
Abstract
Clopidogrel is a widely prescribed prodrug with anti-thrombotic activity through irreversible inhibition of the P2Y12 receptor on platelets. It is FDA-approved for the clinical management of thrombotic diseases like unstable angina, myocardial infarction, stroke, and during percutaneous coronary interventions. Hepatic clopidogrel metabolism generates several distinct metabolites. Only one of these metabolites is responsible for inhibiting the platelet P2Y12 receptor. Importantly, various non-hemostatic effects of clopidogrel therapy have been described. These non-hemostatic effects are perhaps unsurprising, as P2Y12 receptor expression has been reported in multiple tissues, including osteoblasts, leukocytes, as well as vascular endothelium and smooth muscle. While the “inactive” metabolites have been commonly thought to be biologically inert, recent findings have uncovered P2Y12 receptor-independent effects of clopidogrel treatment that may be mediated by understudied metabolites. In this review, we summarize both the P2Y12 receptor-mediated and non-P2Y12 receptor-mediated effects of clopidogrel and its metabolites in various tissues.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Platelets are activated by several endogenous chemical mediators, including adenosine diphosphate (ADP), thrombin, and thromboxane. One therapeutic strategy to inhibit the pathological action of platelets in thrombotic diseases is selective inhibition of one specific receptor pathway, such as ADP-induced activation of purinergic receptor 2Y12 (P2Y12). The P2Y12 receptor is a G-protein coupled receptor (GPCR) that drives platelet activation. Activation of the P2Y12 receptor inhibits adenylyl cyclase thereby decreasing the generation of cAMP within the platelet. Activation of the P2Y1 receptor on platelets increases IP3 production to increase cytoplasmic calcium. P2Y12 and P2Y1 receptor activation increase intracellular calcium concentration which induces platelet shape change and aggregation. Additionally, activation of platelets through other agonists, such as arachidonic acid and collagen, causes secretion of ATP/ADP-containing dense granules resulting in subsequent activation of the P2Y12 receptor, thereby potentiating platelet aggregation [1]. Therefore, it is crucial to inhibit this receptor. However, it is common to combine therapeutic agents to inhibit multiple platelet activation pathways. Dual anti-platelet therapy is commonly used for the treatment of thrombotic diseases and involves the combined administration of low-dose aspirin, which inhibits the cyclooxygenase-dependent activation pathway, with a P2Y12 receptor antagonist. Clopidogrel is the favored P2Y12 receptor antagonist for long-term dual anti-platelet therapy. Indications for its use include percutaneous coronary intervention (balloon angioplasty and stent implantation), acute coronary syndrome, and secondary prevention post-coronary artery bypass graft.
An early thienopyridine anti-platelet agent, ticlopidine, was associated with severe hematological side effects including leucopenia and thrombocytopenia. To address these adverse effects, thousands of analogs of ticlopidine were generated in the hope of identifying novel compounds with improved risk/benefit profiles. One such analog, clopidogrel, underwent preclinical evaluation starting in 1987 with ultimate approval for use in the USA, coming in 1997, and a worldwide launch in 1998 [2]. Interestingly, at the time of its approval, the molecular target of clopidogrel was unknown, although its action was known to be unique from aspirin, sulfinpyrazone, and dipyridamole. Early thienopyridine anti-platelet agents were known to inhibit the platelet ADP receptor [3]. Subsequently, it was recognized that clopidogrel was also a potent inhibitor of ADP-mediated platelet aggregation [4,5,6,7,8,9,10,11]. The discovery of the P2Y12 receptor in 2000 [2, 12, 13] paved the way for its identification as the primary pharmacologic target of clopidogrel in 2001 [14].
Clopidogrel is a prodrug that requires hepatic bioactivation to generate the active metabolite responsible for inhibiting platelets [15]. The active metabolite of clopidogrel (H4) inhibits platelets through covalent interactions, forming disulfide bridges, with cysteine residues within the ligand binding domain (Cys17 and Cys270) of the P2Y12 receptor [16]. Interestingly, the identity of the active metabolite and enzymes (CYP450s) responsible for its formation were a mystery at the time of clopidogrel’s approval. In fact, the clinical pharmacokinetics of clopidogrel were determined using the plasma concentration of the primary circulating metabolite (SR26334) as a surrogate [17]. This carboxylic acid derivative is a product of esterase-dependent metabolism and is not responsible for inhibiting platelet aggregation. The chemical structure of the active metabolite was characterized in 2000 [18], and the important enzymes involved in its formation were systematically identified with the last one proposed in 2011 [2, 19]. The pharmacology of SR26334 and other clopidogrel metabolites has not been extensively investigated, although ample evidence suggests meaningful biological effects of these abundant metabolites.
Clopidogrel metabolism
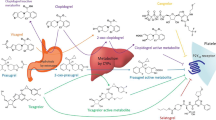
To accelerate the anti-platelet effects, clopidogrel is prescribed at an initial loading dose of 300 mg followed by a maintenance dose of 75 mg/day. Clopidogrel is readily absorbed in the intestine and then converted into several distinct metabolites by a variety of metabolic enzymes, including carboxylic esterase 1 (CES1), members of the cytochrome P450 family (CYP450s), and paraoxonase 1 (PON1). Approximately 85% of the prodrug is converted into the carboxylic acid metabolite (SR26334) by CES1, while only about 5% of the prodrug is ultimately converted into the active metabolite (H4) by a two-step CYP450-mediated process (Fig. 1). The enzymes CYP2C19 and CYP3A4 are critically important for the catalysis of these reactions [20,21,22,23,24]. While it is clear that H4 is responsible for inhibiting the P2Y12 receptor on platelets, the biological effect(s) of the other metabolic products remain unknown.

The proposed bioactivation pathway of the clopidogrel prodrug is dependent upon a complicated, multistep, enzyme-dependent metabolic process. The majority of the ingested prodrug is hydrolyzed by esterases (including CES1) to form the carboxylic acid metabolite (SR26334 or M1). The remaining clopidogrel is metabolized by CYP450 enzymes (primarily CYP2C19 and CYP3A4), producing various chemical products (M2-M17) including M13, the metabolite responsible for inhibiting the P2Y12 receptor. M13 represents a mixture of diastereomers of which only one, H4, has clinical relevance [120]. Metabolism information is summarized from previous reports [2, 118, 121,122,123]
The complex nature of clopidogrel metabolism has significant potential to induce variation in patient responses. A decrease in the anti-platelet activity is observed in patients with loss of function polymorphisms in CYP2C19 (*2 and *3 alleles) [25,26,27]. Patients with CYP2C19 polymorphism have decreased H4 concentrations and increased SR26334 concentrations [26, 28]. Insufficient clopidogrel conversion to H4 results in poor clinical response, and therefore CYP2C19 loss of function carriers have limited protection from thrombotic events when treated with clopidogrel compared to individuals with normal enzyme function [26]. Due to interpatient variability and the potential for lack of clinical response, the United States Food and Drug Administration added a Black Box Warning to (1) warn patients about the reduced effectiveness for those who do not effectively metabolize the prodrug, (2) inform clinicians to evaluate patients for CYP2C19 activity, and (3) instruct clinicians to select other anti-platelet therapeutics for those who do not effectively metabolize clopidogrel [29].
Clopidogrel response is further complicated by various interactions with other drugs. CES1, CYP2C19, and CYP3A4 are commonly involved with the metabolism of other pharmaceuticals. Therefore, induction or inhibition of these enzymes by other pharmaceutical agents could affect the bioactivation and clinical response of clopidogrel. For instance, omeprazole, a commonly prescribed proton pump inhibitor used in the treatment of gastroesophageal reflux disease, competitively inhibits CYP2C19 and, in doing so, modifies the bioactivation of clopidogrel. Previous studies demonstrate that platelet inhibition decreased in patients receiving both omeprazole and clopidogrel compared to the patients receiving clopidogrel alone [25, 30]. Furthermore, ethanol consumption also impacts clopidogrel metabolism by increasing the formation of the H4 metabolite resulting in an increase in platelet inhibition [31]. There is decreased SR26334 metabolite concentration with ethanol (3 g/kg) consumption which appears to result from a shift from the CES1 metabolic pathway toward CYP450-mediated metabolism [32].
Type II diabetes and insulin resistance have been identified as risk factors for diminished clopidogrel response. Generation of H4 is decreased by 40% in diabetic patients compared to non-diabetic patients [33,34,35]. Diet-induced obese (DIO) mice, which are a useful model of human type II diabetes, have a diminished response to the clopidogrel prodrug yet respond normally to a conjugate of the active metabolite that does not require enzymatic activation [36]. Interestingly, DIO IL-1 receptor knockout (IL-1R−/−) mice were able to overcome the clopidogrel resistance as a consequence of increased CYP450 expression and therefore increased H4 generation [36]. Ultimately, it appears diabetes downregulates CYP2C19, resulting in reduced formation of the active metabolite and diminished anti-platelet effects.
