
Abstract
Adenosine signaling plays a complex role in multiple physiological processes in the brain, and its dysfunction has been implicated in pathophysiology of neuropsychiatric diseases such as schizophrenia and affective disorders. In the present study, the coupling between adenosine A1 receptor and G-protein was assessed by means of two [35S]GTPγS binding assays, i.e., conventional filtration method and [35S]GTPγS binding/immunoprecipitation in rat and human brain membranes. The latter method provides information about adenosine A1 receptor-mediated Gαi-3 activation in rat as well as human brain membranes. On the other hand, adenosine-stimulated [35S]GTPγS binding determined with conventional assay derives from functional activation of Gαi/o proteins (not restricted only to Gαi-3) coupled to adenosine A1 receptors. The determination of adenosine concentrations in the samples used in the present study indicates the possibility that the assay mixture under our experimental conditions contains residual endogenous adenosine at nanomolar concentrations, which was also suggested by the results on the effects of adenosine receptor antagonists on basal [35S]GTPγS binding level. The effects of adenosine deaminase (ADA) on basal binding also support the presence of adenosine. Nevertheless, the varied patterns of ADA discouraged us from adding ADA into assay medium routinely. The concentration-dependent increases elicited by adenosine were determined in 40 subjects without any neuropsychiatric disorders. The increases in %Emax values determined by conventional assay according to aging and postmortem delay should be taken into account in future studies focusing on the effects of psychiatric disorders on adenosine A1 receptor/G-protein interaction in postmortem human brain tissue.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Adenosine signaling plays a complex role in multiple physiological and pathophysiological processes in the brain. Among the four known adenosine receptors, referred to as adenosine A1, A2A, A2B, and A3 receptor [1], the A1 and A2A receptors are abundantly and widely distributed in the human central nervous system [2]. All adenosine receptors belong to class A family of G-protein-coupled receptor (GPCR) superfamily, and it has been generally accepted that adenosine A1 receptor is primarily coupled to Gi/o to inhibit adenylyl cyclase whereas adenosine A2A receptor is mainly coupled to Gs to activate the enzyme activity.
Agonist-stimulated guanosine-5′-O-(3-[35S]thio) triphosphate ([35S]GTPγS) binding assay has been widely used to assess functional activation of G-proteins, especially Gi/o proteins, coupled to multiple receptors [3, 4]. Adenosine A1 receptor is one of these receptors, and functional activation of G-proteins coupled to adenosine A1 receptor has been reported by [35S]GTPγS binding in native bovine or rat brain membranes [5,6,7] and by [35S]GTPγS autoradiography [7,8,9]. Although these methods have been widely utilized in neuroscience research to investigate receptor/G-protein interaction between inhibitory receptors and Gi/o proteins, it is not possible to differentiate each G-protein subtype functionally coupled to the receptor by using conventional [35S]GTPγS binding assay.
Recently, we have developed a novel technique, named [35S]GTPγS binding/immunoprecipitation assay, which is an extended [35S]GTPγS binding assay combined with immunoprecipitation using an anti-Gα subtype antibody [10]. By using this method, we have revealed that adenosine A1 receptor is coupled preferentially to Gαi-3 in postmortem human prefrontal cortical membranes. Adenosine signaling dysfunction in the brain has been implicated in pathophysiology of neuropsychiatric diseases such as schizophrenia and affective disorders [11,12,13,14]. However, direct studies on the adenosinergic system in mental disorders are strikingly scare, especially for adenosine A1 receptor-mediated signaling. Based on these considerations, we have a plan to assess possible alterations in adenosine A1 receptor-mediated G-protein activation in psychiatric disorder patients in comparison with control subjects. Ahead of this, the present study aimed at elucidating functional coupling between adenosine A1 receptors and G-proteins in postmortem human brain membranes in a control cohort. In addition, several issues related to the methods of adenosine A1 receptor-mediated G-protein activation have been addressed both in rat and postmortem human brain membranes. Since adenosine deaminase (ADA) is sometimes included routinely in the assay buffer to diminish the possible effects of residual endogenous adenosine in [35S]GTPγS binding assay, especially in autoradiography studies [7, 8], we have tried to elucidate to which extent the residual endogenous adenosine affects the basal and stimulated [35S]GTPγS binding in these measurements.
Materials and methods
Chemicals and reagents
[35S]GTPγS (NEG030H, 1250 Ci/mmol) was purchased from PerkinElmer (Waltham, MA, USA). Adenosine, 1-butyl-3-(3-hydroxypropyl)-8-(3-noradamantyl)xanthine (PSB36), 1,3-dimethyl-8-phenylxanthine, N-(2-methoxyphenyl)-N′-[2-(3-pyridinyl)-4-quinazolinyl]-urea (VUF5574), caffeine, GDP, GTPγS, and Tween 20 were obtained from Sigma-Aldrich (St. Louis, MO, USA). 2-Chloro-N6-cyclopentyladenosine (CCPA), N6-cyclopentyladenosine (CPA), 2-chloro-N-cyclopentyl-2′-methyladenosine (2′-MeCCPA), 4-[2-[[6-amino-9-(N-ethyl-β-D-ribofuranuronamidosyl)-9H-purin-2-yl]amino]ethyl]benzenepropanoic acid hydrochloride (CGS21680), and 8-cyclopentyl-1,3-dipropylxanthine (DPCPX) were from Tocris Cookson (Bristol, UK). Dynabeads Protein A was purchased from ThermoFisher Scientific (Waltham, MA, USA). The rabbit polyclonal antibodies to Gα subtypes (sc-391 for Gαi-1, sc-7276 for Gαi-2, sc-262 for Gαi-3, and sc-387 for Gαo) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Adenosine deaminase (ADA) from calf intestine and Nonidet P40 substitute were obtained from Roche Diagnostics GmbH (Mannheim, Germany). Other chemicals used in this study were of analytical grade.
Animals
Male Sprague-Dawley rats weighing 200–250 g were purchased from Kiwa Laboratory Animals Co. (Wakayama, Japan) and housed in groups under controlled light and humidity conditions with free access to food and water for several days until sacrifice. The experimental protocols were reviewed and approved by the Animal Committee of Saitama Medical University, and the animal care and use procedures conformed to the European Community Guidelines for the use of Experimental Animals (86/609/EEC).
Postmortem human brains
Postmortem human brain samples were obtained at autopsy in the Basque Institute of Legal Medicine (Bilbao, Spain), in accordance with protocols approved by the Human Studies Ethical Committee of each of the institutions involved. Dorsolateral prefrontal cortices (Brodmann’s area 9) dissected from 40 subjects (24 males and 16 females, aged from 16 to 80 years old) without known history of neurological or psychiatric disorders were used. The detailed information on these samples have been described elsewhere [15].
Membrane preparation
Rats were sacrificed, and the cerebral cortex, hippocampus, and striatum were dissected quickly. Rat or human brain tissue was homogenized in 5 mL of ice-cold TED buffer (5 mM Tris–HCl, 1 mM EDTA, 1 mM dithiothreitol; pH 7.4) containing 10% (w/v) sucrose by 20 strokes with a motor-driven Teflon/glass tissue grinder. All of the following centrifuge procedures were performed at 4 °C. Subsequent to centrifugation of the homogenate at 1000 g for 10 min, the supernatant was decanted to another centrifuge tube. The pellet was vortexed in 5 mL of TED/sucrose buffer and centrifuged again at 1000g for 10 min. The combined supernatant (10 mL) was centrifuged at 9000g for 20 min and resuspended in 10 mL of TED buffer. After the same procedure was repeated, the homogenate was kept on ice for 30 min, followed by a final centrifugation at 35,000g for 10 min. The resulting pellet was resuspended in 50 mM Tris–HCl buffer (pH 7.4) to produce the homogenate with a protein concentration of 1.0–2.0 and 2.0–3.0 mg/mL for rat and human brain, respectively. The homogenate was frozen quickly on fine-grained dry ice and stored at − 80 °C until the day of experiment.
