
Abstract
The mitochondrial phylogeography of some conifers shows evidence of introgression from sympatric congeners, with mitochondrial lineages not always reflecting species. This suggests that unique mitochondrial haplotypes previously reported in the ponderosa pines (Pinus subsection Ponderosae) from the USA might be more widespread in taxa not yet sampled. Recent nuclear and plastome phylogenies placed Pinus ponderosa paraphyletic in relation to Ponderosae in Mexico and Central America and confirmed that sympatric Pinus jeffreyi is more closely related to the California big-cone pines (Pinus subsection Sabinianae). We describe a broad survey of the repeated motifs in nad1 intron 2 of Ponderosae and Sabinianae, which revealed that most of the 27 mitochondrial haplotypes were not exclusive to a taxon but showed strong geographic patterns. In surprising contrast to nuclear and plastid phylogenies that resolve a monophyletic P. jeffreyi, unidirectional mitochondrial capture by P. jeffreyi (Sabinianae) from P. ponderosa was observed in all 28 samples of Jeffrey pine. Confirming the paraphyly of P. ponderosa sensu lato, mitochondrial haplotypes found mostly west and those found mostly east of the Great Basin each have more similarity to haplotypes found in Mexican taxa than they have to each other. Two distinctive haplotypes that were terminal nodes on the network were confirmed to be endemic to the Great Basin, USA, suggesting that they arose in place and have been maintained in isolation. Altogether, our results indicate a history of complex and intriguing mitochondrial relationships among the ponderosa pine species, especially between P. ponderosa and P. jeffreyi.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Phylogeography seeks to understand the historical factors (both phylogenetic and geographic) that affected the current diversity and geographical distribution of life on earth (Avise 2009). The understanding gained from these combined analyses of genetic relatedness and biogeographic patterns is crucial for making difficult conservation decisions and for predicting how our present species diversity and species distributions will be restructured in a rapidly changing climate (Nagel et al. 2017; Rehfeldt et al. 2014). Even temperate forests harbor enigmas worthy of further investigation. One of these enigmas is the ponderosa pines, despite being recognizable by even casual forest visitors as “ponderosa pines.” These pine forests often play a dominant role in western North American ecosystems, providing tremendous ecological and economic value (Richardson 1998). Their geographic range is enormous, and their distribution is complicated, encompassing vast continuous forests and uncounted stands of trees that vary in their degree of isolation (Little 1971). This problem of size and complexity is overlain with challenging boundaries (e.g., political between USA and Mexico; disciplinary between foresters and botanists). In retrospect, the sampling schemes for most of the hundreds of ponderosa pine experiments that have been published seem limited, despite some of them being huge undertakings (Conkle and Critchfield 1988; Potter et al. 2013; Smith 1977; Weidman 1939; Wells 1964; Willyard et al. 2017). In spite of this importance and an abundance of published research, the ponderosa pines are still regarded as a poorly understood species complex that lacks a unified phylogeographic framework.
The ponderosa pines are classified in Pinus subsection Ponderosae—section Trifoliae, subgenus Pinus (Pinaceae)—which has 18 commonly recognized taxa (Online Resource 1) that are distributed from southern Canada to Nicaragua. Each of the Ponderosae taxa grows in mixed conifer forests, sometimes intermingled with species of Pinus subsections Australes or Contortae (section Trifoliae). Subsection Sabinianae (section Trifoliae) is a sister clade to the Ponderosae based on nuclear and plastid phylogenies (Gernandt et al. 2009; Willyard et al. 2009, 2021). The Sabinianae includes the California big-cone pines, which are endemic to California, USA, and adjacent regions of Nevada and Oregon, USA, and Baja California, Mexico. Pinus jeffreyi is also in the Sabinianae clade, despite some vegetative similarities that led earlier workers to presume that it was closely related to P. ponderosa (Willyard et al. 2021). Three species of Sabinianae (P. coulteri, P. jeffreyi, and P. sabiniana) have some sympatry with Ponderosae taxa.
Pinus ponderosa in the USA has been treated as one species with three varieties (Haller and Vivrette 2011; Kral 1993) or the subdivisions were named as races or ecotypes (Callaham 2013a; Read 1980; Smith 1977; Weidman 1939; Wells 1964). Evidence from plastid haplotypes (Willyard et al. 2017), mitochondrial haplotypes (Potter et al. 2013), and nuclear and plastome phylogenies (Willyard et al. 2021) support the recognition of three separate species. Pinus ponderosa sensu stricto (s.s.) is distributed mostly west of the Great Basin, USA. On the east side of the Great Basin, USA, the geographic boundaries of P. brachyptera and P. scopulorum overlap. Importantly, P. ponderosa is not sister to the clade that represents P. scopulorum and P. brachyptera (Willyard et al. 2021). Rather than one species that tolerates a remarkably broad range of environmental conditions, these three taxa occupy climate niches that are fundamentally different: P. ponderosa sites receive predominantly winter precipitation while P. brachyptera and P. scopulorum sites mostly receive summer precipitation (Shinneman et al. 2016). Conflict among studies may be due to different sampling where two or more taxa grow intermingled, to incomplete lineage sorting, or to genomic incongruence due to some level of introgression (Willyard et al. 2017). For example, unrecognized sympatric or parapatric distribution may have led earlier workers to combine the ponderosa pines of the Colorado Plateau, which have predominantly 3-needled fascicles (P. brachyptera) with pines of adjacent areas, which have predominantly 2-needled fascicles (P. scopulorum) (Callaham 2013b; Conkle and Critchfield 1988). Similarly, merging P. ponderosa var. ponderosa with the westernmost ponderosa pines (P. ponderosa var. benthamiana) which tend to have longer leaves and cones may be misleading (Haller and Vivrette 2011). The tendency for botanists and foresters to limit their work to one or the other side of the political boundary between Mexico and USA may have contributed to this confusion because the populations in Mexico that Martínez (1948) called P. arizonica were probably additional populations of P. brachyptera (Rehfeldt 1999). Here, we adopted recent recommendations (Haller and Vivrette 2011; Willyard et al. 2021) to treat three ponderosa pine species in the USA as P. ponderosa (with varieties P. ponderosa var. benthamiana, P. ponderosa var. ponderosa, and P. ponderosa var. washoensis), P. brachyptera, and P. scopulorum rather than an unsupported treatment as P. ponderosa sensu lato (s.l.; Online Resource 1; Kral 1993). Similar uncertainty remains with the other taxa of Ponderosae that are native to Mexico and Central America, and we followed recent recommendations for their treatment as well (Online Resource 1;Gernandt et al. 2009; López-Reyes et al. 2015; Willyard et al. 2009, 2021).
Reticulate evolution, created by interspecific hybridization, has been proposed to play an important role in the ancestry of the ponderosa pines (Willyard et al. 2009). Artificial cross-pollinations conducted among 12 taxa of Ponderosae yielded fertile seeds, thus demonstrating their potential to hybridize (Conkle and Critchfield 1988; Critchfield 1966). Pinus jeffreyi was successfully crossed with two other Sabinianae (P. coulteri and P. torreyana), and P. jeffreyi (but not other Sabinianae) was successfully crossed with P. ponderosa s.l., but crosses between species of Ponderosae and Australes and between species of Ponderosae and Contortae all failed. Most of these taxa could not hybridize in nature because they are not sympatric, and the frequency of natural hybridization is likely to be very low (Online Resource 2).
FormalPara Mitochondrial DNAFor plant phylogeographic questions, mitochondrial and plastid data have an advantage over nuclear data in that these organellar genomes are high-copy (so are easily amplified) and that they have a shorter time to coalescence (due to their uniparental inheritance). Plants often have high-copy numbers of nuclear genes, and comparisons of orthologous nuclear genes are even more challenging in the very large nuclear genomes of Pinaceae (Neale et al. 2014). Challenges of assuring orthology in the nuclear genome make organellar data especially important in Pinaceae. Incomplete lineage sorting is also widespread in Pinus, owing in part to the combination of long generation time, large effective population sizes, and their huge nuclear genome (Syring et al. 2005; Willyard et al. 2009). Because plastids are predominantly paternally inherited in Pinaceae (Neale and Sederoff 1989) and wind-dispersed pollen can potentially move great distances (Williams 2010), maternally inherited mitochondria may better reflect more localized population structure in Pinaceae species because they track seed dispersal (Petit and Excoffier 2009). However, capture of resident mitochondria by arriving immigrants of many plant species is well documented and largely explained by maternal inheritance that results in less intraspecific gene flow and hence favors retention of the higher-frequency haplotypes from the resident population (Latta 2006; Rieseberg et al. 1996). The phenomenon of arriving migrants capturing mitochondria has been found in contact zones of Pinus banksiana with Pinus contorta var. latifolia (Godbout et al. 2012) and is widespread in Picea (Pinaceae) (Bouillé et al. 2011; Du et al. 2009; Gérardi et al. 2010; Ran et al. 2015).