Most of the metabolites of clopidogrel are electrophilic species that may unselectively bind to cellular and circulating macromolecules. While not all binding events lead to damaging biological effects, previous reports indicate these reactive metabolites can cause various effects. The primary objective of this review is to present a comprehensive list of all the reported off-target effects of clopidogrel (Fig. 2).
Off-target clopidogrel effects
Non-hemostatic P2Y12 receptor-mediated effects of clopidogrel
Clopidogrel treatment is prescribed for the inhibition of the P2Y12 receptor on platelets to prevent activation and aggregation. While efficacious in that mechanism, several other non-hemostatic P2Y12 receptor effects have been described. Platelets interact with leukocytes and endothelial cells during stress and inflammation [37]. Additionally, platelets express Toll-like receptors, allowing for interactions with neutrophils and monocytes to initiate immune responses [37]. The P2Y12 receptor is also suggested to be expressed in microglia [38,39,40], smooth muscle, and endothelial cells [41,42,43,44]. Furthermore, the P2Y12 receptor is putatively expressed in several other tissues including the brain, reproductive organs, thyroid, lung, adrenal gland, tongue, esophagus, kidney, liver, colon, bladder, heart, skin, spleen, lymph node, pituitary gland, retina, salivary gland, stomach, gull bladder, adipose tissue, tonsil, appendix, and bone marrow [45]. However, it is important to note that tissue-specific expression is difficult to rigorously demonstrate, as samples are often contaminated with platelets. Adequate antibodies for the P2Y12 receptor do not exist; therefore, only RT-PCR is useful for investigating receptor expression. While the focus of clopidogrel actions has been on platelets, the proposed broad tissue-specific expression of the P2Y12 receptor likely enables multiple mechanisms whereby clopidogrel treatment mediates non-hemostatic P2Y12 receptor effects. Due to the complex interactions platelets have with other circulating cells and the vessel wall, as well as the reputed broad expression of the P2Y12 receptor within the body, it is difficult to distinguish which clopidogrel effects are mediated by local P2Y12 receptor inhibition versus indirect effects of platelet inhibition.

Clopidogrel effects have been described in various tissues in both P2Y12 receptor-dependent and independent manners
Modulation of atherogenesis and the progression of atherosclerosis
Atherosclerosis is an immunoinflammatory disease of medium to large arteries that results in the deposition of fatty plaques on the artery wall. Atherosclerosis is not life-threatening itself, but occlusive thrombus formation due to plaque rupture can induce downstream ischemia resulting in unstable angina, stroke, or myocardial infarction. Endothelial cells, leukocytes, and smooth muscle cells are all cellular mediators of atherosclerosis. Lipoprotein particles penetrate the endothelial layer into the subendothelial space, where they become pro-inflammatory. The endothelium is then activated by inflammatory cytokines to express adhesion molecules that recruit blood-borne cells to the atherosclerotic lesion. The inflammation leads to the recruitment of monocytes and T-lymphocytes [46]. As the disease progresses, intimal smooth muscle cells heal and repair the arterial injury. Smooth muscle cells stabilize the plaque, decreasing the chance of rupture but also narrowing the vascular lumen, thereby reducing blood flow [46, 47].
Several studies have demonstrated clopidogrel’s ability to reduce atherosclerosis [48,49,50,51,52]. Activated platelets release platelet-derived growth factor, which causes the secretion of matrix metalloproteinase-2. Matrix metalloproteinases degrade several extracellular matrix proteins to promote inflammation [53]. Platelets also induce monocyte chemotactic protein-1 (MCP-1) and vascular cell adhesion molecule-1 (VCAM-1) expression in endothelial cells, which initiates monocyte recruitment and plays a role in atherosclerotic lesion formation [54, 55]. Indeed, the patients receiving clopidogrel have a significant reduction in atherosclerotic plaque inflammation [56]. Clopidogrel has also been reported to significantly reduce atherosclerotic lesion formation and reduce the inflammatory response in ApoE-deficient mice, a useful animal model for understanding the pathophysiology of atherosclerosis [48, 57]. Bone marrow transplants were performed in P2Y12 receptor knockout mice to differentiate between vessel wall or platelet-derived effects. Atherosclerotic lesions were reduced in vessel wall depleted of the P2Y12 receptor, indicating the vessel wall P2Y12 receptor is involved in the development of atherosclerosis [58]. In a rabbit model of atherosclerosis, animals were fed a high cholesterol diet followed by balloon injury to the iliac artery. The rabbits were treated with clopidogrel throughout the study and monitored for the development of atherosclerosis. Treatment with clopidogrel significantly reduced vascular inflammation and atherosclerotic lesion formation while decreasing the expression of P-selectin, intracellular adhesion molecule-1, VCAM-1, and MCP-1 [49].
Inhibition of hypertension-associated inflammation
Angiotensin II (Ang II) activates the angiotensin type 1 receptor and upregulates Toll-like receptor 4. This activates the myeloid differentiation primary response protein 88 (MyD88) and mitogen-activated protein kinase (MAPK). MAPK activates nuclear factor kappa-light-chain-enhancer of activated B cells (NF-kB), resulting in a release of pro-inflammatory mediators. Ang II also causes the oxidation of NADPH to generate reactive oxygen species (ROS) [59]. Moreover, Ang II-mediated inflammation induces platelet activation and subsequent platelet-monocyte binding leading to monocyte activation. Platelet-monocyte binding enhances vascular inflammatory responses [60]. Both an increase in pro-inflammatory mediators and the generation of ROS lead to hypertension. Therefore, chronic Ang II administration is commonly used as a model of hypertension in rats and mice. Since platelets are involved in Ang II-associated hypertension, the inhibition of platelets was assessed with clopidogrel treatment. Phenylephrine contraction and acetylcholine relaxation were impaired in mesenteric arteries from Ang II mice and rats. Interestingly, clopidogrel treatment prevented these effects in an endothelium-dependent manner [60,61,62]. Additionally, clopidogrel treatment improved the structure of hypertensive arteries. Chronic Ang II-treated mice exhibit vascular remodeling and increased stiffness due to elevated arterial pressure. Aortas collected from these animals have increased wall thickness, increased wall-to-lumen ratio, and exhibit impaired vasodilation. Clopidogrel treatment decreased the hypertension-associated changes in aortic structure. [60, 62]. Ang II-treated mice also develop increased vascular oxidative stress, an effect that is completely abolished by clopidogrel treatment. Vascular NADPH oxidase (NOX)1, NOX2, and NOX4 mRNA and protein levels are increased with Ang II treatment, and concomitant treatment with clopidogrel decreases these levels [60, 62]. The beneficial effects of clopidogrel in hypertension are likely due to reduced infiltration of macrophages in the aorta, since macrophages are the main source of ROS in vessels, as well as decreased platelet-monocyte binding [60, 63].
Inhibition of angiogenesis
The healing of gastric ulcers requires cell proliferation and angiogenesis. Ischemic tissues release leukotriene B to attract leukocytes and macrophages. These cells phagocytize the necrotic tissue and release pro-inflammatory cytokines to activate fibroblasts, endothelial, and epithelial cells [64]. As a result, endothelial cells migrate, proliferate, and re-establish the microvascular network [65]. Rats were subjected to an experimental model of gastric ulceration involving the luminal application of acetic acid. Twenty-four hours later, daily clopidogrel therapy was initiated, and ulcer healing was observed. Clopidogrel treatment increased ulcer size and therefore delayed gastric ulcer healing. Moreover, clopidogrel decreased the number of microvessels at the ulcer base. Protein and mRNA expressions of several angiogenic growth factors (vWF, FGFR2, VEGF, VEGFR2, PDGFRA, and pERK) were significantly decreased with clopidogrel treatment, and angiogenesis was reduced. Clopidogrel inhibited angiogenesis by inhibiting the VEGF-VEGFR2-ERK signaling transduction pathway [66]. The effect of clopidogrel could be partially explained by the inhibition of platelet activation thereby leading to a reduction in the release of platelet-derived growth factor; however, further studies are required to determine the exact mechanism of action.
Inhibition of Ras/Raf/MEK/ERK signaling pathway
Activation of the P2Y12 receptor in human lung epithelial (A549) cells leads to Ras/Raf/mitogen-activated protein kinase (MEK)/extracellular-signal-regulated kinase (ERK) signaling, and inhibition of that pathway has been linked to various biological effects [67,68,69]. The Ras/Raf/MEK/ERK signaling pathway allows for communication between cell surface receptors and downstream transcription factors, which induce cellular proliferation, differentiation, and survival. Several RNA viruses induce the Ras/Raf/MEK/ERK signaling to potentiate their replication [70]. Clopidogrel was examined as a novel treatment for influenza since it targets the same cellular signaling pathway. Calu-3 human bronchial epithelium cells, a common cell line used to study influenza infection, were tested to evaluate the effects of clopidogrel. In vitro clopidogrel treatment decreased the percentage of influenza-infected Calu-3 cells, and pretreatment with clopidogrel reduced viral replication [71]. These results suggest that inhibition of influenza replication is linked to the inhibition of the P2Y12 receptor signaling pathway by clopidogrel. However, the clopidogrel prodrug is not readily metabolized in vitro, and therefore it is unclear whether the effects observed were due to P2Y12 receptor inhibition by the active metabolite or an alternative mechanism.