Conventional [35S]GTPγS binding assay
The [35S]GTPγS binding assay using filtration techniques were performed according to the methods described previously [16]. In brief, brain membranes equivalent to 10–20 μg protein (rat) or 60 μg protein (human) were incubated in duplicate at 30 °C for 60 min in 500 μL of 50 mM Tris–HCl buffer (pH 7.4) containing 0.2 nM [35S]GTPγS, 5 mM MgCl2, 0.1 mM ethylenediaminetetraacetic acid, 0.2 mM ethylene glycolbis(2-aminoethylether)-N,N,N,N-tetraacetic acid (EGTA), 0.2 mM dithiothreitol, 100 mM NaCl, 20 μM (rat) or 50 μM (human) GDP, and the compound of interest at the indicated concentration. After the incubation, the homogenate was filtered under vacuum through glass fiber filters (GF/B; Whatman International, Maidstone, UK) using a Brandel cell harvester with 2 × 5 mL washing with ice-cold 50 mM Tris–HCl buffer (pH 7.4). The nonspecific binding was measured in the presence of 100 μM unlabeled GTPγS.
[35S]GTPγS binding/immunoprecipitation assay
The [35S]GTPγS binding/immunoprecipitation assay was performed according to the methods described previously [10, 16]. The brain membranes were diluted with 50 mM Tris–HCl buffer (pH 7.4) to contain 20 μg protein (rat) or 80 μg protein (human) in 100 μL and were incubated with the compound of interest diluted in 50 μL distilled water at room temperature for 30 min in 1.5 mL polypropylene microtube. Subsequent to the addition of 50 μL assay mixture, the incubation was performed for 60 min in 200 μL of 50 mM Tris–HCl buffer (pH 7.4) containing 2 nM [35S]GTPγS, 20 mM MgCl2, 0.2 mM EGTA, 0.5 mM dithiothreitol, and 300 μM GDP. The membrane homogenate was solubilized with 0.3% Nonidet P40 substitute for 30 min, followed by a 60-min incubation with Dynabeads Protein A, which had been coated with anti-Gα antibody beforehand. The magnetic beads were washed thoroughly with 100 mM phosphate buffer (pH 7.4) containing 0.05% Tween 20 and transferred into a scintillation vial, and the radioactivity of [35S]GTPγS bound to Gα proteins captured by the magnetic beads was counted by a liquid scintillation spectrometer. Nonspecific binding was defined in the presence of 1 mM GTPγS.
Determination of adenosine content
Adenosine content in membrane preparation was examined after acid extraction and converting to fluorescent derivative 1,N6-etheno adenosine. Washed membrane preparation was adjusted to 1 mg protein/mL, and aliquots (100 μL) were mixed with 100 μL of 5% HClO4 for measuring total adenosine content in membrane preparation. To determine whether adenosine release from membrane vesicle to incubation medium during biding experiments, aliquots (150 μL) of membrane fraction were incubated for 30 min at 30 °C, followed by centrifuging at 21,500g for 10 min at 4 °C. The supernatant was mixed with equal volume of 5% HClO4. Acid extracts of membrane and incubated medium were mixed with 1/10 vol. of 4.2 M KOH to neutralize and deposit potassium perchlorate. Adenosine in the acid extracts were converted to 1,N6-etheno derivatives by treating with 1% chloroacetaldehyde at 80 °C for 30 min [17]. The 1,N6-etheno adenine nucleotides were separated using a JASCO HPLC system equipped with an analytical YMC-Pack ODS-A column (S-5, 4.6 × 100 mm, YMC Inc., Kyoto, Japan) [18] and monitored by a fluorescence detection following excitation at 270 nm at the emission wavelength of 410 nm.
Data analysis
The concentration-dependent increase in the specific [35S]GTPγS binding by adenosine was expressed as % over the basal unstimulated binding and analyzed by means of a non-linear regression method using GraphPad Prism (GraphPad Software; La Jolla, CA, USA), to produce the concentration eliciting the half-maximal effect (EC50) and the maximal of percent increase (%Emax). The concentration-response curves for the compounds depicted in Fig. 1 were analyzed, with the stimulatory effect elicited by 100 μM adenosine determined in the same experiment assumed as 100%. The inhibitory effects of adenosine antagonists on the basal binding were also analyzed by a non-linear regression method, with the basal binding regarded as 100%. Results were presented as the mean ± S.E.M. of the values obtained from the indicated number of experiments. The effect of PSB36 on adenosine-stimulated increase in [35S]GTPγS binding to Gαi/o in human prefrontal cortical membranes was analyzed by Schild plot. The stimulatory effects of adenosine on each Gα subtype determined by [35S]GTPγS binding/immunoprecipitation assay in rat brain membranes were analyzed by one-way analysis of variance (ANOVA), and the significant difference between the basal and adenosine-induced binding was determined by Tukey’s post hoc test. Linear regressions were calculated by the method of least squares and Pearson’s coefficient for simple correlation was calculated to test for possible associations between pharmacological parameters (pEC50, %Emax, and slope factor) determined by the two methods in human brain samples.

Effects of adenosine receptor agonists on specific [35S]GTPγS binding to Gαi/o in rat cerebral cortical membranes. Conventional [35S]GTPγS binding assay by means of filtration techniques was performed in the presence of increasing concentrations of CCPA (○), CPA (●), 2′-Me-CCPA (△), adenosine (▲), and CGS21680 (▽). The values represent the mean ± S.E.M. of the percent increase of the maximal stimulation obtained from three independent experiments, each performed in duplicate. In the experiments for the agonists except for adenosine, the binding in the presence of 100 μM adenosine was also determined, which was regarded as a maximal stimulation
Results
Pharmacological characterization of adenosine-induced [35S]GTPγS binding to Gαi/o in rat and postmortem human brain membranes
In rat cerebral cortical membranes, specific [35S]GTPγS binding to Gαi/o determined by the conventional filtration assay was augmented by adenosine in a concentration-dependent manner with a mean EC50 of 200 nM (pEC50 = 6.70 ± 0.02) to %Emax of 54.3 ± 2.3%. This stimulatory effects of adenosine were mimicked by the selective adenosine A1 receptor agonists, CCPA, CPA, and 2′-MeCCPA [19], with a mean EC50 of 8.8 nM (pEC50 = 8.06 ± 0.09), 10 nM (pEC50 = 7.99 ± 0.10), and 180 nM (pEC50 = 6.73 ± 0.23), respectively (Fig. 1). The maximal increases by these three compounds were 90–100% of the value determined in the presence of 100 μM adenosine. On the other hand, the selective adenosine A2A receptor agonist CGS21680 had a stimulatory effects only at the concentrations of micromolar range, resulting in a mean EC50 value of 15 μM (pEC50 = 4.82 ± 0.12). The maximal increase was 83.2 ± 8.7% (N = 3) of the value determined in the presence of 100 μM adenosine.
In postmortem human prefrontal cortical membranes, the effects of PSB36, a selective adenosine A1 receptor antagonist [20], on adenosine-induced [35S]GTPγS binding to Gαi/o were investigated. As exemplified in Fig. 2, the concentration-response curve for adenosine-stimulated [35S]GTPγS binding to Gαi/o was shifted rightward in parallel by the addition of 1, 10, and 100 nM PSB36. Schild regression analysis on three independent experiments resulted in the pA2 value of 8.00 ± 0.22.