Other processes have been discovered that may affect the reliability of mitochondrial markers for species identification. As mitochondrial introns were explored in many plant samples, evidence that unexpected gene transfers have occurred over evolutionary time was observed. While group II introns are the result of vertical descent since the endosymbiotic creation of organelles in ancestral Eukaryotes (Toor et al. 2001), rearrangements have disrupted both mitochondrial introns and mitochondrial gene order. For example, the fragmented group II introns of many plants require trans-splicing to regain their activity (Chaw et al. 2008; Malek et al. 1997) and more recent recombination in Pinus mitochondria due to heteroplasmy has been theorized to explain genetic patterns (Tsutsui et al. 2009; Wang and Ran 2014). Some plant lineages have lost parts of their mitochondrial introns entirely. For example, despite the nad1 intron 2 being widespread in seed plants, it is absent in non-Pinaceae conifers and is inferred to have been lost at least seven times in angiosperms (Gugerli et al. 2001). Horizontal transfer of mitochondrial genes appears to be uncommon, but huge phylogenetic leaps have been documented (Forgione et al. 2019; Wang et al. 2015; Won and Renner 2003). These transfers are rare occurrences and mitochondrial capture has been hard to detect in more closely related taxa. For example, although mitochondrial variation due to transient heteroplasmy was identified in a known zone of hybridization between two species of Picea (Pinaceae), the occurrence of recombination was rare (Jaramillo-Correa and Bousquet 2005).
The extremely low mitochondrial substitution rate in plants (Wolfe et al. 1987) and the structural complexity of their mitochondrial genomes (Jackman et al. 2020; Sullivan et al. 2020) has severely limited previous use of mitochondrial nucleotide sequences for species-level phylogeography. This challenge was overcome in studies of some Pinus species by using length variation in mitochondrial introns due to tandem repeats of motifs that were 6 to 32 bps long (minisatellites; Aizawa et al. 2012; Bastien et al. 2003; Burban and Petit 2003; Godbout et al. 2005, 2008; Jaramillo-Correa et al. 2013; Pyhäjärvi et al. 2008; Soranzo et al. 2000; Sperisen et al. 2001; Strauss et al. 1993). Length variation in the nad1 intron 2 (also named nad1 intron b/c) distinguished P. ponderosa from P. scopulorum at their contact zone near the Continental Divide (Johansen and Latta 2003; Latta and Mitton 1999; Mitton et al. 2000). An enormous range-wide study of P. ponderosa s.l. used haplotype variation in the nad1 intron 2 to create a much clearer phylogeographic pattern than was previously available (Potter et al. 2013).
We wondered how these P. ponderosa s.l. mitochondrial haplotypes would fit into a broader phylogenetic context that includes the Sabinianae and the other Ponderosae taxa. In particular, we investigated whether two related haplotypes found only in California, USA (H9 and H10), might have been introgressed from sympatric Sabinianae species (where the frequency of hybridization between Ponderosae and Sabinianae is not well known), and whether three divergent and geographically isolated haplotypes found in the Great Basin, USA (H2, H4, and H7), might belong to, or be related to, haplotypes from other Ponderosae taxa from Mexico and Central America (Potter et al. 2013). Although some challenges remain in species delimitation within this species complex, phylogenetic analyses of nuclear and plastid DNA sequences (Willyard et al. 2021) provided a useful perspective to frame mitochondrial relationships. We applied a published technique (Potter et al. 2013) by genotyping samples of Sabinianae species and samples of all the other Ponderosae taxa for variation in intron 2 of nad1.
Materials and methods
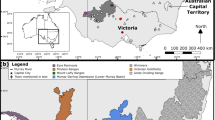
We included 206 samples (one to eight individuals from each of 130 locations), representing each of the generally recognized taxa in subsection Ponderosae (except P. yecorensis) and all four of the species in subsection Sabinianae. We also added 11 nucleotide sequences from GenBank (Fig. 1; Online Resources 1, 3). Each sample was identified based on morphology and geographic location using published taxonomic treatments supplemented with molecular evidence (Gernandt et al. 2009; López-Reyes et al. 2015; Martínez 1948; Willyard et al. 2021; Willyard et al. 2017). We also attempted to align the nucleotide sequences of nad1 intron 2 that were available in GenBank on 14 January 2019 for any species of subsections Australes, Contortae, Ponderosae, or Sabinianae.

Geographic origin of 130 collection locations for 206 individuals representing 22 taxa that were sampled in this study in USA, Mexico, and Guatemala (Online Resource 3) using Albers projection. Inset is nuclear species phylogeny, modified from (Willyard et al. 2021) with taxon symbols that match Fig. 4. USA states: AZ, Arizona, CA, California; CO, Colorado; ID, Idaho; MT, Montana; NE; Nebraska; NM, New Mexico; NV, Nevada; OR, Oregon; SD, South Dakota; TX, Texas; UT, Utah; WA, Washington; WY, Wyoming. Mexican states: AGS, Aguascalientes; BC, Baja California; CDMX, Ciudad de México; CHH, Chihuahua; CHS, Chiapas; COA, Coahuila; COL, Colima; DUR, Durango; EM, Estado de México; GRO, Guerrero; GTO, Guanajuato; HDG, Hidalgo; JAL, Jalisco; MIC, Michoacán; MOR, Morelos; NAY, Nayarit; NL, Nuevo León; OAX, Oaxaca; PUE, Puebla; QRO, Querétaro; SLP, San Luis Potosí; SNL, Sinaloa; SON, Sonora; TAM, Tamaulipas; TLA, Tlaxcala; VER, Veracruz; ZAC, Zacatecas
Leaf tissue was either dried on silica upon collection in the field or taken from herbarium specimens. Genomic DNA was isolated as previously described (López-Reyes et al. 2015; Wofford et al. 2013). The mitochondrial minisatellite region of the second intron in the nad1 gene was PCR-amplified using primers (nad1-in2_F2 and nad1-in2_R2) and conditions previously described (Potter et al. 2013). Because nucleotide sequencing revealed many new haplotypes that would not have been distinguished based on fragment length, we analyzed all samples for this experiment based on nucleotide alignments. PCR products were either purified using QIAquick PCR Purification kit (QIAGEN Germantown, Maryland, USA) and sent to the University of Missouri DNA Core Facility (Columbia, Missouri, USA) for sequencing, or sent to University of Washington High Throughput Genomics Center (Seattle, Washington, USA) for purification and sequencing. At both facilities, we used the PCR amplification primers for sequencing in each direction. The forward and reverse reads were aligned manually using BioEdit (vers. 7.2.5; Hall 1999) to create a consensus sequence for each sample. These consensus sequences were aligned manually with the aid of a template that spanned the largest number of repeats for each motif by inserting motif numbers 1 through 8 into alignment columns (Online Resources 4, 5). Based on the alignment of repeating motifs, each consensus sequence was assigned to one of 10 published haplotypes (Potter et al. 2013) or to one of fourteen newly discovered haplotypes (Fig. 2).

A Nucleotide sequences of Motifs 1 through 8 corresponding to Motifs A through H (Potter et al. 2013). B Motif repeat patterns for 29 mitochondrial haplotypes detected using nucleotide sequence alignment, including two different haplotypes discovered in GenBank (Hj, Hk). Motif x denotes any nucleotide sequence that does not match one of the eight basic motifs. Colors are used in Figs. 3 and 4
We created a binary matrix for the presence/absence of each motif repeat using our alignment for 29 haplotypes: 25 that we observed in our samples, two found in GenBank sequences, and two published for P. coulteri (Hc1 and Hc3) that were not observed in any of our samples (Online Resource 6). We used this binary matrix to calculate distances among haplotypes with the hammingdists function in “cultevo” (vers. 1.0.2 Hamming 1950; Stadler 2018; R Core Team 2021), and Popart (Leigh and Bryant 2015) to create a Median Joining Network with the default option of Epsilon = 0 (Bandelt et al. 1999). The network was redrawn in Inkscape (vers. 0.91; https://inkscape.org) with node sizes roughly proportional to the number of samples and color-coded to match the maps. DIVA-GIS (vers. 7.5; http://www.diva-gis.org) was used to create maps to show the geographic location of samples with each haplotype in our study and haplotypes previously reported.
For the eight locations where we sampled five or more individuals, we calculated haplotype diversity using GENALEX (vers. 6.51b2; Peakall and Smouse 2006). For comparison among taxa, we also calculated haplotype diversity among all samples for each taxon. Among-sample dissimilarity matrices were compared to test whether mitochondrial haplotypes were correlated with their taxonomic assignment and/or with their geographic distance using Mantel tests and partial Mantel tests that account for a third factor. For mitochondrial haplotypes, we used the distances calculated for the network. For taxonomic distance, we used the nuclear phylogenetic distance among taxa on a species tree (Fig. 1 inset; Willyard et al. 2021). This calculation used the patristic option (sum of the branch lengths) in the distTips function of “adephylo” (vers. 1.1–11; Jombart and Dray 2008; R Core Team 2021). For geographic distances, we used the geodist function with the paired distances option and a geodesic measure in “geodist” (vers. 0.0.7; Padgham and Sumner 2020; R Core Team 2021). For each of six pairwise comparisons (Table 1), we reported significance for the null hypothesis that r = 0 using 100,000 permutations of the mantel function, which accounts for a third factor using “ecodist” (vers. 2.0.7; Goslee and Urban 2007; R Core Team 2021).