Ras/Raf/MEK/ERK is also a dominant cancer signaling pathway [72]. Mutations of Ras lead to constitutively active Ras proteins, thereby preventing apoptosis. Since apoptosis regulation has been an attractive chemotherapeutic treatment target, chemical inhibitors of this signaling pathway have been considered as potential treatment candidates for many types of cancers [73]. Breast cancer cells have been demonstrated to cause direct and indirect activation of platelets by ADP, thromboxane A2, and metalloproteinases [74,75,76]. Activated platelets release metalloproteinases, which degrade the vascular basement membrane resulting in tumor growth and metastasis [77]. As a result, clopidogrel is hypothesized to possess anti-cancer properties. In an experimental model of mammary cancer (transplant of mouse mammary adenocarcinoma 4T1 cells into recipient mice), clopidogrel alone did not have significant anti-tumor activity. However, the anti-tumor effects of 5-fluorouracil, cyclophosphamide, and mitoxantrone were potentiated by concomitant clopidogrel treatment [78]. The protective mechanism can be explained by both a decrease in invasive tumor cells and a decrease in the accumulation of platelets within the tumors. In addition, clopidogrel administered with a nitric oxide donor effectively inhibits metastasis by normalizing endothelial function [79]. Conversely, clopidogrel decreased the efficacy of doxorubicin, cisplatin, and tamoxifen [78]. These agents are all CYP3A4 substrates, and therefore clopidogrel may affect the metabolism of these chemotherapeutics reducing their efficacy.
Interestingly, clopidogrel may also have utility as a cancer preventative agent. In a prospective trial investigating cancer prevention, the patients receiving clopidogrel with and without aspirin were monitored for cancer development. Clopidogrel use was associated with a decreased incidence of all cancers, including colorectal cancer, which was reduced by 20–30% [80, 81]. Together with the anti-cancer effects outlined above, these results suggest that clopidogrel may be a beneficial adjunctive agent to existing cancer therapeutic strategies. However, these were observational studies; therefore, they do not provide definitive conclusions to the anti-cancer benefits of clopidogrel.
Regulation of bone homeostasis and bone marrow function
The P2Y12 receptor is suggested to be expressed in the bone and bone marrow of rats and mice [40, 45]. The expression of the P2Y12 receptor in the bone marrow is unsurprising as megakaryocytes are located within the bone marrow. Prolonged exposure to clopidogrel has effects on bone mass and bone cell function. Clopidogrel treatment decreased osteoblast number by 50% and reduced cell viability. Bone formation, bone marrow density, and collagen production decreased after clopidogrel treatment [82]. In addition, adipogenic transcription factor levels increased 4.4-fold, and adipocytes increased by 60% with clopidogrel treatment. The reduction in osteoblast number is likely due to clopidogrel’s action on precursor cells causing them to follow an adipogenic differentiation pathway rather than the osteoblastic pathway, leading to an increase in adipocytes [82]. Since osteoblasts express the P2Y12 receptor, it suggests these effects result from clopidogrel-mediated receptor inhibition, but this has not yet been confirmed. Contrasting to these previous studies; however, clopidogrel also enhanced new bone formation in rabbits and mice [83, 84]. These results highlight how differences in dosage, treatment duration, and species play a crucial role in the effects of clopidogrel.
Non-P2Y12 receptor effects of clopidogrel
Metabolites of clopidogrel are primarily considered “inactive” simply owing to their lack of inhibition of the platelet P2Y12 receptor, the primary target of H4. However, many of the other metabolites are structurally similar to H4. In addition, members of the P2Y receptor family share a high degree of similarity in both sequence and structure, while their expression has been demonstrated in a diverse number of cell types and tissues. Therefore, simply assuming that clopidogrel metabolites are biologically “inactive” due to a lack of platelet inhibition is short-sighted. There may be, in fact, clopidogrel metabolites with understudied but entirely unique pharmacology. Furthermore, several clopidogrel metabolites are electrophilic species, increasing the likelihood that they may unselectively interact with macromolecules like DNA or proteins, leading to idiosyncratic effects.
Regulation of hematopoiesis
In human patients, clopidogrel treatment reduced white blood cell (WBC) count. A month after clopidogrel was discontinued, WBC count increased. Interestingly, ticagrelor treatment (a structurally distinct reversible P2Y12 receptor antagonist) did not alter WBC count. However, when the patients who received ticagrelor were transitioned to clopidogrel treatment, a reduction in WBC count was then observed [85]. No change in WBC count with ticagrelor indicates the mechanism to which clopidogrel decreases WBC counts is P2Y12 receptor-independent. Future studies are required to determine the mechanism underlying clopidogrel’s effect on circulating WBCs.
Inhibition of inflammation
Lipopolysaccharide (LPS) is frequently used to induce experimental inflammation in animals. LPS activates multiple intracellular signaling pathways and transcription factors, including NF-kB [86]. Active NF-kB increases inflammatory cytokines, chemokines, and adhesion molecules while regulating cell proliferation, differentiation, and apoptosis [87]. Clopidogrel inhibits the degradation of IKBα and the phosphorylation of p65, thereby suppressing NF-kB signaling, reducing inflammatory cytokines, and preventing apoptosis [88]. When clopidogrel was administered to LPS-treated rats, inflammatory lung and liver injury were reduced. Clopidogrel also reduced the pro-inflammatory cytokine levels in these animals [89]. To determine if these effects were a consequence of P2Y12 receptor inhibition, LPS was administered to P2Y12 receptor knockout mice. TNF-α, IFN-γ, IL-10, IL-6, IL-4, and keratinocyte-derived chemokine cytokine levels were higher in P2Y12 receptor–knockout LPS-treated mice than wild-type LPS-treated mice suggesting that the P2Y12 receptor is protective in this model of inflammation. Interesting, P2Y12 receptor–knockout LPS-treated mice administered clopidogrel exhibited a decrease in inflammation. These results indicate that clopidogrel has P2Y12 receptor-independent effects capable of reducing inflammation [90]. The effects were also confirmed in a human model of LPS-induced inflammation [91]. Clopidogrel treatment reduced IL-6, TNF-α, and CCL2 in LPS-treated human volunteers. Furthermore, the patients undergoing primary percutaneous coronary intervention who were on clopidogrel avoided increases in high sensitivity C-reactive protein, a marker of systemic inflammation [92]. This beneficial effect of clopidogrel provides evidence towards the use of clopidogrel in the treatment of inflammatory diseases.
Change in vascular function
P2Y1, P2Y2, P2Y4, P2Y6, P2Y11, P2Y12, P2Y13, and P2Y14 receptors are expressed on the vascular smooth muscle and endothelium [44, 93, 94]. The relative expression of P2Y receptors varies between vessel beds, and therefore the actions of purinergic modulators differ depending on the vessel assessed. Clopidogrel affects the vasculature, both acutely and chronically [60,61,62, 66, 95,96,97,98,99,100,101]. Acute responses relate to the modulation of vascular function, including the induction of vasodilation and the inhibition of vasoconstriction. Chronic responses involve changes in vessel structure, including regulation of vascular remodeling and the inhibition of angiogenesis.
Clopidogrel induces acute vascular changes without hepatic metabolism [99, 102]. Clopidogrel was administered to Langendorff-prepared guinea pig isolated hearts that were perfused retrogradely through the aorta. An ultrasonic flowmeter monitored coronary flow. Clopidogrel increased coronary flow in a concentration-dependent manner, mediated by endothelium-derived nitric oxide [97]. Another study evaluated caudal arteries isolated from rats and then treated with clopidogrel. Clopidogrel caused a concentration-dependent increase in vasodilation in these vessels. However, in contradiction to the previous study, this finding was not related to endothelium-derived nitric oxide [99]. The opposing results might be explained by the differences in the specific physiology of each vessel.
Additional studies evaluated tail arteries isolated from clopidogrel-treated rats. Perfusion pressure was measured after adding 2-(methylthio)adenosine 5′-diphosphate (2MeSADP), an analog of ADP that activates the P2Y1, P2Y12, and P2Y13 receptors. Clopidogrel treatment did not impair constriction in response to 2MeSADP [98]. Furthermore, the aorta from clopidogrel-treated mice was analyzed for contraction to 2MeSADP, and again clopidogrel did not affect 2MeSADP contraction [96]. Our group evaluated the 2MeSADP-mediated vasoconstriction in middle cerebral arteries (MCA) from rabbits treated with clopidogrel. Again, clopidogrel did not alter 2MeSADP constriction compared to vehicle-treated rabbits [95]. These data strongly suggest that clopidogrel does not inhibit P2Y1-, P2Y12-, and P2Y13-receptor mediated contraction.