Effects of PSB36 on adenosine-induced [35S]GTPγS binding to Gαi/o in postmortem human prefrontal cortical membranes. Conventional [35S]GTPγS binding assay by means of filtration techniques was performed in the presence of increasing concentrations of adenosine, in the absence (○) and presence of PSB36 at 1 nM (●), 10 nM (▲), and 100 nM (▼). The values represent the mean of duplicate determinations expressed as percent increase over the unstimulated basal binding in a representative experiment
Identification of Gα subtype coupled to adenosine A1 receptor determined by [35S]GTPγS binding/immunoprecipitation assay in rat brain membranes
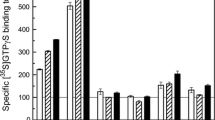
In our previous study on the [35S]GTPγS binding/immunoprecipitation assay, it was shown that adenosine A1 receptor was selectively coupled to Gαi-3 in postmortem human brain membranes [10]. However, species differences in the G-protein selectivity for adenosine A1 receptor have been demonstrated [21]. With possible species differences in mind, adenosine-induced G-protein activation was investigated using several specific anti-Gα subtype antibodies in rat brain membranes. Since the preliminary experiments (N = 3) demonstrated that the increase in specific [35S]GTPγS binding to Gαi-3 elicited by 1 mM adenosine was highest in the cerebral cortex (226 ± 18% basal) and then in the striatum (197 ± 24% basal), but only slight in the hippocampus (118 ± 18% basal), the former two brain regions were further investigated. One-way ANOVA indicated statistically significant effects in rat cerebral cortex [F(4,20) = 14.65, P < 0.0001] as well as in striatum F(4,10) = 49.99, P < 0.0001]. Among Gα subtypes, a significant increase elicited by 1 mM adenosine was obtained only for Gαi-3 (P < 0.001, Tukey’s post hoc test) but not for other Gαi/o subtypes, in both brain regions (Fig. 3).

Effects of adenosine on specific [35S]GTPγS binding to each Gαi/o subtype in rat cerebral cortical and striatal membranes. [35S]GTPγS binding/immunoprecipitation assay was performed for Gαi-1, Gαi-2, Gαi-3, and Gαo, in the absence and presence of 1 mM adenosine in rat cerebral cortical (open bars) and striatal (left hatched bars) membranes. The values represent the mean ± S.E.M. of adenosine-stimulated bindings, expressed as the percent of the unstimulated basal binding, obtained from five (cerebral cortex) and three (striatum) independent experiments, each performed in triplicate. ***p < 0.001, one-way ANOVA followed by Tukey’s post hoc test
Effects of adenosine receptor antagonists on basal [35S]GTPγS binding to Gαi/o
In rat cerebral cortical membranes, the basal specific [35S]GTPγS binding to Gαi/o was inhibited by several adenosine A1 selective or nonselective antagonists partially by approximately 20–30%, with a rank order of potency of DPCPX >> 1,3-dimethyl-8-phenylxanthine > VU5574 > theophylline > caffeine (Fig. 4a). The inhibition by PSB36 reached to the same extent (approximately 20%), and its maximal inhibitory effects were obtained at extraordinarily low concentrations, even at 10−28 M (Fig. 4b). In a representative experiment, specific basal [35S]GTPγS binding was 23,226 ± 171 dpm (mean ± S.E.M. of quadruplicate determinations), whereas the specific binding in the presence of PSB36 at 10−28 M was 18,903 dpm (mean of duplicate determinations).

Effects of adenosine receptor antagonists on basal specific [35S]GTPγS binding to Gαi/o in rat cerebral cortical and postmortem human prefrontal cortical membranes. Conventional [35S]GTPγS binding assay by means of filtration techniques was performed in the presence of increasing concentrations of a DPCPX (○), 1,3-dimethyl-8-phenylxanthine (●), VU5574 (△), theophylline (▲), and caffeine (▽) in rat cerebral cortex, b PSB36 (○) in rat cerebral cortex, and c DPCPX (○) and 1,3-dimethyl-8-phenylxanthine (●) in human prefrontal cortex. The values for all compounds (except for 1,3-dimethyl-8-phenylxanthine in human prefrontal cortex) represent the mean ± S.E.M. of the percent of the unstimulated basal binding obtained from three independent experiments, each performed in duplicate. The values for 1,3-dimethyl-8-phenylxanthine in human prefrontal cortex were the mean of the results obtained from two independent experiments, each performed in duplicate
In human prefrontal cortical membranes, the effects of the two adenosine A1 receptor selective antagonists, DPCPX and 1,3-dimethyl-8-phenylxanthine, were investigated. As shown in Fig. 4c, both compounds inhibited the basal specific [35S]GTPγS binding to Gαi/o to the same extent (approximately 30%). The inhibitory effects of DPCPX were clearly more potent than 1,3-dimethyl-8-phenylxanthine.
Effects of adenosine receptor antagonists on basal [35S]GTPγS binding to Gαi-3
The inhibitory effects of several adenosine receptor antagonists on the basal specific [35S]GTPγS binding to Gαi-3 were determined by [35S]GTPγS binding/immunoprecipitation assay only in rat cerebral cortical membranes. As illustrated in Fig. 5, all the compounds investigated inhibited the basal binding to Gαi-3 by around 20–30%, with a rank order of potency of PSB36 > DPCPX ≈ 1,3-dimethyl-8-phenylxanthine > VUF5574 > theophylline > caffeine.

Effects of adenosine receptor antagonists on basal specific [35S]GTPγS binding to Gαi-3 in rat cerebral cortical membranes. [35S]GTPγS binding/immunoprecipitation assay was performed in the presence of increasing concentrations of PSB36 (○), DPCPX (●), 1,3-dimethyl-8-phenylxanthine (△), VU5574 (▲), theophylline (▽), and caffeine (▼). The values represent the mean ± S.E.M. of the percent of the unstimulated basal binding obtained from 3 to 4 independent experiments, each performed in duplicate
Concentrations of adenosine in membrane preparation and supernatant
Concentrations of adenosine, determined after extraction from the brain membrane preparations adjusted to 1 mg protein/mL, were within a range of 1–2 μM, for all three rat brain regions as well as for postmortem human prefrontal cortex (Table 1). The adenosine concentrations were also determined in supernatant fractions after incubation of the brain membrane preparations at 30 °C for 30 min. In the case of rat brain membranes, the concentrations of adenosine in supernatant were 350 ± 70, 191 ± 15, and 233 ± 40 nM, in the cerebral cortex, hippocampus, and striatum, respectively. Conversely, the supernatant fraction of postmortem human prefrontal cortical membranes contained only one-tenth of the levels found in rat brain membranes (28 ± 6 nM).
Effects of ADA on [35S]GTPγS binding to Gαi/o
The effects of ADA on the basal specific [35S]GTPγS binding to Gαi/o were investigated by conventional [35S]GTPγS binding assay. In rat cerebral cortical membranes, the addition of ADA in incubation buffer resulted in concentration-dependent inhibitory effects (Fig. 6a). In the presence of ADA at 20 U/tube, the basal binding was inhibited to 55.3 ± 2.4% (N = 4) of the binding determined in the absence of ADA. The similar inhibitory effects were also observed in hippocampal and striatal membranes, though to a somewhat smaller extent [72.9 ± 3.9% (N = 3) and 67.2 ± 3.6% (N = 3) in the presence of 20 U/tube ADA in the hippocampus and striatum, respectively] (not shown). In order to ascertain that the influence of ADA derived from its enzymatic activity, we tried to verify whether pretreatment of ADA by heating abolished its inhibitory effects. The enzymatic inactivation of ADA by heating did not reverse the inhibitory effects of ADA, contrary to expectation (Fig. 6a, inset).