New nucleotide sequences were submitted to GenBank (Online resource 3). One specimen voucher per collection location was deposited at the Critchfield Memorial Herbarium (IFGP), Oregon State University (OSC), or Herbario Nacional de México (MEXU).
Results
Our nucleotide alignment of the second intron in the nad1 gene was 1855 bp long; individual sequences ranged from 333 to 792 bps (Online Resource 5). All repeating motifs could be identified, confirming the 10 mitochondrial haplotypes published for P. ponderosa s.l. (H1–H10), 11 new haplotypes in Ponderosae (H11–H21), and three new haplotypes in Sabinianae (Hs1, Ht1, and Ht2; Fig. 2; Online Resource 4). From GenBank, we identified two sequences from P. maximinoi (JN225486 and KM244262) that carried new haplotypes (Hj and Hk). Five GenBank sequences were additional occurrences of H3 or H5, and one documented H3 in P. durangensis, a species that was not included in our own sampling (Online Resource 3). Because these 11 GenBank accessions did not include geographic source information, they were used for haplotype network relationships but not for geographic placement. None of the 18 GenBank nucleotide sequences for nad1 intron 2 that were available in GenBank for species in closely related subsections Australes and Contortae contained any of these motifs.
Most of the samples that carried new haplotypes were species other than P. ponderosa, although a single individual of P. ponderosa var. benthamiana carried new haplotype H11. The three new haplotypes identified in Sabinianae samples—Hs1 (in P. sabiniana), Ht1 and Ht2 (in P. torreyana)—were very divergent from all other haplotypes. We found one P. coulteri individual with the nucleotide sequence for haplotype Hc2 (Potter et al. 2013), but we did not observe the Hc1 or Hc3 haplotypes in our samples.
Haplotypes H9, H10, and our new H11 differed substantially from all other haplotypes by having Motif 8 in place of Motif 2 in some, but not all, repeat regions (Fig. 2). The Ponderosae haplotypes varied twofold in their number of motif repeats, ranging from 10 (H10) to 20 motif repeats (H15), whereas Sabinianae haplotypes varied even more (using our method of coding any dissimilar contiguous indel as one motif), ranging from 7 to 22 motif repeats (Fig. 2). The binary motif presence/absence matrix (Online Resource 6) yielded a Median Joining Network of 29 haplotypes that agreed with the published relationship for H7-H6-H3-H1-H5-H8, but the additional haplotypes from other species dramatically altered the placement of H2, H4, H9, and H10 (Fig. 3). The new haplotypes that we observed in Sabinianae (Hs1, Ht1, and Ht2) were distant from previously reported Hc1, Hc2, and Hc3 from P. coulteri, complicating the question of where this network of Ponderosae haplotypes might connect to Sabinianae. All Sabinianae-specific haplotypes were distant to all Ponderosae haplotypes, with at least four (Hc1 from H13) and up to 11 (Ht1 from H10) differences with the most similar Ponderosae haplotype (Fig. 3).

Median Joining Network using a binary matrix for the presence/absence of motifs in nucleotide alignments of the second intron in the nad1 mitochondrial gene (Fig. 2; Online Resources 4, 6). Connections with more than one mutational step are given with numbers. The size of the nodes is roughly proportional to the number of known samples (Online Resource 7 plus previously reported for P. ponderosa s.l. (Potter et al. 2013)). Haplotypes Hc1, Hc2, Hc3, Hs1, Ht1, and Ht2 have only been observed in P. coulteri, P. sabiniana, and P. torreyana. Haplotypes shown in the shaded area were generally found in the western part of the study area; haplotypes not shaded were generally found in the eastern part of the geographic range. Colors match Fig. 4
All eight individuals of P. douglasiana from one location in Jalisco were fixed for haplotype H13, all eight individuals of P. maximinoi from one location in Jalisco were fixed for haplotype H3, but other locations where we had five or more samples yielded a high haplotype diversity (Table 2). The mean haplotype diversity within each taxon was 0.39, with P. jeffreyi (0.80) and P. pseudostrobus var. pseudostrobus (0.72) containing the most diversity (Table 3). Combining the five taxa sometimes included in P. ponderosa s.l. would yield an even higher haplotype diversity of 0.83 (not shown). The only significant Mantel tests were the correlation of taxonomic with geographic distance (Table 1). All other comparisons, including mitochondrial haplotype with taxonomic, were not significant. Most haplotypes were present in more than one taxon (Online Resource 7).
Results by geographic region
Except for H2 (Fig. 4(b); discussed below) and the haplotypes where few samples were observed (Fig. 4(h)), the geographic distribution of each individual haplotype formed a cohesive pattern (Fig. 4(a, c, d, e, f, g)). Within the USA, there was a general geographic pattern in haplotype similarities that corresponded to P. ponderosa to the west and P. scopulorum plus P. brachyptera to the east (Fig. 1). Haplotypes in the area shown shaded on the network (H1, H5, H8, H9, H10, H11, H12, H13, H14, H21, Hc1, Hc2, Hc3, Hs1, Ht1, and Ht2) were all predominantly observed to the west of (or on the western edge of) the Great Basin. Within the USA, haplotypes H3, H4, H6, and H7 were generally found in (or to the east of) the Great Basin, although the geographically widespread H3 was also observed in five Sabinianae individuals west of the Great Basin (two P. coulteri and three P. jeffreyi) (Fig. 4(c)). In Mexico, H12, H13, H14, H20, and H21 were found only in the Sierra Madre del Sur.


The geographic locations in the United States, Mexico, and Guatemala carrying the 27 mitochondrial haplotypes observed in this study and previously reported as P. ponderosa s.l. (Potter et al. 2013) using Albers projection. Locations where more than one haplotype was observed have a symbol for each haplotype. (a) H1, H5, and H8; (b) H2; (c) H3 and H6; (d) H4; (e) H7; (f) H9, H10, H11, Hc2, Hs1, Ht1, and Ht2; (g) H12, H14, and H21; (h) H13, H15, H16, H17, H18, H19, and H20; Haplotypes Hj, Hk, Hc1, and Hc3 are not included on the maps because geographic source locations are not known. Taxon symbols match Fig. 1; haplotype colors match Fig. 3
Results by taxon
For the taxa where we sampled more than one individual, only four had a single haplotype. Pinus engelmannii and P. montezumae had only the plesiomorphic H3; P. arizonica var. stormiae had only H2; and P. ponderosa var. washoensis had only H1 (Table 3; Online Resource 3). Every other taxon had multiple haplotypes. Some haplotypes were only found in one taxon: H4 and H7 (each observed in a limited portion of the geographic range of P. brachyptera), H13 (only in P. douglasiana), H14 (only in P. maximinoi), H15 (only in P. pseudostrobus var. apulcensis), and H18 and H20 (each only in P. pseudostrobus var. pseudostrobus). In contrast, H3 was found in 15 different taxa (Online Resource 7).
None of the more divergent haplotypes that were discovered in Sabinianae (Hc1, Hc2, Hc3, Hs1, Ht1, and Ht2; Fig. 3) were found in a subsection Ponderosae taxon but two species of these California big-cone pines carried Ponderosae haplotypes. Of four individuals of P. coulteri that were sampled from one location, one carried H1, two carried H3, and one had a haplotype previously reported for this species (Hc2; Online Resource 3). Our observation of H1 and H3 at this location in southern California, USA, represented a large disjunct from any other observation of these haplotypes (Fig. 4(a, c)).
None of our 28 P. jeffreyi individuals carried any of the expected Sabinianae-specific haplotypes or other unique haplotypes; rather, P. jeffreyi individuals had one of six different Ponderosae haplotypes (Online Resource 3). All but one location where H1, H2, H5, H10, or H11 were found in a P. jeffreyi individual was within the known geographic range of that haplotype for a Ponderosae taxon (Fig. 4(a, b, f)). The only exception was the plesiomorphic H3 that was found in three P. jeffreyi individuals (Fig. 4(c)). The Baja California, Mexico, occurrence of H3 in P. jeffreyi could also be compatible with regional sharing because a P. coulteri individual from Fry Creek Campground, California, USA, also had H3. However, no other H3 occurrences were found near the P. jeffreyi in western Nevada (Fig. 4(c)). The geographic location was not published for GenBank JN225485 which documents a potential third population where a P. jeffreyi individual carried H3.
Discussion
Our main findings, each discussed further below, were a confirmation of the separation of P. ponderosa s.s. mitochondrial haplotype similarities west of the Great Basin, USA, from P. arizonica var. stormiae, P. brachyptera, and P. scopulorum mostly east of the Great Basin that was evident in nuclear and plastid phylogenies (Fig. 1 inset; Figs. 3, 4; Potter et al. 2013; Willyard et al. 2021). We confirmed that H3 is plesiomorphic within subsection Ponderosae based on the large proportion of samples that carry H3, its central position on the network, its large geographic distribution, and its presence in many taxa. Some new haplotypes identified in Mexican taxa (H12, H13, H14, H18, and H21) were most similar to P. ponderosa s.s. haplotype H5, while most other new haplotypes were more similar to the ancestral H3 (Fig. 3). There was extensive sharing of haplotypes among species, notably by the distantly related but sympatric P. jeffreyi with P. ponderosa. We identified several endemic haplotypes. Except for two sites with only one haplotype, we found haplotype diversity at locations where we sampled more than 5 individuals to be at least twofold higher than the mean population diversity of 0.08 observed for Pinus ponderosa s.l. (Table 2; Potter et al. 2013).