To further evaluate the effects of clopidogrel on purinergic receptors in the vasculature, our group subsequently analyzed P2Y2-, P2Y4-, P2Y6-, P2Y11-, and P2Y14-mediated vasoconstriction in rabbit MCA. P2Y11 did not produce a response in MCA and P2Y4-, P2Y6-, and P2Y14-mediated vasoconstriction was not impaired by clopidogrel pretreatment. However, clopidogrel significantly inhibited endothelium-dependent P2Y2-mediated vasoconstriction (Fig. 3) [95].

Modified from Kuszynski et al. [95]
Clopidogrel modulates P2Y2 activation in rabbit MCAs. The effects of clopidogrel on P2Y2-mediated contraction were assessed in rabbit MCAs. Constriction response was evaluated by pressure myography. A Activation by MRS 2768, a P2Y2-selective agonist, was significantly inhibited with clopidogrel (3 mg/kg and 10 mg/kg) treatment. B P2Y2-mediated contraction is endothelium-dependent in rabbit MCAs. The data are presented as the mean ± SEM, n = 5. **p < 0.01 and ****p < 0.0001 when compared with the vehicle‐treated group by two‐way ANOVA followed by Dunnett’s post hoc test.
Changes in vascular structure
Changes in vascular structure are commonly associated with interventional vascular procedures. Intimal hyperplasia is a complication from stent placement, endarterectomy, and vascular reconstruction procedures. To evaluate preventative therapeutics, rats were subjected to carotid endarterectomy. Coadministration of clopidogrel and pravastatin, a hydroxymethylglutaryl coenzyme-A (HMG Co-A) reductase inhibitor used to treat dyslipidemia, significantly decreased intimal hyperplasia and serum cholesterol levels. Pravastatin treatment alone did not reduce intimal hyperplasia [101]. Interestingly, clopidogrel alone also did not decrease intimal hyperplasia [100]. Further studies are required to identify the synergistic mechanism underlying simultaneous administration of pravastatin and clopidogrel. It remains to be determined whether this treatment strategy could be a valuable therapy for reducing intimal hyperplasia in a clinical setting.
Clopidogrel-associated bleeding cannot be explained by platelet inhibition alone
Clopidogrel is associated with adverse bleeding, particularly cerebral microbleeds and intracerebral hemorrhages [103,104,105,106,107]. For instance, 30–40% of the patients who have received clopidogrel for at least 1 year had cerebral microbleeds, and the patients who received clopidogrel for more than 5 years have an increased risk of not only cerebral microbleeds but also macroscopic bleeding [106,107,108,109]. Cerebral microbleeds increase the likelihood of recurrent intracerebral hemorrhage [110]. This is a significant public health concern because dual anti-platelet therapy increases the risk of intracerebral hemorrhage by 42% [111].
Most attribute the adverse bleeding observed with clopidogrel to the anti-platelet properties of the drug. However, several groups have recently discovered that this is not the case. The patients with CYP2C19 polymorphisms, who cannot form the H4 active metabolite, have indistinguishable bleeding events compared to those with normal CYP2C19 function [112]. Selatogrel is a reversible antagonist of the P2Y12 receptor that produces comparable anti-thrombotic effects to clopidogrel, albeit with a wider therapeutic window. Crescence and colleagues compared tail blood loss and bleeding time in selatogrel- and clopidogrel-treated mice. Their results revealed that bleeding time in clopidogrel-treated animals was more than eightfold longer than in selatogrel-treated animals. Additionally, clopidogrel treatment increased blood loss 34-fold, while selatogrel treatment only induced a fourfold increase in blood loss [113]. To further characterize the effect(s) of selatogrel treatment, calcium mobilization was quantified in the endothelial cell layer from cremaster muscle arterioles after damage by laser injury. Calcium mobilization was unchanged in P2Y12 receptor–knockout mice compared to wild-type mice indicating calcium mobilization is a P2Y12 receptor-independent mechanism. Selatogrel did not alter calcium mobilization in P2Y12 receptor–knockout mice compared to vehicle treatment, concluding that selatogrel was a highly selective P2Y12 receptor antagonist devoid of off-target effects [113]. However, the inhibitory effects of clopidogrel on calcium release in P2Y12 receptor–knockout mice were not determined.
Subsequently, André and colleagues evaluated clopidogrel treatment in P2Y12 receptor–knockout mice and found a significant increase in blood loss compared to vehicle-treated P2Y12 receptor–knockout mice [114]. This study represented one of the first reports of the potentiation of bleeding by clopidogrel in P2Y12 receptor–knockout animals and strongly suggests that the bleeding effects associated with this drug are mediated, in part, by P2Y12 receptor-independent effects.
To further characterize the adverse bleeding associated with clopidogrel, our group previously reported the development of a conjugate of the H4 metabolite. In the presence of glutathione, the conjugate releases H4 without the requirement of CYP450-mediated metabolism [115, 116]. At the doses required for platelet inhibition, the H4 conjugate does not significantly increase tongue template bleeding time in rabbits. However, clopidogrel induced a > twofold increase in bleeding time at equally effective anti-platelet dosages [117]. Collectively, the results suggest that the H4 metabolite is not entirely responsible for the increase in bleeding observed with clopidogrel treatment, and that P2Y12 receptor-independent effects of clopidogrel metabolites may exacerbate or directly cause bleeding.
The key to appreciating these P2Y12 receptor-independent effects is likely a complete map of the structure and pharmacology of clopidogrel metabolites. The M15 metabolite was the first to be assessed for non-platelet, non-P2Y12 receptor effects in the body. The M15 endo metabolite of clopidogrel undergoes spontaneous hydrolysis to release hydrogen sulfide (H2S) [118]. H2S is an important regulator of the cardiovascular system and mediates intracellular signal transduction, much like nitric oxide or carbon dioxide. It regulates the cell cycle, apoptosis, and oxidative stress. H2S donors reduce thrombus formation and occlusion [119]. To test the ability of the M15 metabolite to minimize thrombus formation, FeCl3-mediated carotid artery injury was induced in mice and time to occlusion was recorded. The M15 metabolite was shown to prolong time to occlusion in mice significantly. This result provides evidence that a previously classified “inactive” metabolite, M15, may be pharmacologically active through the release of H2S, thereby interfering with hemostasis [118]. These findings suggest that while specific clopidogrel metabolites have not been evaluated for off target bleeding, M15 represents an exciting candidate that might be responsible, in part, for these adverse effects. The understudied metabolites of clopidogrel must be further analyzed to evaluate this possibility.
Conclusion
Clopidogrel is an effective anti-platelet agent used to treat and prevent a variety of thrombotic diseases. It is clinically indicated for the prevention of myocardial infarction, stroke, and transient ischemic attacks in high-risk individuals. Clopidogrel requires extensive metabolism for activation and is affiliated with frequent drug-drug interactions and interpatient variability. A significant concern with clopidogrel metabolism is interpatient variability due to genetic mutations that affect CYP450 function. Additionally, diabetic patients also exhibit reduced CYP450 expression and are associated with decreased formation of H4. Due to these factors, the patients’ CYP450 activity should be evaluated before initiating clopidogrel therapy.
Despite clopidogrel’s extensive use in the clinical management of patients, a comprehensive understanding of the drug’s action in the body is still being uncovered. The P2Y12 receptor is proposed to be expressed in numerous tissues in addition to platelets, including osteoblasts, microglia, and the vasculature. If the P2Y12 receptor is in fact expressed in various tissues, it is unsurprising that clopidogrel has been reported to possess platelet-independent effects. The structural similarity of H4 to the additional metabolites and the similarity of P2Y receptor family members make referring to these metabolites as “inactive” imprudent. Given the extensive studies supporting non-platelet and non-P2Y12 receptor effects in animal models, clopidogrel should be further evaluated clinically as it may be more than just a platelet P2Y12 receptor antagonist.
Data availability
All the data analyzed in this review are available in the cited references.