Effects of ADA on basal specific [35S]GTPγS binding to Gαi/o in rat cerebral cortical and postmortem human prefrontal cortical membranes. Conventional [35S]GTPγS binding assay by means of filtration techniques was performed in the presence of increasing concentrations of ADA in rat cerebral cortex (a) and human prefrontal cortex (b). The open symbols represent the mean ± S.E.M. of the percent of the unstimulated basal binding obtained from four independent experiments (depicted as thin lines), each performed in duplicate or triplicate. (Insets) Effects of pretreatment of ADA by heating in rat cerebral cortical (a) and postmortem human prefrontal cortical (b) membranes. Conventional [35S]GTPγS binding assay by means of filtration techniques was performed in the presence of 15 U/tube or 1 mU/tube ADA in rat cerebral cortex and human prefrontal cortex, respectively, either non-treated (open bar) or pretreated by boiling at 95 °C for 15 min (rat) or for 5 min (human) (left hatched bar). The values represent the mean ± S.E.M. of the percent of the unstimulated basal binding determined in the absence of ADA, obtained from 3 to 4 independent experiments, each performed in duplicate
In postmortem human prefrontal cortical membranes, ADA inhibited the basal specific [35S]GTPγS binding to Gαi/o (Fig. 6b). The inhibitory effects of ADA were observed at very low concentrations as compared to those in rat brain membranes. Thus, in the presence of ADA at 1 mU/tube, the basal binding was inhibited to 82.4 ± 2.4% (N = 4). This inhibitory effect of ADA was canceled when ADA was inactivated by boiling (95 °C for 5 min) beforehand (Fig. 6b, inset).
Effects of ADA on [35S]GTPγS binding to Gαi-3
The effects of ADA on the basal specific [35S]GTPγS binding were also studied in [35S]GTPγS binding/immunoprecipitation experiments. In rat cerebral cortical membranes, the effects of ADA appeared inconsistent (Fig. 7a). The inhibitory effects of ADA were observed in some experiments, whereas ambiguous or even stimulatory effects were detected in other experiments. In the rat brain membranes in which the stimulatory effects of ADA were observed, these effects were not altered by the pretreatment of ADA by heating (not shown).

Effects of ADA on basal specific [35S]GTPγS binding to Gαi-3 in rat cerebral cortical and postmortem human prefrontal cortical membranes. [35S]GTPγS binding/immunoprecipitation assay was performed in the presence of increasing concentrations of ADA in rat cerebral cortex (a) and human prefrontal cortex (b). a The open symbols represent the mean of duplicate determinations of each experiment, expressed as the percent of the respective unstimulated basal binding. b The open symbols represent the mean ± S.E.M. of the percent of the unstimulated basal binding obtained from four independent experiments (depicted as thin lines), each performed in duplicate
In postmortem prefrontal cortical membranes, the basal specific [35S]GTPγS binding to Gαi-3 was inhibited by ADA in a concentration-dependent manner (Fig. 7b). In the presence of ADA at 5 U/tube, the basal binding was inhibited to 66.0 ± 2.1% (N = 4).
Effects of adenosine on [35S]GTPγS binding to Gαi/o in postmortem human brain membranes
As shown in Fig. 8, the %Emax values of adenosine-stimulated [35S]GTPγS binding to Gαi/o determined by conventional [35S]GTPγS binding assay ranged from 133 to 422%, with a mean value of 272 ± 12%. The mean EC50 value, derived from pEC50 values (6.34 ± 0.02, ranging from 5.98 to 6.52), was 454 nM. Hill coefficient ranged from 0.71 to 1.06, with a mean value of 0.86 ± 0.01.

Stimulatory effect of adenosine on the specific [35S]GTPγS binding to Gαι/ο in postmortem human prefrontal cortical membranes. Conventional [35S]GTPγS binding assay by means of filtration techniques was performed in the presence of increasing concentrations of adenosine in human prefrontal cortex. The open symbols represent the mean ± S.E.M. of the percent of the unstimulated basal binding obtained from independent experiments determined in 40 subjects (depicted as thin lines), each performed in duplicate
Effects of adenosine on [35S]GTPγS binding to Gαi-3- in postmortem human brain membranes
The concentration-dependent increases in specific [35S]GTPγS binding to Gαi-3-were also determined by means of [35S]GTPγS binding/immunoprecipitation assay in the same 40 subjects (Fig. 9). The specific [35S]GTPγS binding to Gαi-3-was increased by the addition of adenosine in a concentration-dependent manner, with the %Emax value of 160 ± 9% (ranging from 58 to 269%) and a slope value of 0.93 ± 0.04 (ranging from 0.49 to 1.72). The mean EC50 value, derived from pEC50 values (6.09 ± 0.06, ranging from 5.39 to 6.98), was 822 nM.

Stimulatory effect of adenosine on the specific [35S]GTPγS binding to Gαι-3 in postmortem human prefrontal cortical membranes. [35S]GTPγS binding/immunoprecipitation assay was performed in the presence of increasing concentrations of adenosine in human prefrontal cortex. The open symbols represent the mean ± S.E.M. of the percent of the unstimulated basal binding obtained from independent experiments determined in 40 subjects (depicted as thin lines), each performed in duplicate
Effects of sex, drug(s) detected in toxicological screening, age, postmortem delay, storage period, and tissue pH on adenosine-stimulated [35S]GTPγS binding in postmortem human brain membranes
When 40 subjects were divided into two groups based on sex (26 males/14 females) and presence or absence of any drug(s) in their blood (12 presence/28 absence), there were no statistically significant differences between the two groups in any of the tested parameters, in either [35S]GTPγS binding experiment. In addition, no statistically significant correlation was obtained between each pharmacological parameter (%Emax, pEC50, and slope) and age (range 16–80 years), postmortem delay (range 3–64 h), freezing storage period (range 30–257 and 35–244 months for conventional [35S]GTPγS binding and [35S]GTPγS binding/immunoprecipitation, respectively), or tissue pH (range 5.8–6.8, available in 22 subjects), except for the following two correlations. One significant correlation was obtained between age and %Emax values determined in adenosine-induced [35S]GTPγS binding to Gαi/o determined by conventional [35S]GTPγS binding assay (r = 0.38, p < 0.05) (Fig. 10a). The significant positive correlation was kept in male subjects (r = 0.54, p < 0.05), but not in females. Another one was a significant correlation between postmortem delay and %Emax values in adenosine-induced [35S]GTPγS binding to Gαi/o (r = 0.31, p < 0.05) (Fig. 10b). The correlation was still significant in the male subjects (r = 0.43, p < 0.05), but not in the female group.

Effects of age and postmortem delay on adenosine-induced [35S]GTPγS binding to Gαi/o in postmortem human prefrontal cortical membranes. a The values represent individual %Emax of the adenosine-stimulated [35S]GTPγS binding to Gαi/o in male (○) or female (●) subjects, scattered as a function of age. The regression lines are depicted for all (solid line) or male data (broken line). (b) The values represent individual %Emax of the adenosine-stimulated [35S]GTPγS binding to Gαi/o in male (○) or female (●) subjects, scattered as a function of postmortem delay. The regression lines are depicted for all (solid line) or male data (broken line)
Interrelation between adenosine-stimulated [35S]GTPγS bindings to Gαi/o and adenosine-stimulated [35S]GTPγS bindings to Gαi-3 in postmortem human brain membranes
Interrelation of each pharmacological parameter (%Emax, pEC50, and slope) between adenosine-stimulated [35S]GTPγS binding to Gαi/o determined by conventional [35S]GTPγS binding assay and adenosine-stimulated [35S]GTPγS binding to Gαi-3 determined by [35S]GTPγS binding/immunoprecipitation assay was investigated by means of linear regression analysis by the method of least squares. No significant correlation was obtained for any parameter between both measures (r = −0.01, p > 0.05 for %Emax; r = − 0.03, p > 0.05 for pEC50; and r = − 0.28, p > 0.05 for slop factor).