Haplotype relationships
The new haplotypes created some reticulations that prevent the network from being resolved without an outgroup (Huson and Scornavacca 2011). For example, H5 (observed only in California and Oregon, USA) is one motif distant from H18 and from H13 (both found only in central and southern Mexico) and two motifs distant from H1 and from H8 (both partially sympatric with H5; Fig. 4(a)). There were as many as 16 motif differences among Ponderosae-type haplotypes. Because the mutational rate of these repeats must be relatively rapid to create the high level of variation, it is also likely that at least a few of our samples had a haplotype that arose via convergent evolution. Nonetheless, the mostly cohesive geographic patterns (Fig. 4) strongly infer a tendency for long-term stability of this repeat region.
The six haplotypes from three Sabinianae species (Hc1, Hc2, Hc3, Hs1, Ht1, and Ht2) contained a partial set of the Ponderosae motifs but intervening genomic regions were too divergent to align with the Ponderosae nucleotide sequences (Online Resource 4). The Sabinianae haplotypes were also very dissimilar to each other. In contrast with nuclear and plastome phylogenies that resolved a monophyletic four-taxon Sabinianae (Fig. 1 inset; Willyard et al. 2021), our mitochondrial haplotype network connected the P. coulteri haplotypes (Hc1, Hc2, and Hc3) to H13, but the other three Sabinianae haplotypes (Hs1, Ht1, and Ht2) were connected to H10 (Fig. 3). Because of these conflicting placements, we did not use any of the Sabinianae to root the Ponderosae haplotype network. The putative similarity of the H9, H10, and H11 haplotypes to Sabinianae haplotypes Hs1, Ht1, and Ht2 lent some support to the possibility that the mitochondrial ancestor of H9, H10, and H11 was carried forward from an ancestor shared with the Sabinianae in California, USA. For this explanation to hold, Motif 8 had to have been lost in the six extant Sabinianae haplotypes that we observed. A simpler interpretation is that Motif 8 was gained after the divergence of this mitochondrial haplotype from the shared Sabinianae ancestor.
These repeating motifs appear to have originated in section Trifoliae. There were five exact matches to Motif 2 (32 bp long; Fig. 2) in the complete mitochondrial nucleotide sequence of P. taeda (subsection Australes; NC_039746), but none were annotated as being in a nad1 intron and none were tandemly repeated. None of the nad1 intron 2 sequences of Australes or Contortae from GenBank contain any of the Ponderosae-style motifs. One accession of the more distantly related P. pinaster (subgenus Pinus, section Pinus) had one tandem repeat of Motif 1 (AJ509805) but lacked the other Ponderosae-style motifs. Because the subsection Sabinianae haplotypes have a partial set of these repeat motifs (Online resource 4), these motifs are likely to have begun being duplicated after the divergence of the Australes and Contortae or, alternatively, these motif repeats were lost in the Australes and Contortae. The expansion of this complex repeat pattern took place mostly within Ponderosae, presumably with additions and deletions of repeated motifs operating in a manner analogous to the stepwise mutations of simple sequence repeats (Ellegren 2004). The primers that we used for PCR amplification did not acquire nucleotides from flanking exonic regions that would allow us to determine if the Ponderosae nad1 intron 2 repeats had increased the substitution rate of nearby regions as some repeats have been shown to do (Jaramillo-Correa et al. 2013), but this intron is certainly variable in the Ponderosae.
Haplotypes shared among taxa
Our results made it clear that mitochondrial haplotype relationships in the Ponderosae cross species boundaries, as observed in maternally inherited oak plastomes (Petit et al. 2002). Mitochondrial and nuclear taxonomic dissimilarity matrices among our samples were not correlated (Table 1). What is more, the Mantel r values were nearly an order of magnitude lower than reported from a wide survey of symbionts with their hosts, even when using a partial Mantel test that took geographic distance into account (Table 1; Mazel 2018). We observed extensive interspecific sharing of haplotypes, some between sympatric samples and some across huge geographic disjunctions (Fig. 4; Online Resource 7). Some of these shared haplotypes could be due to incomplete lineage sorting of ancestral polymorphism, to convergent evolution, or to unclearly delimited taxa (López-Reyes et al. 2015). Although our sampling of Mexican Ponderosae taxa (supplemented with GenBank nucleotide sequences) included all of the commonly recognized taxa except P. yecorensis, our low sample sizes for some taxa were unlikely to clearly show geographic patterns. Nonetheless, some of the geographic clustering of shared haplotypes was between species that can be clearly diagnosed morphologically and are mutually monophyletic on nuclear and plastome phylogenies (e.g., P. ponderosa and P. jeffreyi; see below), implying the capture of mitochondria from other species. Mitochondrial DNA capture has been reported much more broadly than at the inter-subsection level that our results suggested, including in Pinaceae. For example, mitochondrial transfer was inferred between sympatric species of Pinus (Godbout et al. 2012; Wang and Wang 2014) and widely among species of Picea (Bouillé et al. 2011; Du et al. 2009; Ran et al. 2015). The ability of many Ponderosae species to be artificially crossed with each other (Conkle and Critchfield 1988; Critchfield 1966), along with reports that P. jeffreyi can hybridize naturally at low frequency with P. coulteri and with P. ponderosa (Haller 1962; Mirov 1967; Smith 1977), means that mitochondrial transfer via hybridization is possible. We also note that an adaptive value has been suggested for mitochondrial genes (Bock et al. 2014; Houliston and Olson 2006) and evidence for a selective sweep of mitochondrial haplotypes across species was inferred in Salix (Salicaceae; Percy et al. 2014), suggesting that the interspecific transfer and retention that we observed might indicate that adaptive alleles are present in these captured mitochondrial lineages as well. More informative mitochondrial nucleotide sequences are theoretically possible from high-throughput sequencing (Straub et al. 2012), but a better understanding of the complexity of mitochondrial genomes (Jackman et al. 2020) will be needed to distinguish convergent evolution in the nad1 intron 2 repeats from shared mitochondrial ancestry and to detect adaptive effects for mitochondrial nucleotide sequences in the Ponderosae. As in any phylogeographic study of present-day genotypes in wild populations, there remain important and unresolved questions of what mechanism — interspecific hybridization or some form of vector-mediated process (Won and Renner 2003) — facilitated the transfers as well as whether their maintenance in a population is due to chance, to demographic factors, or to adaptive advantage.
Despite the interspecific sharing, our mitochondrial haplotype results provide additional evidence for separate taxa within P. ponderosa s.l. that were supported by nuclear and plastome phylogenies (Willyard et al. 2021). Haplotype H5 was predominantly observed in P. ponderosa var. benthamiana; H1 and H8 were strongly associated with P. ponderosa var. ponderosa and P. ponderosa var. washoensis (Online Resource 7). Most samples of P. brachyptera had the plesiomorphic H3, while most H6 were found in P. scopulorum (Fig. 4; Online Resource 7). However, some mitochondrial haplotypes conflict with morphological assignments. At 12 locations that were not observed to be morphologically mixed, we found multiple haplotypes represented among the sampled individuals. What’s more, three of these locations with multiple haplotypes had haplotypes that were genetically distant to each other—P. brachyptera from Spring Mountains, Nevada, USA, with H2, H3, and H4; P. coulteri from Fry Creek Campground, California, USA with H1, H3, and Hc2; and P. jeffreyi at Palomar Mountain, California, USA with H1, H2, and H10 (Fig. 3; Online Resource 3). Because the unexpected haplotypes found at these three locations were also present in sympatric or parapatric taxa, admixture is a better explanation for these intermingled haplotypes than convergent evolution.
Because plastids have greater intraspecific gene flow than mitochondria in pines, plastid markers would generally be expected to be better at delimiting species (Petit et al. 2005; Petit and Excoffier 2009; Rieseberg et al. 1996). In the Ponderosae, a phylogeny using nearly complete plastome sequences was largely congruent with a phylogeny based on hundreds of nuclear gene trees from high-throughput sequencing (Willyard et al. 2021), but with several interesting conflicts that are worth comparing with this mitochondrial haplotype evidence. For example, one plastome clade had five P. ponderosa var. benthamiana samples from the western Sierra Nevada, USA, that resolved with their conspecifics on the nuclear tree, but the clade was sister to the Sabinianae using plastome data, suggesting an ancient plastid capture (labeled P1 in their Fig. 1; Willyard et al. 2021). We found mitochondrial haplotypes H2, H3, H5, H9, and H10 in these same populations, suggesting that introgression in this geographic region included both organelles. Another clade with four P. ponderosa var. ponderosa from Idaho, USA, resolved sister to P. scopulorum using nuclear genes despite their placement as expected on the plastome phylogeny (labeled N4 in their Fig. 1; Willyard et al. 2021), suggesting interspecific nuclear introgression. We found mitochondrial haplotypes H1 and H8 in these populations, which is congruent with their morphological and plastome resolution as P. ponderosa var. ponderosa. This suggests introgression might be limited to the nuclear genome in this geographic region. Denser sampling and data from the same individuals for all three genomes will be needed to test alternative explanations of admixture and intermingled individuals that have migrated into sympatry, but our findings are in agreement with previous reports of asymmetric introgression of nuclear, plastid, and mitochondrial genomes in Pinaceae contact zones (Godbout et al. 2012).