References
Dangelmaier C, Jin J, Smith JB, Kunapuli SP (2001) Potentiation of thromboxane A2-induced platelet secretion by Gi signaling through the phosphoinositide-3 kinase pathway. Thromb Haemost 85:341–348. https://doi.org/10.1055/s-0037-1615690
Maffrand J-P (2012) The story of clopidogrel and its predecessor, ticlopidine: could these major antiplatelet and antithrombotic drugs be discovered and developed today? C R Chim 15:737–743. https://doi.org/10.1016/j.crci.2012.05.006
Lee H, Paton RC, Ruan C, Caen JP (1981) The in vitro effect of ticlopidine on fibrinogen and factor VIII binding to human platelets. Thromb Haemost 46:590–592
Savi P, Laplace MC, Maffrand JP, Herbert JM (1994) Binding of [3H]-2-methylthio ADP to rat platelets–effect of clopidogrel and ticlopidine. J Pharmacol Exp Ther 269:772–777
Mills DC, Puri R, Hu CJ, Minniti C, Grana G, Freedman MD et al (1992) Clopidogrel inhibits the binding of ADP analogues to the receptor mediating inhibition of platelet adenylate cyclase. Arterioscler Thromb 12:430–436. https://doi.org/10.1161/01.atv.12.4.430
Feliste R, Simon MF, Chap H, Douste-Blazy L, Defreyn G, Maffrand JP (1988) Effect of PCR 4099 on ADP-induced calcium movements and phosphatidic acid production in rat platelets. Biochem Pharmacol 37:2559–2564. https://doi.org/10.1016/0006-2952(88)90246-8
Gachet C, Cazenave JP, Ohlmann P, Bouloux C, Defreyn G, Driot F et al (1990) The thienopyridine ticlopidine selectively prevents the inhibitory effects of ADP but not of adrenaline on cAMP levels raised by stimulation of the adenylate cyclase of human platelets by PGE1. Biochem Pharmacol 40:2683–2687. https://doi.org/10.1016/0006-2952(90)90587-b
Féliste R, Delebassée D, Simon MF, Chap H, Defreyn G, Vallée E et al (1987) Broad spectrum anti-platelet activity of ticlopidine and PCR 4099 involves the suppression of the effects of released ADP. Thromb Res 48:403–415. https://doi.org/10.1016/0049-3848(87)90398-7
Defreyn G, Gachet C, Savi P, Driot F, Cazenave JP, Maffrand JP (1991) Ticlopidine and clopidogrel (SR 25990C) selectively neutralize ADP inhibition of PGE1-activated platelet adenylate cyclase in rats and rabbits. Thromb Haemost 65:186–190. https://doi.org/10.1055/s-0038-1647481
Gachet C, Stierlé A, Cazenave JP, Ohlmann P, Lanza F, Bouloux C et al (1990) The thienopyridine PCR 4099 selectively inhibits ADP-induced platelet aggregation and fibrinogen binding without modifying the membrane glycoprotein IIb-IIIa complex in rat and in man. Biochem Pharmacol 40:229–238. https://doi.org/10.1016/0006-2952(90)90683-c
Damas J, Grek V, Remacle-Volon G (1987) Inhibition of the thrombocytopenic effect of exogenous and endogenous thrombin by PCR 4099 (d,1)methyl 2-(2-chlorophenyl)-2(4,5,6, 7-tetrahydrothieno (3,2-c)pyridin-5-yl) acetate.hydrochloride.monohydrate. Thromb Res 48:585–589. https://doi.org/10.1016/0049-3848(87)90390-2
Hollopeter G, Jantzen HM, Vincent D, Li G, England L, Ramakrishnan V et al (2001) Identification of the platelet ADP receptor targeted by antithrombotic drugs. Nature 409:202–207. https://doi.org/10.1038/35051599
Takasaki J, Kamohara M, Saito T, Matsumoto M, Matsumoto S, Ohishi T et al (2001) Molecular cloning of the platelet P2T(AC) ADP receptor: pharmacological comparison with another ADP receptor, the P2Y(1) receptor. Mol Pharmacol 60:432–439
Savi P, Labouret C, Delesque N, Guette F, Lupker J, Herbert JM (2001) P2y(12), a new platelet ADP receptor, target of clopidogrel. Biochem Biophys Res Commun 283:379–383. https://doi.org/10.1006/bbrc.2001.4816
Coukell AJ, Markham A (1997) Clopidogrel. Drugs 54:745–50. https://doi.org/10.2165/00003495-199754050-00006 (discussion 751)
Ding Z, Kim S, Dorsam RT, Jin J, Kunapuli SP (2003) Inactivation of the human P2Y12 receptor by thiol reagents requires interaction with both extracellular cysteine residues, Cys17 and Cys270. Blood 101:3908–3914. https://doi.org/10.1182/blood-2002-10-3027
Drug Approval Package: Plavix/Clopidogrel bisulfate NDA 20839. [cited 2 Jun 2022]. Available: https://www.accessdata.fda.gov/drugsatfda_docs/nda/97/020839_plavix_toc.cfm
Savi P, Pereillo JM, Uzabiaga MF, Combalbert J, Picard C, Maffrand JP et al (2000) Identification and biological activity of the active metabolite of clopidogrel. Thromb Haemost 84:891–896. https://doi.org/10.1055/s-0037-1614133
Bouman HJ, Schömig E, van Werkum JW, Velder J, Hackeng CM, Hirschhäuser C et al (2011) Paraoxonase-1 is a major determinant of clopidogrel efficacy. Nat Med 17:110–116. https://doi.org/10.1038/nm.2281
Silvestro L, Gheorghe M, Iordachescu A, Ciuca V, Tudoroniu A, Rizea Savu S et al (2011) Development and validation of an HPLC-MS/MS method to quantify clopidogrel acyl glucuronide, clopidogrel acid metabolite, and clopidogrel in plasma samples avoiding analyte back-conversion. Anal Bioanal Chem 401:1023–1034. https://doi.org/10.1007/s00216-011-5147-4
Lins R, Broekhuysen J, Necciari J, Deroubaix X (1999) Pharmacokinetic profile of 14C-labeled clopidogrel. Semin Thromb Hemost 25(Suppl 2):29–33
Caplain H, Donat F, Gaud C, Necciari J (1999) Pharmacokinetics of clopidogrel. Semin Thromb Hemost 25(Suppl 2):25–28
Hagihara K, Kazui M, Kurihara A, Yoshiike M, Honda K, Okazaki O et al (2009) A possible mechanism for the differences in efficiency and variability of active metabolite formation from thienopyridine antiplatelet agents, prasugrel and clopidogrel. Drug Metab Dispos 37:2145–2152. https://doi.org/10.1124/dmd.109.028498
Tang M, Mukundan M, Yang J, Charpentier N, LeCluyse EL, Black C et al (2006) Antiplatelet agents aspirin and clopidogrel are hydrolyzed by distinct carboxylesterases, and clopidogrel is transesterificated in the presence of ethyl alcohol. J Pharmacol Exp Ther 319:1467–1476. https://doi.org/10.1124/jpet.106.110577
Funck-Brentano C, Szymezak J, Steichen O, Ducint D, Molimard M, Remones V et al (2013) Effects of rabeprazole on the antiplatelet effects and pharmacokinetics of clopidogrel in healthy volunteers. Arch Cardiovasc Dis 106:661–671. https://doi.org/10.1016/j.acvd.2013.09.002
Lin R, Zhang L, Zhang P, Zhou L, Liu T, Li Y et al (2015) Influence of CYP2C19 loss-of-function variants on the metabolism of clopidogrel in patients from north-western China. J Clin Pharm Ther 40:308–314. https://doi.org/10.1111/jcpt.12254
Wang Y, Zhao X, Lin J, Li H, Johnston SC, Lin Y et al (2016) Association between CYP2C19 loss-of-function allele status and efficacy of clopidogrel for risk reduction among patients with minor stroke or transient ischemic attack. JAMA 316:70–78. https://doi.org/10.1001/jama.2016.8662
Zhou H, Meng S, Zhao J, Dong J, Xu A, Wang F et al (2013) Influence of genetic and non-genetic factors on the plasma concentrations of the clopidogrel metabolite (SR26334) among Chinese patients. Clin Chim Acta 416:50–53. https://doi.org/10.1016/j.cca.2012.11.022
FDA Drug Safety Communication: Reduced effectiveness of Plavix (clopidogrel) in patients who are poor metabolizers of the drug | FDA. [cited 29 Nov 2021]. Available: https://www.fda.gov/drugs/postmarket-drug-safety-information-patients-and-providers/fda-drug-safety-communication-reduced-effectiveness-plavix-clopidogrel-patients-who-are-poor
Angiolillo DJ, Gibson CM, Cheng S, Ollier C, Nicolas O, Bergougnan L et al (2011) Differential effects of omeprazole and pantoprazole on the pharmacodynamics and pharmacokinetics of clopidogrel in healthy subjects: randomized, placebo-controlled, crossover comparison studies. Clin Pharmacol Ther 89:65–74. https://doi.org/10.1038/clpt.2010.219