Discussion
In the present study, we utilized two [35S]GTPγS binding methods, i.e., conventional [35S]GTPγS binding assay using filtration techniques [16] and [35S]GTPγS binding/immunoprecipitation assay [10, 16], in rat and postmortem human brain membranes. In postmortem human prefrontal cortical membranes, the receptor subtype involved in adenosine-induced [35S]GTPγS binding to Gαi-3 was pharmacologically characterized as adenosine A1 receptor [10]. The experiments using conventional [35S]GTPγS binding assay performed in the present study also indicated the involvement of adenosine A1 receptor. Adenosine itself stimulated the specific [35S]GTPγS binding to Gαi/o in rat cerebral cortical membranes with a mean EC50 of 200 nM. These stimulatory effects were mimicked by several selective adenosine A1 receptor agonists, CCPA, CPA, and 2′-MeCCPA [19], at submicromolar concentrations. The results depicted in Fig. 1 indicate that all of these three compounds behave as almost full agonists, with intrinsic activities of 90–100%. Although it is exceedingly difficult to determine the affinity of the endogenous ligand adenosine to adenosine receptors [19], one study using a functional assay for inhibitory effects on adenylate cyclase in rat fat cell membranes has indicated that its potency to adenosine A1 receptor is 73 nM [22]. Also, it was reported that adenosine inhibited forskolin-stimulated cyclic AMP formation in Chinese hamster ovary (CHO) cells stably transfected with human adenosine A1 receptor with an EC50 of 310 and 54 nM in the absence and presence of nitrobenzylthioinosine (adenosine transport inhibitor), respectively [23]. The stimulatory effects of CGS21680, a selective adenosine A2A receptor agonist, at higher concentrations are likely attributable to its property as a weak agonist at adenosine A1 receptor [24,25,26]. The intrinsic activity of CGS21680 (83.2 ± 8.7%) appeared somewhat lower than other three compounds. However, the exact intrinsic activity of this compound is difficult to determine due to a lack of enough saturability at the concentrations investigated in the present study. According to the previous report [25], CGS21680 has been reported to act as a full agonist at human adenosine A1 receptor expressed in CHO cells. In postmortem human prefrontal cortical membranes, adenosine-stimulated [35S]GTPγS binding to Gαi/o was inhibited by the selective adenosine A1 receptor antagonist PSB36 in a competitive manner, with a mean pA2 value of 8.00. The Ki value of this compound for human adenosine A1 receptor is reported to be 0.7 nM, based on the data obtained from [3H]CCPA binding experiments [20].
Differences in G-protein coupling with adenosine A1 receptors from rat, human, and bovine brain have been demonstrated [21, 27]. In the present study, we verified whether selective coupling between adenosine A1 receptor and Gαi-3 observed in postmortem human brain membranes [10] was also detected in rat brain membranes. As in human brain membranes, adenosine-induced [35S]GTPγS binding through adenosine A1 receptors was detectable only for Gαi-3, but not for other Gαi/o subtypes, in [35S]GTPγS binding/immunoprecipitation experiments, at least in rat cerebral cortical and striatal membranes. We performed this type of experiments in these two brain regions, but not in hippocampus, since the preceding results indicated the magnitudes of adenosine-stimulated [35S]GTPγS binding to Gαi-3 were prominent in cortex and striatum, but faint in hippocampus. The reason of scarce response in hippocampus in [35S]GTPγS binding/immunoprecipitation experiments is unclear at the moment. The autoradiographic as well as immunohistochemical studies have shown that adenosine A1 receptors are distributed widespread throughout the brain including hippocampal formation [2, 28, 29]. Although an immunohistochemical study indicates the existence of Gαi-3-peptide-positive neurons in the hippocampus [30], the expression level of Gαi-3 proteins may be lower in hippocampus than in other brain regions such as the cerebral cortex and striatum. Alternatively, coupling efficiency between adenosine A1 receptor and Gαi-3 protein may be weak in the hippocampus compared to the two other brain regions.
Several adenosine receptor antagonists inhibited the basal [35S]GTPγS binding to Gαi/o in rat and human brain membranes. Since it has been shown that some adenosinergic ligands including DPCPX act as an inverse agonist, but not as a neutral antagonist [31], it is possible to regard the data obtained in the present study as the effects of inverse agonists on constitutive active adenosine A1 receptors in the brain membranes. However, since the constitutive activity is mainly evidenced for recombinant receptors overexpressed and/or mutated [32, 33], an alternative explanation for the phenomena is that the negative intrinsic activities of these compounds derive from pseudo basal binding levels due to the presence of residual endogenous adenosine in the assay buffer [7, 34]. In either way, the rank order of potencies of these antagonists in rat cerebral cortical membranes, i.e., DPCPX >> 1,3-dimethyl-8-phenylxanthine > VUF5574 > theophylline > caffeine, indicates the involvement of adenosine A1 receptor subtype [19, 35]. Similar inhibition of the basal [35S]GTPγS binding to Gαi/o via adenosine A1 receptor subtype was also observed in human prefrontal cortical membranes (DPCPX > 1,3-dimethyl-8-phenylxanthine). To our surprise, the maximal inhibitory effects of PSB36 on the basal [35S]GTPγS binding to Gαi/o in rat cerebral cortical membranes (approximately 20%) were obtained even at 10−28 M. The inhibitory effects of this compound were concentration-independent and constant at wide range of concentrations from 10−28 to 10−4 M (data not shown). The Ki value of this compound at rat and human adenosine A1 receptor is reported to be 0.12 and 0.7 nM, respectively [20]. The inhibitory effects of PSB36 at unusually low concentrations may be pharmacologically irrelevant artifact.
In contrast with the peculiar inhibitory effects of PSB36 in [35S]GTPγS binding experiments in rat cerebral cortical membranes, PSB36 inhibited the basal [35S]GTPγS binding to Gαi-3 in rat cerebral cortical membranes determined by [35S]GTPγS binding/immunoprecipitation assay in an ordinary way, with an IC50 value of nanomolar order. This compound is the most potent among the ligands investigated, and the rank order of potencies as an antagonist (PSB36 > DPCPX ≈ 1,3-dimethyl-8-phenylxanthine > VUF5574 > theophylline > caffeine) indicates the involvement of adenosine A1 receptor subtype again in these inhibitory effects.
The question of whether residual endogenous adenosine exists in incubation buffer under the experimental conditions in the present study was addressed by direct determination of adenosine concentrations. The results indicate that the membrane preparations used in the present study contain substantial contents of adenosine. Moreover, endogenous adenosine is detectable in the supernatant fraction (350 and 28 nM in rat and human cortex, respectively) subsequent to the incubation of the membranes (1 mg protein/mL). If it is assumed that the rate of endogenous adenosine production by membranes is proportional to the concentration of membrane protein, it follows that the assay buffer in the conventional [35S]GTPγS binding experiments using rat (10–20 μg protein/500 μL) and human (60 μg protein/500 μL) cortical membranes contains 7–14 and 3 nM adenosine, respectively. In the [35S]GTPγS binding/immunoprecipitation study, the assay was performed with the membranes prepared from rat (20 μg protein/200 μL) and human (80 μg protein/200 μL), and adenosine concentration in the assay buffer was calculated to be 35 and 11 nM, respectively. These concentrations of endogenous adenosine are very low as compared with the EC50 values of adenosine, and thus, it is unlikely that the basal [35S]GTPγS binding is elevated to a considerable extent by the existence of residual adenosine in the experimental conditions adopted in the present study.
Although ADA has been included in experimental systems frequently in order to remove endogenous adenosine in radioligand binding and functional assays for adenosine receptors, little information is available as to how much ADA is necessary and enough [34]. In the present study, the results indicate that ADA affects the basal [35S]GTPγS binding levels with different sensitivity and varied pattern, depending on species (rat vs. human) as well as experimental designs (conventional assay vs. [35S]GTPγS binding/immunoprecipitation). In human prefrontal cortical membranes, the basal specific [35S]GTPγS binding determined by conventional filtration assay was inhibited by the addition of ADA at very low concentrations (< 1 mU/tube), compared to the higher concentrations (~ 20 U/tube) of ADA needed in rat cerebral cortical membranes. The inhibitory effects of ADA in human prefrontal cortical membranes were likely originated from its enzymatic activity, since the effects of ADA were canceled subsequent to its deactivation by heating. The basal binding was also inhibited by ADA (~ 5 U/tube) in the [35S]GTPγS binding/immunoprecipitation experiments using human brain membranes. On the other hand, the results from the same experiments using rat cerebral cortical membranes were perplexing. The effects of ADA on the basal [35S]GTPγS binding to Gαi-3 were inhibitory as anticipated in some samples, but even stimulatory in others. These stimulatory effects of ADA appear to be irrelevant to its activity as an enzyme, because the pretreatment of ADA by heating failed to counteract its effects (not shown).
