinson LP, da Silva Rosa B, Bonadiman MM, Pillat PBD, Gonçalves H, Ulrich MRC, Schetinger MR, Morsch VM (2019) 1α, 25-Dihydroxyvitamin D3 alters ectonucleotidase expression and activity in human cutaneous melanoma cells. J of Cell Biochem 120:9992
Jacobson KA, Müller CE (2016) Medicinal chemistry of adenosine, P2Y and P2X receptors. Neuropharmacology 104:31
Adinolfi E, Giuliani AL, De Marchi E, Pegoraro A, Orioli E, Di Virgilio F (2018) The P2X7 receptor: a main player in inflammation. Biochem Pharmacol 151:234
Bian S, Sun X, Bai A, Zhang C, Li L, Enjyoji K, Junger WG, Robson SC, Wu Y (2013) P2X7 Integrates PI3K/AKT and AMPK-PRAS40-mTOR signaling pathways to mediate tumor cell death. PLoS One 8:e60184
Kepp O, Bezu L, Yamazaki T, Di Virgilio F, Smyth MJ, Kroemer G, Galluzzi L (2021) ATP and cancer immunosurveillance. EMBO J 40
Chen J-C, Hwang J-H (2016) Effects of caffeine on cell viability and activity of histone deacetylase 1 and histone acetyltransferase in glioma cells. Tzu Chi Med J 28:103
Cheng Y-C, Ding Y-M, Hueng D-Y, Chen J-Y, Chen Y (2016) Caffeine suppresses the progression of human glioblastoma via cathepsin B and MAPK signaling pathway. The J Nutri Biochem 33:63
Valastyan S, Weinberg RA (2011) Tumor metastasis: molecular insights and evolving paradigms. Cell 147:275
Trepat X, Chen Z, Jacobson K (2012) In: Terjung R (ed) Cell migration in comprehensive physiology, 1st edn. (Wiley, pp 2369–2392
Meisaprow P, Aksorn N, Vinayanuwattikun C, Chanvorachote P, Sukprasansap M (2021) Caffeine induces G0/G1 cell cycle arrest and inhibits migration through integrin αv, β3, and FAK/Akt/c-Myc signaling pathway. Molecules 26:7659
Bhatia S, Tykodi SS, Lee SM, Thompson JA (2015) Systemic therapy of metastatic melanoma: on the road to cure. Oncology 29:126
Pourahmad J, Amirmostofian M, Kobarfard F, Shahraki J (2009) Biological reactive intermediates that mediate dacarbazine cytotoxicity. Cancer Chemother Pharmacol 65:89
Amirmostofian M, Pourahmad Jaktaji J, Soleimani Z, Tabib K, Tanbakosazan F, Omrani M, Kobarfard F (2013) Synthesis and molecular-cellular mechanistic study of pyridine derivative of dacarbazine. Iran. J Pharm Res 12:255
Shukla V, Gude RP (2003) Potentiation of lipid peroxidation in B16F10 and B16F1 melanoma cells by caffeine, a methylxanthine derivative: relationship to intracellular glutathione. Chemotherapy 49:71
Caini S et al (2017) Coffee, tea and melanoma risk: findings from the European Prospective Investigation into Cancer and Nutrition: coffee, tea and melanoma risk. Int J Cancer 140:2246
Funding
This study is funded by grants from Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) (MDB), Fundação de Amparo à Pesquisa e Inovação do Estado de Santa Catarina (FAPESC) (MDB). MDB is 2 CNPq Research Fellow. DM and GBS are thankful to the Fundo de Apoio à Manutenção e ao Desenvolvimento da Educação Superior for the scholarships.
Author information
Authors and Affiliations
Contributions
Conceptualisation: DM, GBS, JL, JC, MDB and MM.
Methodology: DM and GBS.
Investigation: DM, GBS, JL, JC, PD and RAN.
Visualisation: DM, GBS, JL, JC, PD, RAN, MDB and MM.
Funding acquisition: MDB and MM.
Project administration: MDB and MM.
Supervision: MDB and MM.
Writing—original draft: DM and GBS.
Writing—review and editing: DM and GBS.
All authors reviewed and approved the final version.
Corresponding authors
Ethics declarations
Ethical approval
Not applicable.
Conflicts of interest
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Highlights
- Several researchers have demonstrated the antineoplastic effects of caffeine and its involvement in some cell signaling pathways.
- We found that caffeine has antineoplastic effects on SK-MEL-28 cells, thereby reducing cell viability and migration, inducing apoptosis and modulating purinergic signalling.
- Caffeine downregulates expression of CD39 and CD73 and ATPase/ADPase/AMPase enzyme activity of ectonucleotidases in cutaneous melanoma cells.
- The CD39/CD73 axis was displaced, resulting in greater extracellular ATP accumulation.
- Caffeine may be an interesting coffee-derived compound for future clinical trials with the CD39/CD73 axis as a target.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Manica, ., da Silva, G.B., de Lima, J. et al. Caffeine reduces viability, induces apoptosis, inhibits migration and modulates the CD39/CD73 axis in metastatic cutaneous melanoma cells. Purinergic Signalling 20, 385–397 (2024). https://doi.org/10.1007/s11302-023-09967-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11302-023-09967-6