Ge P-X, Jiang L-P, Tai T, Zhu T, Ji J-Z, Li Y-F et al (2021) Short-term standard alcohol consumption enhances platelet response to clopidogrel through inhibition of Nrf2/Ces1 pathway and induction of Cyp2c in mice. Life Sci 279:119268. https://doi.org/10.1016/j.lfs.2021.119268
Laizure SC, Hu Z-Y, Potter PM, Parker RB (2020) Inhibition of carboxylesterase-1 alters clopidogrel metabolism and disposition. Xenobiotica 50:245–251. https://doi.org/10.1080/00498254.2019.1612535
Price MJ, Murray SS, Angiolillo DJ, Lillie E, Smith EN, Tisch RL et al (2012) Influence of genetic polymorphisms on the effect of high- and standard-dose clopidogrel after percutaneous coronary intervention: the GIFT (genotype information and functional testing) study. J Am Coll Cardiol 59:1928–1937. https://doi.org/10.1016/j.jacc.2011.11.068
Angiolillo DJ, Jakubowski JA, Ferreiro JL, Tello-Montoliu A, Rollini F, Franchi F et al (2014) Impaired responsiveness to the platelet P2Y12 receptor antagonist clopidogrel in patients with type 2 diabetes and coronary artery disease. J Am Coll Cardiol 64:1005–1014. https://doi.org/10.1016/j.jacc.2014.06.1170
Erlinge D, Varenhorst C, Braun OO, James S, Winters KJ, Jakubowski JA et al (2008) Patients with poor responsiveness to thienopyridine treatment or with diabetes have lower levels of circulating active metabolite, but their platelets respond normally to active metabolite added ex vivo. J Am Coll Cardiol 52:1968–1977. https://doi.org/10.1016/j.jacc.2008.07.068
Sun Y, Venugopal J, Guo C, Fan Y, Li J, Gong Y et al (2020) Clopidogrel resistance in a murine model of diet-induced obesity is mediated by the interleukin -1 receptor and overcome with DT-678. Arterioscler Thromb Vasc Biol 40:1533–1542. https://doi.org/10.1161/ATVBAHA.120.314146
Koupenova M, Clancy L, Corkrey HA, Freedman JE (2018) Circulating platelets as mediators of immunity, inflammation, and thrombosis. Circ Res 122:337–351. https://doi.org/10.1161/CIRCRESAHA.117.310795
Keren-Shaul H, Spinrad A, Weiner A, Matcovitch-Natan O, Dvir-Szternfeld R, Ulland TK et al (2017) A unique microglia type associated with restricting development of Alzheimer’s disease. Cell 169:1276-1290.e17. https://doi.org/10.1016/j.cell.2017.05.018
Burnstock G (2018) Purine and purinergic receptors. Brain Neurosci Adv 2:239821281881749. https://doi.org/10.1177/2398212818817494
Orriss IR, Burnstock G, Arnett TR (2010) Purinergic signalling and bone remodelling. Curr Opin Pharmacol 10:322–330. https://doi.org/10.1016/j.coph.2010.01.003
Uehara K, Uehara A (2011) P2Y1, P2Y6, and P2Y12 receptors in rat splenic sinus endothelial cells: an immunohistochemical and ultrastructural study. Histochem Cell Biol 136:557–567. https://doi.org/10.1007/s00418-011-0859-2
Gdula AM, Swiatkowska M (2020) Effects of dual purinoceptor-dependent approach on release of vascular endothelial growth factor from human microvascular endothelial cell (HMEC-1) and endothelial cell condition. J Cardiovasc Pharmacol 76:349–359. https://doi.org/10.1097/FJC.0000000000000866
Li F, Xu D, Hou K, Gou X, Li Y (2020) The role of P2Y12 receptor inhibition in ischemic stroke on microglia, platelets and vascular smooth muscle cells. J Thromb Thrombolysis 50:874–885. https://doi.org/10.1007/s11239-020-02098-4
Burnstock G (2017) Purinergic signaling in the cardiovascular system. Circ Res 120:207–228. https://doi.org/10.1161/CIRCRESAHA.116.309726
Tissue expression of P2RY12 - summary - the Human Protein Atlas. [cited 29 Nov 2021]. Available: https://www.proteinatlas.org/ENSG00000169313-P2RY12/tissue
Libby P (2002) Inflammation in atherosclerosis. Nature 420:868–874. https://doi.org/10.1038/nature01323
Falk E (2006) Pathogenesis of atherosclerosis. J Am Coll Cardiol 47:C7-12. https://doi.org/10.1016/j.jacc.2005.09.068
Heim C, Gebhardt J, Ramsperger-Gleixner M, Jacobi J, Weyand M, Ensminger SM (2016) Clopidogrel significantly lowers the development of atherosclerosis in ApoE-deficient mice in vivo. Heart Vessels 31:783–794. https://doi.org/10.1007/s00380-015-0696-7
Li M, Zhang Y, Ren H, Zhang Y, Zhu X (2007) Effect of clopidogrel on the inflammatory progression of early atherosclerosis in rabbits model. Atherosclerosis 194:348–356. https://doi.org/10.1016/j.atherosclerosis.2006.11.006
Halim H, Pinkaew D, Chunhacha P, Sinthujaroen P, Thiagarajan P, Fujise K (2019) Ticagrelor induces paraoxonase-1 (PON1) and better protects hypercholesterolemic mice against atherosclerosis compared to clopidogrel. PLoS ONE 14:e0218934. https://doi.org/10.1371/journal.pone.0218934
Zhang J, Shi Q, Hu Y, Li X (2021) Silibinin augments the effect of clopidogrel on atherosclerosis in diabetic ApoE deficiency mice. Clin Hemorheol Microcirc. https://doi.org/10.3233/CH-211279
Evans DJW, Jackman LE, Chamberlain J, Crosdale DJ, Judge HM, Jetha K et al (2009) Platelet P2Y(12) receptor influences the vessel wall response to arterial injury and thrombosis. Circulation 119:116–122. https://doi.org/10.1161/CIRCULATIONAHA.107.762690
Lindemann S, Krämer B, Seizer P, Gawaz M (2007) Platelets, inflammation and atherosclerosis. J Thromb Haemost 5(Suppl 1):203–211. https://doi.org/10.1111/j.1538-7836.2007.02517.x
Gawaz M, Neumann FJ, Dickfeld T, Koch W, Laugwitz KL, Adelsberger H et al (1998) Activated platelets induce monocyte chemotactic protein-1 secretion and surface expression of intercellular adhesion molecule-1 on endothelial cells. Circulation 98:1164–1171. https://doi.org/10.1161/01.cir.98.12.1164
Cybulsky MI, Iiyama K, Li H, Zhu S, Chen M, Iiyama M et al (2001) A major role for VCAM-1, but not ICAM-1, in early atherosclerosis. J Clin Invest 107:1255–1262. https://doi.org/10.1172/JCI11871
Oh M, Lee CW, Lee HS, Chang M, Ahn J-M, Park D-W et al (2016) Similar impact of clopidogrel or ticagrelor on carotid atherosclerotic plaque inflammation. Clin Cardiol 39:646–652. https://doi.org/10.1002/clc.22575
Getz GS, Reardon CA (2016) ApoE knockout and knockin mice: the history of their contribution to the understanding of atherogenesis. J Lipid Res 57:758–766. https://doi.org/10.1194/jlr.R067249
West LE, Steiner T, Judge HM, Francis SE, Storey RF (2014) Vessel wall, not platelet, P2Y12 potentiates early atherogenesis. Cardiovasc Res 102:429–435. https://doi.org/10.1093/cvr/cvu028
Biancardi VC, Bomfim GF, Reis WL, Al-Gassimi S, Nunes KP (2017) The interplay between angiotensin II, TLR4 and hypertension. Pharmacol Res 120:88–96. https://doi.org/10.1016/j.phrs.2017.03.017
An X, Jiang G, Cheng C, Lv Z, Liu Y, Wang F (2018) Inhibition of platelets by clopidogrel suppressed Ang II-Induced Vascular Inflammation, Oxidative Stress, and Remodeling. J Am Heart Assoc 7:e009600. https://doi.org/10.1161/JAHA.118.009600
Giachini FRC, Osmond DA, Zhang S, Carneiro FS, Lima VV, Inscho EW et al (2010) Clopidogrel, independent of the vascular P2Y12 receptor, improves arterial function in small mesenteric arteries from AngII-hypertensive rats. Clin Sci 118:463–471. https://doi.org/10.1042/CS20090392
Giachini FR, Leite R, Osmond DA, Lima VV, Inscho EW, Webb RC et al (2014) Anti-platelet therapy with clopidogrel prevents endothelial dysfunction and vascular remodeling in aortas from hypertensive rats. PLoS ONE 9:e91890. https://doi.org/10.1371/journal.pone.0091890
Mittal M, Siddiqui MR, Tran K, Reddy SP, Malik AB (2014) Reactive oxygen species in inflammation and tissue injury. Antioxid Redox Signal 20:1126–1167. https://doi.org/10.1089/ars.2012.5149