All of the abovementioned results, i.e., decrease in basal [35S]GTPγS binding by adenosine receptor antagonists through adenosine A1 receptor, detection of adenosine at nanomolar concentrations in supernatant fraction subsequent to incubation of the membranes, and inhibitory effects of ADA on basal [35S]GTPγS binding, suggest the possibility that residual endogenous adenosine is present in the assay mixture in the present study. However, it is said that it is difficult to remove endogenous adenosine completely with ADA [34]. In fact, it has been reported that lipophilic adenosine receptor antagonists such as DPCPX and the neutral antagonist N-0840 inhibited basal [35S]GTPγS binding even in the presence of ADA in rat cerebellar membranes, indicative of ADA-resistant adenosine pool [36]. The diverse influences of ADA on basal [35S]GTPγS binding assessed by [35S]GTPγS binding/immunoprecipitation assay in rat brain membranes prompted us to hesitate to include ADA routinely in assay buffers. It has been reported that ADA is a moonlighting protein, with multifunctional properties (e.g., as an allosteric modulator of adenosine receptors) in addition to its enzymatic activity [37]. The perplexing results of ADA in the present study may be ascribed to its functions other than catalytic action. Moreover, the presumed concentrations of adenosine in incubation mixture in the present study are only nanomolar order, much lower in comparison with the reported potencies of adenosine at adenosine A1 receptor [22, 23] and with the EC50 values determined in the present study (200 and 450 nM in rat and human brain membranes, respectively, in conventional [35S]GTPγS binding assay; 820 nM in human membranes in [35S]GTPγS binding/immunoprecipitation assay). Taking these results into account, it was ultimately decided that ADA was not routinely added in the following experiments in the present study.
By utilizing conventional [35S]GTPγS binding and [35S]GTPγS binding/immunoprecipitation assays in the absence of ADA [10], adenosine-stimulated G-protein activation was determined in prefrontal cortical membranes prepared from 40 subjects with no psychiatric and/or neurological disorders (16~80 years old). There have been lots of reports dealing with alterations in the number of adenosine A1 receptors according to aging. Most of such studies using radioligand binding assay or quantitative autoradiography in rodent brains [38,39,40,41,42] as well as positron emission tomography (PET) in living humans [43, 44] have indicated consistently age-related decline of adenosine A1 receptors in the brain. However, conflicting results as to the declining effects of aging on adenosine A1 receptor density have also been reported by several studies [28, 45,46,47,48,49,50]. Ułas et al. [51] demonstrated that coupling of adenosine A1 receptors to G-proteins remained unaltered in spite of remarkable reduction of their density in hippocampus of patients with Alzheimer’s disease. Even if there may be a tendency of age-dependent reduction of adenosine A1 receptors, the coupling efficiency between adenosine A1 receptors and G-proteins is enhanced, rather than reduced, according to aging, likely by compensatory mechanisms.
The %Emax values of adenosine-stimulated [35S]GTPγS binding determined by conventional assay were also correlated with postmortem delay in the present study. The effects of postmortem delay on receptor-mediated [35S]GTPγS bindings were reported in the previous study [52], which indicated that postmortem delay with a range of 8–92 h had no effects on basal levels of [35S]GTPγS binding or stimulation mediated through α2-adrenergic, μ-opioid, 5-HT1A, GABAB, or muscarinic acetylcholine receptor. Although the decrease in receptor binding associated with prolonged postmortem delay has been reported for many receptors [53,54,55,56,57,58], this rule is not generalized to all receptors. Indeed, the unaltered or even increased receptor binding induced by postmortem delay has been reported for some receptors [54,55,56]. Since the information on how postmortem delay affects adenosine A1 receptor binding is unavailable, it is unknown whether the results obtained in the present study are attributable to increase in adenosine A1 receptors or to strengthened coupling efficiency between adenosine A1 receptor and G-protein by prolonged postmortem period.
A lack of correlation between parameters such as %Emax values of adenosine-stimulated [35S]GTPγS binding determined by conventional assay and [35S]GTPγS binding/immunoprecipitation assay was against our expectation. These results may indicate that the two functional measures reflect distinct biochemical consequences, although mediated commonly through adenosine A1 receptor. Although adenosine A1 receptor-mediated G-protein activation was detectable only for Gαi-3 in [35S]GTPγS binding/immunoprecipitation experiments, adenosine A1 receptor-mediated [35S]GTPγS binding determined by conventional filtration assay derives not only from Gαi-3 but also from other Gαi/o proteins.
The coupling between adenosine A1 receptor and G-proteins was assessed by means of both [35S]GTPγS binding assays, i.e., conventional filtration method and [35S]GTPγS binding/immunoprecipitation in the present study. In conclusion, the latter method provided information about adenosine A1 receptor-mediated Gαi-3 activation in rat as well as human brain membranes. On the other hand, adenosine-stimulated [35S]GTPγS binding determined with conventional assay derives from functional activation of Gαi/o proteins (not restricted only to Gαi-3) coupled to adenosine A1 receptors. The determination of adenosine concentrations in the samples used in the present study indicates the possibility that the assay mixture under our experimental conditions contains residual endogenous adenosine at nanomolar concentrations, which was also suggested by the results on the effects of adenosine receptor antagonists on basal [35S]GTPγS binding level. The effects of ADA on basal binding also support the presence of adenosine. Nevertheless, the varied patterns of ADA discouraged us from adding ADA into assay medium routinely. The concentration-dependent increases elicited by adenosine were determined in 40 subjects without any neuropsychiatric disorders. The increases in %Emax values determined by conventional assay according to aging and postmortem delay should be taken into account in future studies focusing on the effects of psychiatric disorders on adenosine A1 receptor/G-protein interaction in postmortem human brain tissue.