Phylogeographic patterns
Glacial refugia have been inferred for P. ponderosa in California, USA, along the coastal ranges; in lower elevations of the Sierra Nevada, Cascade Range, Siskiyou Mountains, and Wasatch Range; in the Central Valley; and in the Klamath-Siskiyou region. For a combination of P. scopulorum and P. brachyptera, refugia have been inferred in southern Arizona and New Mexico, USA; the Mogollon Plateau (Arizona, USA); the Rio Grande basin, and the Sierra Madre Occidental of Mexico. These patterns have incorporated fossil evidence (Norris et al. 2016), niche modeling (Shinneman et al. 2016), intraspecific mitochondrial haplotypes (Potter et al. 2013), and nuclear SSRs (Potter et al. 2015). The interspecific mitochondrial haplotype distributions presented here (Fig. 4) and our broader perspective on the similarity of these haplotypes to each other (Fig. 3) generally support those inferred refugia. Our sampling of taxa in Mexico and Central America was too sparse to extend those refugium projections in any substantial way, but we were able to confirm that H3 remains widely distributed in other Ponderosae taxa. Haplotype H6, which overlaps geographically with H3 in the Rocky Mountains and Great Plains, is generally found north and east of H3 but was also found sporadically in the Sierra Madre Occidental (Fig. 4(c)). We confirmed that mitochondrial haplotype H4 is endemic to a very limited geographic region in the Great Basin, USA (Fig. 4(d)). Because our GenBank search did not reveal any other Sabinianae-style haplotypes, we can infer that the mitochondrial lineage in the Sabinianae clade (with the apparent exception of captured mitochondria in P. jeffreyi; see below) is autochthonous to California, USA, and adjacent Baja California, Mexico, where those species are now endemic (Fig. 4(f)).
Our mitochondrial haplotype phylogeography shows several zones where taxa may have migrated into relatively recent contact, and not all of these contact zones agree with published hypotheses (Callaham 2013b; Conkle and Critchfield 1988; Latta and Mitton 1999; Read 1980; Wells 1964). As discussed above, we found both H1 and H8 (4 motifs distant from each other; Fig. 3) in Idaho, USA, in a region where nuclear, but not plastome, introgression has been reported (Willyard et al. 2021). A contact zone between H1 and H5 is evident in the region where the Klamath, Cascade, and Sierra Nevada mountain ranges meet in northern California and southern Oregon, USA (Fig. 4(a)). In the contiguous ponderosa pine forest that spans these ecoregions (Buck et al. 1970; Kitzmiller 2005), H5 predominates in the Klamath Range, while H1 (two motifs distant) is mostly found in the Cascade Range in California (Fig. 4(a)). However, we found two locations that had both H1 and H5 (Fig. 4(a); Online Resource 3). A contact zone between H2 and H4 is apparent in southern California, where P. ponderosa var. benthamiana may have migrated into secondary contact with populations carrying H2, perhaps in a Pleistocene refugium shared with Mojave Desert populations of P. brachyptera (Fig. 4(b)). The Mojave Desert populations themselves appear to be complicated, with distantly related haplotypes H2 and H4 found in the same populations (Fig. 4(b, d); Online Resource 3). There is also intermingling of H3 and H6 in Colorado, Wyoming, and northern New Mexico, USA (Fig. 4(c)). Researchers using different types of data have approximated several conflicting boundaries between P. scopulorum and P. brachyptera (Conkle and Critchfield 1988; Read 1980; Smith 1977; Wells 1964). Mitochondrial haplotype phylogeography suggests that populations carrying H3 and populations carrying H6 have migrated into contact and that there is a broad contact zone in the central Rocky Mountains (Fig. 4(c)). Contact zones have certainly moved over time, adding to their complexity (Godbout et al. 2012). As more details emerge about these and other secondary contact zones, a useful working hypothesis for future research would be that geographically proximal stands or individuals within the same stand of ponderosa pine trees may have different ancestry.
California taxa are related to Mexican taxa
The shaded area on the network highlights similarities between haplotypes H1, H5, and H8 plus the Sabinianae haplotypes (found only in central California, USA, and to the north) with H13, with H18, and with the H21/H12/H14 cluster (found only in central Mexico and to the south; Figs. 3 and 4). Although the western haplotype H1 is one motif distant from the plesiomorphic H3, the western haplotype H5 is also only one motif distant to H13 and to H18 (Fig. 3). Previous speculations for a connection between the pines of these two disjunct geographic regions were based on several different types of evidence. A plastid haplotype network placed P. maximinoi from Honduras with the Sabinianae species, and P. coulteri was more similar to species from the northern Sierra Madre, Mexico, than other Sabinianae (Willyard et al. 2009). Axelrod speculated that a Miocene fossil from western Nevada, P. truckensis Axelrod, was similar to both the Sabinianae and P. pseudostrobus based on its large, asymmetrical, ovulate cone (Axelrod 1986). Among the ponderosa pines of the USA, heptane is found only in the monoterpenes of the Sabinianae—as a major component in P. jeffreyi and as a minor component in P. coulteri, P. sabiniana, and P. torreyana (Mirov 1953). But Mirov also reported heptane in P. pseudostrobus var. apulcensis (which he considered P. oaxacana Mirov) and in some other subdivisions of the P. pseudostrobus species complex (Mirov 1953). It is noteworthy that some P. pseudostrobus trees in Oaxaca have, as a variable character, “strongly hooked apophyses” on their ovulate cone scales that have reminded some of the Sabinianae (Mirov 1958). Whether or not an ancestral relationship between these western populations (P. ponderosa plus subsection Sabinianae) with some Mexican taxa is confirmed, these mitochondrial haplotypes agree with nuclear and plastid phylogenies that P. ponderosa var. ponderosa shares a more recent ancestry with other Ponderosae taxa than with P. brachyptera and P. scopulorum.
Jeffrey pine
Pinus jeffreyi has been considered an enigmatic species that was presumed to be closely related to P. ponderosa because it superficially resembles this partially sympatric taxon in some vegetative morphological characters (Conkle and Critchfield 1988). However, a monophyletic four-taxon clade (subsection Sabinianae) that includes P. jeffreyi is now strongly supported by nuclear and plastid phylogenies, and P. jeffreyi shares synapomorphies for seed size and seed wing proportions with P. coulteri, P. sabiniana, and P. torreyana (Willyard et al. 2021). Wind dispersal of P. ponderosa seeds is effective for regeneration, although this species can also benefit from occasional long-distance dispersal of seeds by birds (Lesser et al. 2013; Parchman and Benkman 2007). In contrast, the Sabinianae species (including P. jeffreyi) rely on rodents or jays who gather and hoard seeds (Briggs et al. 2009; Vander Wall 1993; Vander Wall et al. 2012; Willyard et al. 2021). Our mitochondrial haplotype results for P. jeffreyi—all of our 28 samples carrying an apparently Ponderosae-style haplotype—were unexpected and are apparently an extreme example of unidirectional mitochondrial capture between phylogenetic clades. Because the geographic locations where we observed P. jeffreyi individuals with H1, H2, and H5 correspond to sympatric or parapatric Ponderosae with those haplotypes (Fig. 4(a, b, f)), interspecific mitochondrial transfer rather than retention of ancestral polymorphism is the simplest explanation for this pattern. What’s more, in order to have all 28 P. jeffreyi samples in our study carry a mitochondrial haplotype of Ponderosae species while none of our 52 Ponderosae samples that are sympatric with P. jeffreyi carried a Sabinianae mitochondrial haplotype, there must have been an advantage or an historical circumstance that favored repeated unidirectional transfer and retention of mitochondria from Ponderosae into P. jeffreyi. We know that Jeffrey pine was able to serve as either the seed parent or the pollen parent in successful test crosses (Critchfield 1966), yet our results would require a Ponderosae seed parent as an ancestor of each of our samples. The mechanism generally proposed for unidirectional introgression of mitochondria is that when taxa migrate into secondary contact, the fewer arriving migrants are more likely to acquire the abundant local mitochondrial lineage (Rieseberg et al. 1996). Thus, the simplest explanation is that P. jeffreyi more recently migrated into contact with a number of different already-established Ponderosae populations (Fig. 4(a, b, f)), perhaps acquiring advantageous mitochondrial adaptations that allowed it to succeed in the higher-elevation or nutrient-poor soils where it now predominates (Furnier and Adams 1986). An alternative is that P. jeffreyi mitochondrial diversity (and the H9/H10/H11 haplotype group that contains Motif 8) is plesiomorphic. Even if it could be demonstrated that the seven P. jeffreyi individuals with haplotypes containing Motif 8 (H9, H10, and H11) retained those haplotypes since their divergence in the ancestor of the Sabinianae, the direction of transfer would still be unbalanced. There would remain 21 samples of P. jeffreyi with four different haplotypes that clearly belong to Ponderosae and there would be 15 Ponderosae individuals with H9, H10, or H11 (Online Resource 7).