Tarnawski AS (2005) Cellular and molecular mechanisms of gastrointestinal ulcer healing. Dig Dis Sci 50(Suppl 1):S24-33. https://doi.org/10.1007/s10620-005-2803-6
Tarnawski AS, Ahluwalia A, Jones MK (2014) Angiogenesis in gastric mucosa: an important component of gastric erosion and ulcer healing and its impairment in aging. J Gastroenterol Hepatol 29(Suppl 4):112–123. https://doi.org/10.1111/jgh.12734
Luo J-C, Peng Y-L, Chen T-S, Huo T-I, Hou M-C, Huang H-C et al (2016) Clopidogrel inhibits angiogenesis of gastric ulcer healing via downregulation of vascular endothelial growth factor receptor 2. J Formos Med Assoc 115:764–772. https://doi.org/10.1016/j.jfma.2015.07.022
Ludwig S, Wolff T, Ehrhardt C, Wurzer WJ, Reinhardt J, Planz O et al (2004) MEK inhibition impairs influenza B virus propagation without emergence of resistant variants. FEBS Lett 561:37–43. https://doi.org/10.1016/S0014-5793(04)00108-5
Pleschka S, Wolff T, Ehrhardt C, Hobom G, Planz O, Rapp UR et al (2001) Influenza virus propagation is impaired by inhibition of the Raf/MEK/ERK signalling cascade. Nat Cell Biol 3:301–305. https://doi.org/10.1038/35060098
Orr-Burks N, Murray J, Todd KV, Bakre A, Tripp RA. (2021) G-protein-coupled receptor and ion channel genes used by influenza virus for replication. J Virol. 95. https://doi.org/10.1128/JVI.02410-20
Pleschka S (2008) RNA viruses and the mitogenic Raf/MEK/ERK signal transduction cascade. Biol Chem 389:1273–1282. https://doi.org/10.1515/BC.2008.145
Orr-Burks N, Murray J, Todd KV, Bakre A, Tripp RA (2021) Drug repositioning of clopidogrel or triamterene to inhibit influenza virus replication in vitro. PLoS ONE 16:e0259129. https://doi.org/10.1371/journal.pone.0259129
Degirmenci U, Wang M, Hu J (2020) Targeting aberrant RAS/RAF/MEK/ERK signaling for cancer therapy. Cells 9 https://doi.org/10.3390/cells9010198
Asati V, Mahapatra DK, Bharti SK (2016) PI3K/Akt/mTOR and Ras/Raf/MEK/ERK signaling pathways inhibitors as anticancer agents: structural and pharmacological perspectives. Eur J Med Chem 109:314–341. https://doi.org/10.1016/j.ejmech.2016.01.012
Suzuki-Inoue K (2011) Essential in vivo roles of the platelet activation receptor CLEC-2 in tumour metastasis, lymphangiogenesis and thrombus formation. J Biochem 150:127–132. https://doi.org/10.1093/jb/mvr079
Medina C, Jurasz P, Santos-Martinez MJ, Jeong SS, Mitsky T, Chen R et al (2006) Platelet aggregation-induced by caco-2 cells: regulation by matrix metalloproteinase-2 and adenosine diphosphate. J Pharmacol Exp Ther 317:739–745. https://doi.org/10.1124/jpet.105.098384
Danckwardt S, Hentze MW, Kulozik AE (2013) Pathologies at the nexus of blood coagulation and inflammation: thrombin in hemostasis, cancer, and beyond. J Mol Med 91:1257–1271. https://doi.org/10.1007/s00109-013-1074-5
Gresele P, Falcinelli E, Sebastiano M, Momi S (2017) Matrix metalloproteinases and platelet function. Prog Mol Biol Transl Sci 147:133–165. https://doi.org/10.1016/bs.pmbts.2017.01.002
Denslow A, Świtalska M, Jarosz J, Papiernik D, Porshneva K, Nowak M et al (2017) Clopidogrel in a combined therapy with anticancer drugs-effect on tumor growth, metastasis, and treatment toxicity: studies in animal models. PLoS ONE 12:e0188740. https://doi.org/10.1371/journal.pone.0188740
Porshneva K, Papiernik D, Psurski M, Nowak M, Matkowski R, Ekiert M et al (2018) Combination therapy with DETA/NO and clopidogrel inhibits metastasis in murine mammary gland cancer models via improved vasoprotection. Mol Pharm 15:5277–5290. https://doi.org/10.1021/acs.molpharmaceut.8b00781
Rodríguez-Miguel A, García-Rodríguez LA, Gil M, Montoya H, Rodríguez-Martín S, de Abajo FJ (2019) Clopidogrel and low-dose aspirin, alone or together, reduce risk of colorectal cancer. Clin Gastroenterol Hepatol 17:2024-2033.e2. https://doi.org/10.1016/j.cgh.2018.12.012
Leader A, Zelikson-Saporta R, Pereg D, Spectre G, Rozovski U, Raanani P et al (2017) The effect of combined aspirin and clopidogrel treatment on cancer incidence. Am J Med 130:826–832. https://doi.org/10.1016/j.amjmed.2017.01.022
Syberg S, Brandao-Burch A, Patel JJ, Hajjawi M, Arnett TR, Schwarz P et al (2012) Clopidogrel (Plavix), a P2Y12 receptor antagonist, inhibits bone cell function in vitro and decreases trabecular bone in vivo. J Bone Miner Res 27:2373–2386. https://doi.org/10.1002/jbmr.1690
Mediero A, Wilder T, Reddy VSR, Cheng Q, Tovar N, Coelho PG et al (2016) Ticagrelor regulates osteoblast and osteoclast function and promotes bone formation in vivo via an adenosine-dependent mechanism. FASEB J 30:3887–3900. https://doi.org/10.1096/fj.201600616R
Lillis T, Veis A, Sakellaridis N, Tsirlis A, Dailiana Z (2019) Effect of clopidogrel in bone healing-experimental study in rabbits. World J Orthop 10:434–445. https://doi.org/10.5312/wjo.v10.i12.434
Nelson TA, Parker WAE, Ghukasyan Lakic T, Westerbergh J, James SK, Siegbahn A, et al. (2021) Differential effect of clopidogrel and ticagrelor on leukocyte count in relation to patient characteristics, biomarkers and genotype: a PLATO substudy. Platelets. 1–7 https://doi.org/10.1080/09537104.2021.1934667
Hamesch K, Borkham-Kamphorst E, Strnad P, Weiskirchen R (2015) Lipopolysaccharide-induced inflammatory liver injury in mice. Lab Anim 49:37–46. https://doi.org/10.1177/0023677215570087
Liu T, Zhang L, Joo D, Sun S-C. (2017) NF-κB signaling in inflammation. Signal Transduct Target Ther. 2 https://doi.org/10.1038/sigtrans.2017.23
Jia Z, Huang Y, Ji X, Sun J, Fu G (2019) Ticagrelor and clopidogrel suppress NF-κB signaling pathway to alleviate LPS-induced dysfunction in vein endothelial cells. BMC Cardiovasc Disord 19:318. https://doi.org/10.1186/s12872-019-01287-1
Hagiwara S, Iwasaka H, Hasegawa A, Oyama M, Imatomi R, Uchida T et al (2011) Adenosine diphosphate receptor antagonist clopidogrel sulfate attenuates LPS-induced systemic inflammation in a rat model. Shock 35:289–292. https://doi.org/10.1097/SHK.0b013e3181f48987
Liverani E, Rico MC, Yaratha L, Tsygankov AY, Kilpatrick LE, Kunapuli SP (2014) LPS-induced systemic inflammation is more severe in P2Y12 null mice. J Leukoc Biol 95:313–323. https://doi.org/10.1189/jlb.1012518
Thomas MR, Outteridge SN, Ajjan RA, Phoenix F, Sangha GK, Faulkner RE et al (2015) Platelet P2Y12 inhibitors reduce systemic inflammation and its prothrombotic effects in an experimental human model. Arterioscler Thromb Vasc Biol 35:2562–2570. https://doi.org/10.1161/ATVBAHA.115.306528
Malek LA, Grabowski M, Spiewak M, Filipiak KJ, Szpotanska M, Imiela T et al (2007) Relation between impaired antiplatelet response to clopidogrel and possible pleiotropic effects. J Thromb Thrombolysis 24:301–305. https://doi.org/10.1007/s11239-007-0026-8
Wihlborg A-K, Malmsjö M, Eyjolfsson A, Gustafsson R, Jacobson K, Erlinge D (2003) Extracellular nucleotides induce vasodilatation in human arteries via prostaglandins, nitric oxide and endothelium-derived hyperpolarising factor. Br J Pharmacol 138:1451–1458. https://doi.org/10.1038/sj.bjp.0705186
Haanes KA, Edvinsson L (2014) Expression and characterization of purinergic receptors in rat middle meningeal artery-potential role in migraine. PLoS ONE 9:e108782. https://doi.org/10.1371/journal.pone.0108782
Kuszynski DS, Christian BD, Dorrance AM, Lauver DA (2021) Clopidogrel treatment inhibits P2Y2-mediated constriction in the rabbit middle cerebral artery. Eur J Pharmacol 911:174545. https://doi.org/10.1016/j.ejphar.2021.174545