Abbreviations
- GPCR:
-
G-protein-coupled receptor
- [35S]GTPγS:
-
Guanosine-5′-O-(3-[35S]thio) triphosphate
- PSB36:
-
1-Butyl-3-(3-hydroxypropyl)-8-(3-noradamantyl)xanthine
- VUF5574:
-
1,3-Dimethyl-8-phenylxanthine, N-(2-Methoxyphenyl)-N′-[2-(3-pyridinyl)-4-quinazolinyl]-urea
- CPA:
-
N6-Cyclopentyladenosine
- CCPA:
-
2-Chloro-N6-cyclopentyladenosine
- 2′-MeCCPA:
-
2-Chloro-N-cyclopentyl-2′-methyladenosine
- CGS21680:
-
4-[2-[[6-Amino-9-(N-ethyl-β-D-ribofuranuronamidosyl)-9H-purin-2-yl] amino]ethyl]benzenepropanoic acid hydrochloride
- DPCPX:
-
8-Cyclopentyl-1,3-dipropylxanthine
- ADA:
-
Adenosine deaminase
- TED:
-
5 mM Tris–HCl, 1 mM EDTA, 1 mM dithiothreitol; pH 7.4
- EGTA:
-
Ethylene glycolbis(2-aminoethylether)-N,N,N,N-tetraacetic acid
- EC50 :
-
The concentration eliciting the half-maximal effect
- %E max :
-
The maximal percent increase
- ANOVA:
-
Analysis of variance
- PET:
-
Positron emission tomography
References
Fredholm BB, IJzerman AP, Jacobson KA, Klotz K-N, Linden J (2001) International Union of Pharmacology. XXV. Nomenclature and classification of adenosine receptors. Pharmacol Rev 53:527–552
Svenningsson P, Hall H, Sedvall G, Fredholm BB (1997) Distribution of adenosine receptors in the postmortem human brain: an extended autoradiographic study. Synapse 27:322–335
Harrison C, Traynor JR (2003) The [35S]GTPγS binding assay: approaches and applications in pharmacology. Life Sci 74:489–508
Strange PG (2010) Use of the GTPγS ([35S]GTPγS and Eu-GTPγS) binding assay for analysis of ligand potency and efficacy at G protein-coupled receptor. Br J Pharmacol 161:1238–1249. https://doi.org/10.1111/j.1476-5381.2010.00963.x
Lorenzen A, Fuss M, Vogt H, Schwabe U (1993) Measurement of guanine nucleotide-binding protein activation by A1 adenosine receptor agonists in bovine brain membranes: stimulation of guanosine-5’-O-(3-[35S]thio)triphosphate binding. Mol Pharmacol 44:115–123
Lorenzen A, Guerra L, Vogt H, Schwabe U (1996) Interaction of full and partial agonists of the A1 adenosine receptor with receptor/G protein complexes in rat brain membranes. Mol Pharmacol 49:915–926
Moore RJ, Xiao R, Sim-Selley LJ, Childers SR (2000) Agonist-stimulated [35S]GTPγS binding in brain. Modulation by endogenous adenosine. Neuropharmacology 39:282–289
Laitinen JT (1999) Selective detection of adenosine A1 receptor-dependent G-protein activity in basal and stimulated conditions of rat brain [35S]guanosine 5’-(γ-thio)triphosphate autoradiography. Neuroscience 90:1265–1279
Childers SR, Li X, Xiao R, Eisenach JC (2005) Allosteric modulation of A1 receptor coupling to G-proteins in brain. J Neurochem 2005:715–723. https://doi.org/10.1111/j.1471-4159.2005.03044.x
Odagaki Y, Kinoshita M, Ota T, Meana JJ, Callado LF, García-Sevilla JA (2015) Adenosine A1 receptors are selectively coupled to Gαi-3 in postmortem human brain cortex: Guanosine-5’-O-(3-[35S]thio)triphosphate ([35S]GTPγS) binding/immunoprecipitation study. Eur J Pharmacol 764:592–598. https://doi.org/10.1016/j.ejphar.2015.07.049
Lara DR, Dall’Igna OP, Ghisolfi ES, Brunstein MG (2006) Involvement of adenosine in the neurobiology of schizophrenia and its therapeutic implications. Prog Neuro-Psychopharmacol Biol Psychiatry 30:617–629. https://doi.org/10.1016/j.pnpbp.2006.02.002
Boison D, Singer P, Shen H-Y, Feldon J, Yee BK (2012) Adenosine hypothesis of schizophrenia—opportunities for pharmacotherapy. Neuropharmacology 62:1527–1543. https://doi.org/10.1016/j.neuropharm.2011.01.048
Hirota T, Kishi T (2013) Adenosine hypothesis in schizophrenia and bipolar disorder: a systematic review and meta-analysis of randomized controlled trial of adjuvant purinergic modulators. Schizoph Res 149:88–95. https://doi.org/10.1016/j.schres.2013.06.038
Krügel U (2016) Purinergic receptors in psychiatric disorders. Neuropharmacology 104:212–225. https://doi.org/10.1016/j.neuropharm.2015.10.032
Odagaki Y, Kinoshita M, Ota T, Meana JJ, Callado LF, García-Sevilla JA (2017) Functional activation of Gαq coupled to 5-HT2A receptor and M1 muscarinic acetylcholine receptor in postmortem human cortical membranes. J Neural Transm 124:1123–1133. https://doi.org/10.1007/s00702-017-1749-0
Odagaki Y, Kinoshita M, Ota T (2016) Comparative analysis of pharmacological properties of xanomeline and N-desmethylclozapine in rat brain membranes. J Psychopharmacol 30:896–912. https://doi.org/10.1177/0269881116658989
Kawamoto Y, Shinozuka K, Kunitomo M, Haginaka J (1998) Determination of ATP and its metabolites released from rat caudal artery by isocratic ion-pair reversed-phase high-performance liquid chromatography. Anal Biochem 262:33–38. https://doi.org/10.1006/abio.1998.2729
Matsuoka I, Zhou Q, Ishimoto H, Nakanishi H (1995) Extracellular ATP stimulates adenylyl cyclase and phospholipase C through distinct purinoceptors in NG108-15 cells. Mol Pharmacol 47:855–862
Fredholm BB, IJzerman AP, Jacobson KA, Linden J, Müller CE (2011) International Union of Basic and Clinical Pharmacology. LXXXI. Nomenclature and classification of adenosine receptors—an update. Pharmacol Rev 63:1–34. https://doi.org/10.1124/pr.110.003285
Weyler S, Fülle F, Diekmann M, Schumacher B, Hinz S, Klotz K-N, Müller CE (2006) Improving potency, selectivity, and water solubility of adenosine A1 receptor antagonists: xanthines modified at position 3 and related pyrimido[1,2,3-cd]purinediones. ChemMedChem 1:891–902. https://doi.org/10.1002/cmdc.200600066
Jockers R, Linder ME, Hohenegger M, Nanoff C, Bertin B, Strosberg AD, Marullo S, Freissmuth M (1994) Species difference in the G protein selectivity of the human and bovine A1-adenosine receptor. J Biol Chem 269:32077–32084
Daly JW, Padgett WL (1992) Agonist activity of 2- and 5’-substituted adenosine analogs and their N 6-cycloalkyl derivatives at A1- and A2-adenosine receptors coupled to adenylate cyclase. Biochem Pharmacol 43:1089–1093
Fredholm BB, Irenius E, Kull B, Schulte G (2001) Comparison of the potency of adenosine as an agonist at human adenosine receptors expressed in Chinese hamster ovary cells. Biochem Pharmacol 61:443–448
Lupica CR, Cass WA, Zahniser NR, Dunwiddle TV (1990) Effects of the selective adenosine A2 receptor agonist CGS21680 on in vitro electrophysiology, cAMP formation and dopamine release in rat hippocampus and striatum. J Pharmacol Exp Ther 252:1134–1141
Gao Z-G, Mamedova LK, Chen P, Jacobson KA (2004) 2-Substituted adenosine derivatives: affinity and efficacy at four subtypes of human adenosine receptors. Biochem Pharmacol 68:1985–1993. https://doi.org/10.1016/j.bcp.2004.06.011
Casadó V, Barrondo S, Spasic M, Callado LF, Mallol J, Canela E, Lluís C, Meana J, Cortés A, Sallés J, Franco R (2010) Gi protein coupling to adenosine A1-A2A receptor heteromers in human brain caudate nucleus. J Neurochem 114:972–980. https://doi.org/10.1111/j.1471-4159.2010.06810.x
Nanoff C, Mitterauer T, Roka F, Hohenegger M, Freissmuth M (1995) Species differences in A1 adenosine receptor/G protein coupling: identification of a membrane protein that stabilizes the association of the receptor/G protein complex. Mol Pharmacol 48:806–817
Fastbom J, Pazos A, Probst A, Palacios JM (1987) Adenosine A1 receptors in the human brain: a quantitative autoradiographic study. Neuroscience 22:827–839
Rivkees SA, Price SL, Zhou FC (1995) Immunohistochemical detection of A1 adenosine receptors in rat brain with emphasis on localization in the hippocampal formation, cerebral cortex, cerebellum, and basal ganglia. Brain Res 677:193–203