The evolution of morphological features that P. jeffreyi apparently shares with P. ponderosa vs. those that it shares with its common ancestors in Sabinianae would have different histories under the alternatives of migration-and-capture vs. mitochondrial plesiomorphy. Coevolution with different animals and different fire regimes may have furthered the “big-cone” features (e.g., heavier cones with larger out curved prickles) seen in the other Sabinianae (Haller 1986; Johnson et al. 2003; Schwilk and Keeley 2006) while the convergent evolution of Jeffrey pine vegetative and reproductive features to superficially resemble P. ponderosa may have been affected by the different animals that compete for seed in these habitats (Siepielski and Benkman 2010). Alternatively, P. jeffreyi retained some plesiomorphic mitochondrial haplotypes as well as morphological characters it shared with the ancestors of Ponderosae and Sabinianae, while P. coulteri, P. sabiniana, and P. torreyana developed divergent mitochondrial haplotypes, and other characters that are useful for interactions with different animals in frequent-fire foothills or islands.
We estimated the likelihood that the Ponderosae-style mitochondrial haplotypes observed in P. jeffreyi might have been retained due to incomplete lineage sorting. The data we used here do not support a direct calculation of effective population size, but a rough estimate of the time for coalescence can be made using the range of 4 to 28 million years that was inferred for nuclear genes for these taxa (Willyard et al. 2009). Given the expected fourfold faster coalescence for organelle markers (Birky et al. 1983), mitochondrial haplotypes should be expected to display monophyly on the order of 1 to 7 million years. The earliest known fossils attributed to Sabinianae were dated to the Miocene in the western Mojave Desert (Axelrod 1986). Based on an estimate that subsection Ponderosae diverged from subsection Australes between 8 and 15 million years ago (Willyard et al. 2007), the more recent divergence of Sabinianae from Ponderosae could encompass a time span where mitochondrial haplotypes would not be expected to have coalesced. This alternative has some support based on our observation of H3 in five Sabinianae samples, but it seems less likely overall because it would not account for our observation that the plesiomorphic H3 and all of the Sabinianae-style haplotypes are absent in all 69 of our Ponderosae samples west of the Great Basin in the USA.
Great Basin
Previously, three divergent mitochondrial haplotypes (H2, H4, and H7) were reported in the Great Basin (Potter et al. 2013). Our additional data allowed us to conclude that these haplotypes are not disjunct remnants shared with taxa currently distributed south of the Mexico/USA border. Rather, long-term isolation in the Great Basin allowed their localized divergence.
We found H2 in three geographic regions in the USA—southern California (in the Transverse Range), the Guadalupe and Davis Mountains (New Mexico and Texas), and the sky islands of the Mojave Desert in southern Nevada. The southern California, USA, populations of P. ponderosa var. benthamiana were proposed as a geographic race based on monoterpene diversity (Critchfield 1984; Smith 1977), but they have not been allied with P. brachyptera from the Great Basin. Haplotype H2 was most similar to the eastern group of haplotypes and to haplotypes found only in Mexico (Fig. 3). Except for a few widely distributed individuals with the plesiomorphic H3, H2 is the only haplotype found in both P. ponderosa and P. brachyptera. Because the known locations for H2 do not share similar ecological parameters (Shinneman et al. 2016) and H2 is only one motif different than H3, we cannot rule out convergent evolution of repeat patterns. The more likely scenario seems to be that these southern California populations are another example of mitochondrial capture that occurred when they shared a Pleistocene refugium with taxa from the Great Basin rather than sharing a refugium with the ancestors of western populations that carried H1 or H5. The main geographic distribution of P. arizonica var. stormiae is in Nuevo León, Mexico, with several smaller populations in Coahuila, Mexico, and in the Chisos, Davis, and Guadalupe Mountains of Texas, USA (Perry 1991). In addition to our Guadalupe and Davis Mountains collections, we also tentatively genotyped H2 in the Chisos Mountains using fragment lengths, but did not obtain clear nucleotide sequences from the Chisos Mountains that we could include in this study. Our two P. pseudostrobus var. pseudostrobus samples from Mexico with H2 were not within the expected geographic range of P. arizonica var. stormiae. Additional samples of P. arizonica var. stormiae individuals from the main area of their distribution in Mexico will be needed to determine whether the ponderosa pines with H2 in the Great Basin are related to P. arizonica var. stormiae or whether their shared H2 haplotypes arose from mitochondrial capture or from convergent evolution of repeat motifs.
Despite our increased sampling, H4 remained a terminal node on the network, indicating that it is narrowly endemic on two sky islands of the Mojave Desert region of southern Nevada: the Spring Mountains and the Sheep Range (Figs. 3, 4(d); Charlet 2007). Niche modeling suggested that ponderosa pines in this area were uniquely adapted to winter-spring dominated precipitation (Shinneman et al. 2016). Individuals with H4 were found near or intermingled within a single population with individuals that carry H2 (three motifs distant) and with individuals that carry H3 (four motifs different). However, H4 is more similar to haplotypes found only in other taxa whose ranges do not reach northern Mexico: H4 is one motif distant from Hj and two motifs distant from H18. Because of its apparent endemism and network distance, H4 most likely arose in place from an ancestral haplotype rather than from haplotypes H2, H3, H6, or H7 that are currently maintained nearby. These populations in the Spring Mountains and Sheep Range may have maintained their long-term diversity of haplotypes H2, H3, and H4 due to their isolation. Alternatively, H4, Hj, and H18 may have been more widespread during Pleistocene glacial cycles and H4 may yet be discovered in denser sampling of the ponderosa pine taxa, especially in northern Mexico.
Haplotype H7 also formed a terminal node on the network, but is only similar to H6 (three motifs distant) and H2 (three motifs distant; Fig. 3). In the present day, H7 is geographically fairly close to populations that carry H2, and some individual samples with H7 were found near the range of H6. Although H7 is mostly limited to the Great Basin sky islands (Charlet 1996), it extends eastward into western Utah (with a single outlier in Colorado) and southward into far northwest Arizona, USA (Fig. 4(e)). Haplotype H7 has a much wider distribution than H4 and H2. Ecological niche models for previously known locations of H7 inferred compatible climate in Colorado (Shinneman et al. 2016). Because the oldest ponderosa pine fossils in Colorado are younger than those in the Great Basin (Norris et al. 2016), the eastward expansion of H7 into the Rocky Mountains might be relatively recent. Although H7 may be an ancient lineage (Shinneman et al. 2016), our haplotype relationships suggest this haplotype arose in place in the isolated sky islands of the Great Basin and then moved out onto the northern Colorado Plateau and into the Rocky Mountains.
Conclusions
Because P. scopulorum was inferred to migrate at a very fast pace—500 km northward in less than 5000 years (Norris et al. 2016)—and P. ponderosa was able to establish new populations more than 75 km distant from the source population (Lesser et al. 2013), it might be considered surprising that mitochondrial geographic patterns in the Ponderosae are as stable as our observations have shown (Fig. 4). However, introgression can progress rapidly into a few individuals who migrate into sympatry with another species (Currat et al. 2008), especially for mitochondria that have low levels of intraspecific gene flow (Rieseberg et al. 1996). Although the various repeated motifs in this intron are not expected to function differently, it may be possible that they act as a marker for adaptive mitochondrial alleles, though, as noted above, a better understanding of the complexity of the mitochondrial genome is necessary to address the validity of this hypothesis. Retention of introgressed mitochondria may be favored if the resident population has mitochondrial genes that are well adapted to local climate conditions. Tree species have been shown to be capable of rapid local adaptation, including along altitudinal gradients of temperature and moisture (Petit and Hampe 2006; Rehfeldt et al. 2014), and perhaps adaptive mitochondria alleles in Ponderosae contribute to this local adaptation. Thus, the geographically localized mitochondrial haplotypes that we observed may be displaying a “footprint” of ancient mitochondrial haplotypes that are currently shared by more recent immigrants, perhaps including the distantly related P. jeffreyi.
It is important to note that these haplotypes were based solely on a series of complex repeats within a single mitochondrial gene. Therefore, the genetic distances among these haplotypes are only rough estimates that are not expected to correspond to mitochondrial point mutation rates because polymerase slippage and rearrangements (Jaramillo-Correa and Bousquet 2005) might both create variation in the number of repeats. Our PCR-amplification method would not have detected mitochondrial heteroplasmy, which would facilitate unequal crossing over. Although PCR primer bias can mask heteroplasmy, our preponderance of clear nucleotide sequences suggests that mitochondrial heteroplasmy is not the dominant pattern in these taxa. The absence of incomplete repeats and the strong geographic pattern together suggest that these mitochondrial haplotypes are reasonably stable reflections of the genetic history of these pine populations.