Högberg C, Svensson H, Gustafsson R, Eyjolfsson A, Erlinge D (2010) The reversible oral P2Y12 antagonist AZD6140 inhibits ADP-induced contractions in murine and human vasculature. Int J Cardiol 142:187–192. https://doi.org/10.1016/j.ijcard.2008.12.091
Jakubowski A, Chlopicki S, Olszanecki R, Jawien J, Lomnicka M, Dupin JP et al (2005) Endothelial action of thienopyridines and thienopyrimidinones in the isolated guinea pig heart. Prostaglandins Leukot Essent Fatty Acids 72:139–145. https://doi.org/10.1016/j.plefa.2004.10.011
Grzesk G, Kozinski M, Navarese EP, Krzyzanowski M, Grzesk E, Kubica A et al (2012) Ticagrelor, but not clopidogrel and prasugrel, prevents ADP-induced vascular smooth muscle cell contraction: a placebo-controlled study in rats. Thromb Res 130:65–69. https://doi.org/10.1016/j.thromres.2011.12.029
Froldi G, Bertin R, Dorigo P, Montopoli M, Caparrotta L (2011) Endothelium-independent vasorelaxation by ticlopidine and clopidogrel in rat caudal artery. J Pharm Pharmacol 63:1056–1062. https://doi.org/10.1111/j.2042-7158.2011.01313.x
Bledsoe SL, Brown AT, Davis JA, Chen H, Eidt JF, Moursi MM (2005) Effect of clopidogrel on platelet aggregation and intimal hyperplasia following carotid endarterectomy in the rat. Vascular 13:43–49. https://doi.org/10.1258/rsmvasc.13.1.43
Bledsoe SL, Barr JC, Fitzgerald RT, Brown AT, Faas FH, Eidt JF et al (2006) Pravastatin and clopidogrel combined inhibit intimal hyperplasia in a rat carotid endarterectomy model. Vasc Endovascular Surg 40:49–57. https://doi.org/10.1177/153857440604000107
Erga KS, Seubert CN, Liang HX, Wu L, Shryock JC, Belardinelli L (2000) Role of A(2A)-adenosine receptor activation for ATP-mediated coronary vasodilation in guinea-pig isolated heart. Br J Pharmacol 130:1065–1075. https://doi.org/10.1038/sj.bjp.0703386
Killory BD, Kilbourn KJ, Ollenschleger M (2015) A Novel use of direct platelet application during surgery for clopidogrel-associated intracerebral hemorrhage. World Neurosurg 84(2078):e1-4. https://doi.org/10.1016/j.wneu.2015.08.016
Cordina SM, Hassan AE, Ezzeddine MA (2009) Prevalence and clinical characteristics of intracerebral hemorrhages associated with clopidogrel. J Vasc Interv Neurol 2:136–138
DiNicolantonio JJ, D’Ascenzo F, Tomek A, Chatterjee S, Niazi AK, Biondi-Zoccai G (2013) Clopidogrel is safer than ticagrelor in regard to bleeds: a closer look at the PLATO trial. Int J Cardiol 168:1739–1744. https://doi.org/10.1016/j.ijcard.2013.06.135
Darweesh SKL, Leening MJG, Akoudad S, Loth DW, Hofman A, Ikram MA et al (2013) Clopidogrel use is associated with an increased prevalence of cerebral microbleeds in a stroke-free population: the Rotterdam study. J Am Heart Assoc 2:e000359. https://doi.org/10.1161/JAHA.113.000359
Ge L, Ouyang X, Ban C, Yu H, Wu Q, Wu H et al (2019) Cerebral microbleeds in patients with ischemic cerebrovascular disease taking aspirin or clopidogrel. Medicine (Baltimore) 98:e14685. https://doi.org/10.1097/MD.0000000000014685
Vernooij MW, Haag MDM, van der Lugt A, Hofman A, Krestin GP, Stricker BH et al (2009) Use of antithrombotic drugs and the presence of cerebral microbleeds: the Rotterdam Scan study. Arch Neurol 66:714–720. https://doi.org/10.1001/archneurol.2009.42
Lovelock CE, Cordonnier C, Naka H, Al-Shahi Salman R, Sudlow CLM, Edinburgh Stroke Study Group et al (2010) Antithrombotic drug use, cerebral microbleeds, and intracerebral hemorrhage: a systematic review of published and unpublished studies. Stroke 41:1222–1228. https://doi.org/10.1161/STROKEAHA.109.572594
Lee J, Sohn EH, Oh E, Lee AY (2018) Characteristics of cerebral microbleeds. Dement Neurocognitive Disord 17:73–82. https://doi.org/10.12779/dnd.2018.17.3.73
Ha ACT, Bhatt DL, Rutka JT, Johnston SC, Mazer CD, Verma S (2021) Intracranial hemorrhage during dual antiplatelet therapy: JACC review topic of the week. J Am Coll Cardiol 78:1372–1384. https://doi.org/10.1016/j.jacc.2021.07.048
Mega JL, Close SL, Wiviott SD, Shen L, Hockett RD, Brandt JT et al (2009) Cytochrome p-450 polymorphisms and response to clopidogrel. N Engl J Med 360:354–362. https://doi.org/10.1056/NEJMoa0809171
Crescence L, Darbousset R, Caroff E, Hubler F, Riederer MA, Panicot-Dubois L et al (2021) Selatogrel, a reversible P2Y12 receptor antagonist, has reduced off-target interference with haemostatic factors in a mouse thrombosis model. Thromb Res 200:133–140. https://doi.org/10.1016/j.thromres.2021.01.026
André P, DeGuzman F, Haberstock-Debic H, Mills S, Pak Y, Inagaki M et al (2011) Thienopyridines, but not elinogrel, result in off-target effects at the vessel wall that contribute to bleeding. J Pharmacol Exp Ther 338:22–30. https://doi.org/10.1124/jpet.110.178574
Zhang H, Lauver DA, Lucchesi BR, Hollenberg PF (2013) Formation, reactivity, and antiplatelet activity of mixed disulfide conjugates of clopidogrel. Mol Pharmacol 83:848–856. https://doi.org/10.1124/mol.112.084392
Zhang H, Lauver DA, Wang H, Sun D, Hollenberg PF, Chen YE et al (2016) Significant improvement of antithrombotic responses to clopidogrel by use of a novel conjugate as revealed in an arterial model of thrombosis. J Pharmacol Exp Ther 359:11–17. https://doi.org/10.1124/jpet.116.236034
Lauver DA, Kuszynski DS, Christian BD, Bernard MP, Teuber JP, Markham BE et al (2019) DT-678 inhibits platelet activation with lower tendency for bleeding compared to existing P2Y12 antagonists. Pharmacol Res Perspect 7:e00509. https://doi.org/10.1002/prp2.509
Zhu Y, Romero EL, Ren X, Sanca AJ, Du C, Liu C et al (2018) Clopidogrel as a donor probe and thioenol derivatives as flexible promoieties for enabling H2S biomedicine. Nat Commun 9:3952. https://doi.org/10.1038/s41467-018-06373-0
Olas B (2015) Hydrogen sulfide in signaling pathways. Clin Chim Acta 439:212–218. https://doi.org/10.1016/j.cca.2014.10.037
Tuffal G, Roy S, Lavisse M, Brasseur D, Schofield J, Delesque Touchard N et al (2011) An improved method for specific and quantitative determination of the clopidogrel active metabolite isomers in human plasma. Thromb Haemost 105:696–705. https://doi.org/10.1160/TH10-09-0582
Zhu Y, Zhou J (2013) In vitro biotransformation studies of 2-oxo-clopidogrel: multiple thiolactone ring-opening pathways further attenuate prodrug activation. Chem Res Toxicol 26:179–190. https://doi.org/10.1021/tx300460k
Pereillo J-M, Maftouh M, Andrieu A, Uzabiaga M-F, Fedeli O, Savi P et al (2002) Structure and stereochemistry of the active metabolite of clopidogrel. Drug Metab Dispos 30:1288–1295. https://doi.org/10.1124/dmd.30.11.1288
Zhu Y, Zhou J (2012) Identification of the significant involvement and mechanistic role of CYP3A4/5 in clopidogrel bioactivation. ACS Med Chem Lett 3:844–849. https://doi.org/10.1021/ml3002067
Acknowledgements
The authors wish to thank Dr. James Luyendyk, Michigan State University, for proofreading and helpful suggestions.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of interest
Author 1 declares that he/she has no conflict of interest.
Author 2 declares that he/she has no conflict of interest.
Ethical approval
Not applicable.
Informed consent
Not applicable.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Kuszynski, D.S., Lauver, D.A. Pleiotropic effects of clopidogrel. Purinergic Signalling 18, 253–265 (2022). https://doi.org/10.1007/s11302-022-09876-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11302-022-09876-0