Cortés R, Hökfelt T, Schalling M, Goldstein M, Goldsmith P, Spiegel A, Unson C, Walsh J (1988) Antiserum raised against residues 159-168 of the guanine nucleotide-binding protein Gi3-α reacts with ependymal cells and some neurons in the rat brain containing cholecystokinin- or cholecystokinin- and tyrosine 3-hydroxylase-like immunoreactivities. Proc Natl Acad USA 85:9351–9355
Shryock JC, Ozeck MJ, Belardinelli L (1998) Inverse agonists and neutral antagonists of recombinant human A1 adenosine receptors stably expressed in Chinese hamster ovary cells. Mol Pharmacol 53:886–893
Lefkowiz RJ, Cotecchia S, Samama P, Costa T (1993) Constitutive activity of receptors coupled to guanine nucleotide regulatory proteins. Trends Pharmacol Sci 14:303–307
Milligan G, Bond RA, Lee M (1995) Inverse agonism: pharmacological curiosity or potential therapeutic strategy? Trends Pharmacol Sci 16:10–13
Linden J (1989) Adenosine deaminase for removing adenosine: how much is enough? Trends Pharmacol Sci 10:260–262
Alnouri MW, Jepards S, Casari A, Schiedel AC, Hinz S, Müller CE (2015) Selectivity is species-dependent: characterization of standard agonists and antagonists at human, rat, and mouse adenosine receptors. Purinergic Signal 11:389–407. https://doi.org/10.1007/s11302-015-9460-9
Savinainen JR, Saario SM, Niemi R, Järvinen T, Laitinen JT (2003) An optimized approach to study endocannabinoid signaling: evidence against constitutive activity of rat brain adenosine A1 and cannabinoid CB1 receptors. Br J Pharmacol 140:1451–1459. https://doi.org/10.1038/sj.bjp.0705577
Cortés A, Gracia E, Moreno E, Mallol J, Lluís C, Canela EI, Csadó V (2015) Moonlighting adenosine deaminase: a target protein for drug development. Med Res Rev 35:85–125. https://doi.org/10.1002/med.21324
Pagonopoulou O, Angelatou F (1992) Reduction of A1 adenosine receptors in cortex, hippocampus and cerebellum in ageing mouse brain. Neuro Report 3:735–737
Cunha RA, Constantino MD, Sebastião AM, Ribeiro JA (1995) Modification of A1 and A2a adenosine receptor binding in aged striatum, hippocampus and cortex of the rat. Neuroreport 6:1583–1588
Sperlágh B, Zsilla G, Baranyi M, Kékes-Szabó A, Vizi ES (1997) Age-dependent changes of presynaptic neuromodulation via A1-adenosine receptors in rat hippocampal slices. Int J Dev Neurosci 15:739–747
Ekonomou A, Pagonopoulou O, Angelatou F (2000) Age-dependent changes in adenosine A1 receptor and uptake site binding in the mouse brain: an autoradiographic study. J Neurosci Res 60:257–265. https://doi.org/10.1002/(SICI)1097-4547(20000415)60:2<257::AID-JNR15>3.0.CO;2-U
Meerlo P, Roman V, Farkas E, Keijser JN, Nyakas C, Luiten PGM (2004) Ageing-related decline in adenosine A1 receptor binding in the rat brain: an autoradiographic study. J Neurosci Res 78:742–748. https://doi.org/10.1002/jnr.20314
Meyer PT, Elmenhorst D, Boy C, Winz O, Matusch A, Zilles K, Bauer A (2007) Effect of aging on cerebral A1 adenosine receptors: a [18F]CPFPX PET study in humans. Neurobiol Aging 28:1914–1924. https://doi.org/10.1016/j.neurobiolaging.2006.08.005
Mishina M, Kimura Y, Naganawa M, Ishii K, Oda K, Sakata M, Toyohara J, Kobayashi S, Katayama Y, Ishikawa K (2012) Differential effects of age on human striatal adenosine A1 and A2A receptors. Synapse 66:832–839. https://doi.org/10.1002/syn.21573
Corradetti R, Kiedrowski L, Nordström O, Pepeu G (1984) Disappearance of low affinity adenosine binding sites in aging rat cerebral cortex and hippocampus. Neurosci Lett 49:143–146
Virus RM, Baglajewsky T, Radulovacki M (1984) [3H]N6-(L-Phenylisopropyl) adenosine binding in brains from young and old rats. Neurobiol Aging 5:61–62
Hara H, Onodera H, Kato H, Kogure K (1992) Effects of aging on signal transmission and transduction systems in the gerbil brain: morphological and autoradiographic study. Neuroscience 46:475–488
Araki T, Kato H, Kanai Y, Kogure K (1993) Selective changes of neurotransmitter receptors in middle aged gerbil brain. Neurochem Int 23:541–548
Glass M, Faull RLM, Bullock JY, Jansen K, Mee EW, Walker EB, Synek BJL, Dragunow M (1996) Loss of A1 adenosine receptors in human temporal lobe epilepsy. Brain Res 710:56–68
Fredholm BB, Johansson B, Lindström K, Wahlström G (1998) Age-dependent changes in adenosine receptors are not modified by life-long intermittent alcohol administration. Brain Res 791:177–185
Ułas J, Brunner LC, Nguyen L, Cotman CW (1993) Reduced density of adenosine A1 receptors and preserved coupling of adenosine A1 receptors to G proteins in Alzheimer hippocampus: a quantitative autoradiographic study. Neuroscience 52:843–854
González-Maeso J, Torre I, Rodríguez-Puertas R, García-Sevilla JA, Guimon J, Meana JJ (2002) Effects of age, postmortem delay and storage time on receptor-mediated activation of G-proteins in human brain. Neuropsychopharmacology 26:468–478. https://doi.org/10.1016/S0893-133X(01)00342-6
Perry EK, Perry RH (1983) Human brain neurochemistry—some postmortem problems. Life Sci 33:1733–1743
Whitehouse PJ, Lynch D, Kuhar MJ (1984) Effects of postmortem delay and temperature on neurotransmitter receptor binding in a rat model of the human autopsy process. J Neurochem 43:553–559
Burke RE, Greenbaum D (1987) Effect of postmortem factors on muscarinic receptor subtypes in rat brain. J Neurochem 49:592–596
Kontur PJ, Al-Tikriti M, Innis RB, Roth RH (1994) Postmortem stability of monoamines, their metabolites, and receptor binding in rat brain regions. J Neurochem 62:282–290
Paul D, Gauthier CA, Minor LD, Gonzales GR (1997) The effects of postmortem delay on mu, delta and kappa opioid receptor subtypes in rat brain and guinea pig cerebellum evaluated by radioligand receptor binding. Life Sci 61:1993–1997
Ma JK, Zhu H, Piletz JE (2003) Effects of postmortem delay on imidazoline receptor-binding proteins in human and mouse brain. Ann N Y Acad Sci 1009:341–346
Funding
This work was supported by the Saitama Medical University Internal Grant 16B-1-11 to Y.O., the Spanish MINECO-FEDER (SAF 2009-08460, SAF 2013-48586-R, and SAF 2011-29918 to J.J.M., L.F.C., and J.A.G-S., respectively), and the Basque Government (IT-616-13). The authors thank the collaboration of the staff members of the Basque Institute of Legal Medicine.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of interest
Yuji Odagaki declares no conflict of interest.
Masakazu Kinoshita declares no conflict of interest.
Toshio Ota declares no conflict of interest.
J. Javier Meana declares no conflict of interest.
Luis F. Callado declares no conflict of interest.
Isao Matsuoka declares no conflict of interest.
Jesús A. García-Sevilla declares no conflict of interest.
Ethical approval
All animals received care according to institutional guidelines, and all procedures were according to the European Community Guidelines for the use of Experimental Animals (86/609/EEC) after approval by the Animal Committee of Saitama Medical University. All procedures regarding postmortem human brains were in accordance with protocols approved by the Human Studies Ethical Committee of each of the institutions involved.
Rights and permissions
About this article

Cite this article
Odagaki, Y., Kinoshita, M., Ota, T. et al. Functional coupling between adenosine A1 receptors and G-proteins in rat and postmortem human brain membranes determined with conventional guanosine-5′-O-(3-[35S]thio)triphosphate ([35S]GTPγS) binding or [35S]GTPγS/immunoprecipitation assay. Purinergic Signalling 14, 177–190 (2018). https://doi.org/10.1007/s11302-018-9603-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11302-018-9603-x