Intriguing questions remain. Did the unique haplotypes with Motif 8 (H9, H10, and H11) arise in the ancestor of subsections Ponderosae and Sabinianae? A range-wide survey of P. coulteri, P. sabiniana, and P. jeffreyi would be helpful to determine the diversity of mitochondrial lineages in relation to the Ponderosae using longer mitochondrial sequences (Wang and Wang 2014). Are P. jeffreyi continuing to acquire Ponderosae mitochondria or are these organelle lineages being passed on from the founding individuals once the population has been established? Population-level sampling of P. jeffreyi across its range along with nearby P. ponderosa populations would help unravel this question. Because hybridization levels in natural ponderosa pines have only been inferred based on intermediate morphology or intermediate monoterpene composition, a comparison of nuclear, plastid, and mitochondrial genomic sequences as well as morphological traits from the same individual samples would be valuable. Are the ponderosa pines from southern California that carry the H2 haplotype distinct from P. ponderosa var. benthamiana and are they similar to the trees in the Spring Mountains of Nevada, USA, or the Guadalupe, Davis, and Chisos Mountains of Texas, USA, and New Mexico, USA? To pursue this question, three taxa from Mexico would need to be sampled more broadly for comparison: P. arizonica var. arizonica, P. arizonica var. stormiae, and the populations that have been called P. ponderosa in the Sierra Madre Occidental of northern Mexico. Finally, we suggest that future studies acknowledge the potential for distinct ponderosa pine taxa to exist in sympatry, including intermingled in what might appear to be a continuous forest with variable morphological characters. Sampling molecular markers from all three genomes and morphological characters from the same individuals will be critical to draw conclusions regarding any admixture. Despite the appeal of polygons drawn on maps to document the geographic range of taxa, our results, along with many other experiments, have shown that trying to assign each area on a map to a single taxon of ponderosa pines over-simplifies a reality that is often more complex and interesting.
Code availability
Not applicable.
References
Aizawa M, Kim Z-S, Yoshimaru H (2012) Phylogeography of the Korean pine (Pinus koraiensis) in northeast Asia: inferences from organelle gene sequences. J Plant Res 125:713–723
Avise JC (2009) Phylogeography: retrospect and prospect. J Biogeography 36:3–15
Axelrod D (1986) Cenozoic history of some western American pines. Ann Missouri Bot Gard 73:565
Bandelt H-J, Forster P, Röhl A (1999) Median-joining networks for inferring intraspecific phylogenies. Mol Biol Evol 16:37–48
Bastien D, Favre JM, Collignon AM, Sperisen C, Jeandroz S (2003) Characterization of a mosaic minisatellite locus in the mitochondrial DNA of Norway spruce (Picea abies (L.) Karst.). Theor Appl Gen 107:574–580
Birky CW Jr, Maruyama T, Fuerst P (1983) An approach to population and evolutionary genetic theory for genes in mitochondria and chloroplasts, and some results. Genet 103:513–527
Bock DG, Andrew RL, Rieseberg LH (2014) On the adaptive value of cytoplasmic genomes in plants. Mol Ecol 23:4899–4911
Bouillé M, Senneville S, Bousquet J (2011) Discordant mtDNA and cpDNA phylogenies indicate geographic speciation and reticulation as driving factors for the diversification of the genus Picea. Tree Gen Genomes 7:469–484
Briggs JS, Vander Wall SB, Jenkins SH (2009) Forest rodents provide directed dispersal of Jeffrey pine seeds. Ecology 90:675–687. https://doi.org/10.1890/07-0542.1
Buck JM, Adams RS, Cone J, Conkle MT, Libby WJ, Eden CJ, Knight MJ (1970) California tree seed zones. USDA Forest Service San Francisco, California, USA
Burban C, Petit R (2003) Phylogeography of maritime pine inferred with organelle markers having contrasted inheritance. Mol Ecol 12:1487–1495
Callaham RZ (2013a) Pinus ponderosa: a taxonomic review with five subspecies in the United States. USDA Forest Service, Pacific Southwest Research Station PSW-RP 264:1–53
Callaham RZ (2013b) Pinus ponderosa: geographic races and subspecies based on morphological variation. USDA Forest Service, Pacific Southwest Research Sation PSW-RP-265:1–54
Charlet D (1996) Atlas of Nevada conifers: a phytogeographic reference. University of Nevada Press, Reno, Nevada, USA
Charlet D (2007) Distribution patterns of Great Basin conifers: implications of extinction and immigration. Aliso 24:31–61
Chaw S-M, Shih AC-C, Wu Y-W, Liu S-M, Chou T-Y, Wang D (2008) The mitochondrial genome of the gymnosperm Cycas taitungensis contains a novel family of short interspersed elements, Bpu sequences, and abundant RNA editing sites. Mol Biol Evol 25:603–615
Conkle M, Critchfield WB (1988) Genetic variation and hybridization of ponderosa pine. In: Baumgartner D, Lotan J (eds) Ponderosa pine: the species and its management. Washington State University, Pullman, Washington, USA, pp 27–44
Critchfield WB (1966) Crossability and relationships of the California big-cone pines USDA Forest Service Research Paper NC-6:36–44
Critchfield WB (1984) Crossability and relationships of Washoe pine. Madroño 31:144–170
Currat M, Ruedi M, Petit RJ, Excoffier L (2008) The hidden side of invasions: massive introgression by local genes. Evolution 62:1908–1920
Du FK, Petit RJ, Liu JQ (2009) More introgression with less gene flow: chloroplast vs. mitochondrial DNA in the Picea asperata complex in China, and comparison with other conifers. Mol Ecol 18:1396–1407
Ellegren H (2004) Microsatellites: simple sequences with complex evolution. Nat Rev Genet 5:435–445. https://doi.org/10.1038/nrg1348
Forgione I, Bonavita S, Rosaria Regina TM (2019) Mitochondria of Cedrus atlantica and allied species: a new chapter in the horizontal gene transfer history. Plant Sci 281:93–101
Furnier GR, Adams WT (1986) Geographic patterns of allozyme variation in Jeffrey pine. Am J Bot 73:1009–1015
Gérardi S, Jaramillo-Correa J, Beaulieu J, Bousquet J (2010) From glacial refugia to modern populations: new assemblages of organelle genomes generated by differential cytoplasmic gene flow in transcontinental black spruce. Mol Ecol 19:5265–5280
Gernandt DS, Hernández-León S, Salgado-Hernández E, Pérez de la Rosa JA, Jardón-Barbolla L (2009) Phylogenetic relationships of Pinus subsection Ponderosae inferred from rapidly evolving cpDNA regions. Sys Bot 34:481–491
Godbout J, Fazekas A, Newton CH, Yeh FC, Bousquet J (2008) Glacial vicariance in the Pacific Northwest: evidence from a lodgepole pine mitochondrial DNA minisatellite for multiple genetically distinct and widely separated refugia. Mol Ecol 17:2463–2475
Godbout J, Jaramillo-Correa JP, Beaulieu J, Bousquet J (2005) A mitochondrial DNA minisatellite reveals the postglacial history of jack pine (Pinus banksiana), a broad-range North American conifer. Mol Ecol 14:3497–3512
Godbout J, Yeh FC, Bousquet J (2012) Large-scale asymmetric introgression of cytoplasmic DNA reveals Holocene range displacement in a North American boreal pine complex. Ecol Evol 2:1853–1866
Goslee SC, Urban DL (2007) The ecodist package for dissimilarity-based analyses of ecological data. J Stat Software 22:1–19
Gugerli F, Senn J, Anzidei M, Madaghiele A, Buchler U, Sperisen C, Vendramin G (2001) Chloroplast microsatellites and mitochondrial nad1 intron2 sequences indicate congruent phylogenetic relationships among Swiss stone pine (Pinus cembra), Siberian stone pine (Pinus sibirica), and Siberian dwarf pine (Pinus pumila). Mol Ecol 10:1489–1497
Hall TA (1999) BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT Nucleic acids symposium series 41:95–98
Haller JR (1962) Variation and hybridization in ponderosa and Jeffrey pines. Univ Calif Publ Bot 34:123–165
Haller JR (1986) Taxonomy and relationships of the mainland and island populations of Pinus torreyana (Pinaceae). Sys Bot 1:39–50
Haller JR, Vivrette NJ (2011) Ponderosa pine revisited. Aliso 29:53–57
Hamming RW (1950) Error detecting and error correcting codes The. Bell Syst Tech J 29:147–160
Houliston GJ, Olson MS (2006) Nonneutral evolution of organelle genes in Silene vulgaris. Genet 174:1983–1994
Huson DH, Scornavacca C (2011) A survey of combinatorial methods for phylogenetic networks. Genome Biol Evol 3:23–25
Jackman SD, Coombe L, Warren RL, Kirk H, Trinh E, MacLeod T, Pleasance S, Pandoh P, Zhao Y, Coope R, Bousquet J, Bohlmann J, Jones SJM, Birol I (2020) Complete mitochondrial genome of a gymnosperm, Sitka spruce (Picea sitchensis) indicates a complex physical structure. Genome Biol Evol 12:1174–1179
Jaramillo-Correa JP, Aguirre-Planter E, Eguiarte LE, Khasa DP, Bousquet J (2013) Evolution of an ancient microsatellite hotspot in the conifer mitochondrial genome and comparison with other plants. J Mol Evol 76:146–157
Jaramillo-Correa JP, Bousquet J (2005) Mitochondrial genome recombination in the zone of contact between two hybridizing conifers. Genet 171:1951–1962
Johansen AD, Latta RG (2003) Mitochondrial haplotype distribution, seed dispersal and patterns of postglacial expansion of ponderosa pine. Mol Ecol 12:293–298
Johnson M, Vander Wall SB, Borchert M (2003) A comparative analysis of seed and cone characteristics and seed-dispersal strategies of three pines in the subsection Sabinianae. Plant Ecol 168:69–84
Jombart T, Dray S (2010) adephylo: exploratory analyses for the phylogenetic comparative method. Bioinformatics 26:1907–1909
Kitzmiller JH (2005) Provenance trials of ponderosa pine in Northern California. Forest Sci 51:595–607
Kral R (1993) Pinus. In: Flora of North America Editorial Committee (ed) Flora of North America, vol 2. Oxford University Press, New York, New York, USA, pp 373–398
Latta RG (2006) Integrating patterns across multiple genetic markers to infer spatial processes. Landscape Ecol 21:809–820
Latta RG, Mitton JB (1999) Historical separation and present gene flow through a zone of secondary contact in ponderosa pine. Evolution 53:769–774
Leigh JW, Bryant D (2015) Popart: full-feature software for haplotype network construction. Meth Ecol Evol 6:1110–1116
Lesser MR, Jackson ST (2013) Contributions of long-distance dispersal to population growth in colonising Pinus ponderosa populations. Ecol Letters 16:380–389. https://doi.org/10.1111/ele.12053
Lesser MR, Parchman TL, Jackson ST (2013) Development of genetic diversity, differentiation and structure over 500 years in four ponderosa pine populations. Mol Ecol 22:2640–2652. https://doi.org/10.1111/mec.12280
Little EL Jr (1971) Atlas of United States trees, vol 1. District of Columbia, USA, Washington
López-Reyes A, Pérez de la Rosa J, Ortiz E, Gernandt DS (2015) Morphological, molecular, and ecological divergence in Pinus douglasiana and P. maximinoi. Sys Bot 40:658–670. https://doi.org/10.1600/036364415X689384
Malek O, Axel B, Knoop V (1997) Evolution of trans-splicing plant mitochondrial introns in pre-Permian times. Proc Nat Acad Sci USA 94:553–558
Martínez M (1948) Los pinos mexicanos, segunda edicion. Ediciones Botas, Mexico City, Mexico
Mazel F, Davis KM, Loudon A, Kwong WK, Groussin M, Parfrey LW (2018) Is host filtering the main driver of phylosymbiosis across the tree of life?. mSystems 3:1–15.
Mirov NT (1953) Chemical aspects of diploxylon pines. Silv Gen 2:93–96
Mirov NT (1958) Pinus oaxacana, a new species from Mexico. Madroño 14:145–176
Mirov NT (1967) The genus Pinus. The Ronald Press Company, New York, New York, USA
Mitton JB, Kreiser BR, Rehfeldt GE (2000) Primers designed to amplify a mitochondrial nad1 intron in ponderosa pine, Pinus ponderosa, limber pine, P. flexilis, and Scots pine P Sylvestris. Theor Appl Gen 101:1269–1272
Nagel LM et al (2017) Adaptive silviculture for climate change: a national experiment in manager-scientist partnerships to apply an adaptation framework. J Forest 115:167–178. https://doi.org/10.5849/jof.16-039
Neale D, Sederoff R (1989) Paternal inheritance of chloroplast DNA and maternal inheritance of mitochondrial DNA in loblolly pine. Theor Appl Gen 77:212–216
Neale DB et al (2014) Decoding the massive genome of loblolly pine using haploid DNA and novel assembly strategies. Genome Biol 15:1–29. https://doi.org/10.1186/gb-2014-15-3-r59
Norris JR, Betancourt JL, Jackson ST (2016) Late Holocene expansion of ponderosa pine (Pinus ponderosa) in the Central Rocky Mountains USA. J Biogeography 134:778–790
Padgham M and Sumner MD (2020) Geodist: fast, dependency-free geodesic distance calculations. R package version 0.0.4
Parchman TL, Benkman C (2007) The geographic selection mosaic for ponderosa pine and crossbills: a tale of two squirrels. Evolution 62:348–360. https://doi.org/10.1111/j.1558-5646.2007.00295.x
Peakall R, Smouse PE (2006) GENALEX 6: genetic analysis in Excel. Population Genetic Software for Teaching and Research Mol Ecol Notes 6:288–295
Percy DM et al (2014) Understanding the spectacular failure of DNA barcoding in willows (Salix): does this result from a trans-specific selective sweep? Mol Ecol 23:4737–4756
Perry JP Jr (1991) The pines of Mexico and Central America. Timber Press, Portland, Oregon, USA
Petit RJ et al (2002) Identification of refugia and post-glacial colonisation routes of European white oaks based on chloroplast DNA and fossil pollen evidence. For Ecol Mgmt 156:49–74
Petit RJ, Duminil J, Fineschi S, Hampe A, Salvini D, Vendramin GG (2005) Comparative organization of chloroplast, mitochondrial and nuclear diversity in plant populations. Mol Ecol 14:689–701
Petit RJ, Excoffier L (2009) Gene flow and species delimitation. Trends Ecol Evol 24:386–393
Petit RJ, Hampe A (2006) Some evolutionary consequences of being a tree. Ann Rev Ecol Evol Syst 37:187–214. https://doi.org/10.1146/annurev.ecolsys.37.091305.110215
Potter KM, Hipkins VD, Mahalovich MF, Means RE (2013) Mitochondrial DNA haplotype distribution patterns in Pinus ponderosa (Pinaceae): range-wide evolutionary history and implications for conservation. Am J Bot 100:1562–1579
Potter KM, Hipkins VD, Mahalovich MF, Means RE (2015) Nuclear genetic variation across the range of ponderosa pine (Pinus ponderosa): Phylogeographic, taxonomic, and conservation implications. Tree Gen Genomes 11:1–23. https://doi.org/10.1007/s11295-015-0865-y
Pyhäjärvi T, Salmela MJ, Savilainen O (2008) Colonization routes of Pinus sylvestris inferred from distribution of mitochondrial DNA variation. Tree Gen Genomes 4:247–254
Ran J-H, Shen T-T, Liu W-J, Wang P-P, Wang X-Q (2015) Mitochondrial introgression and complex biogeographic history of the genus Picea. Mol Phylogen Evol 93:63–76
R Core Team (2021) R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria https://www.R-project.org/
Read R (1980) Genetic variation in seedling progeny of ponderosa pine provenances. Society of American Foresters, Washington, District of Columbia, USA
Rehfeldt GE (1999) Systematics and genetic structure of Ponderosae taxa (Pinaceae) inhabiting the mountain islands of the Southwest. Am J Bot 86:741–752
Rehfeldt GE, Jaquish BC, López-Upton J, Sáenz-Romero C, St Clair JB, Leites LP, Joyce DG (2014) Comparative genetic responses to climate for the varieties of Pinus ponderosa and Pseudotsuga menziesii: realized climate niches. Forest Ecol Manage 324:126–137
Richardson D (1998) Ecology and Biogeography of Pinus. Cambridge University Press, New York, New York, USA
Rieseberg LH, Whitton J, Linder CR (1996) Molecular marker incongruence in plant hybrid zones and phylogenetic trees. Acta Bot Neerlandica 45:243–262
Schwilk DW, Keeley JE (2006) The role of fire refugia in the distribution of Pinus sabiniana (Pinaceae) in the southern Sierra Nevada. Madroño 53:364–372
Shinneman DJ, Means RE, Potter KM, Hipkins VD (2016) Exploring climate niches of ponderosa pine (Pinus ponderosa Douglas ex Lawson) haplotypes in the western United States: Implications for evolutionary history and conservation PLOS ONE 11 https://doi.org/10.1371/journal.pone.0151811
Siepielski AM, Benkman CW (2010) Conflicting selection from an antagonist and a mutualist enhances phenotypic variation in a plant. Evolution 64:1120–1128. https://doi.org/10.1111/j.1558-5646.2009.00867.x
Smith RH (1977) Monoterpenes of ponderosa pine xylem resin in western United States. USDA. For Serv Tech Bull 1532
Soranzo N, Alia R, Provan J, Powell W (2000) Patterns of variation at a mitochondrial sequence-tagged-site locus provides new insights into the postglacial history of European Pinus sylvestris populations. Mol Ecol 9:1205–1211
Sperisen C, Buchler U, Gugerli F, Matyas G, Geburek T, Vendramin GG (2001) Tandem repeats in plant mitochondrial genomes: application to the analysis of population differentiation in the conifer Norway spruce. Mol Ecol 10:257–263
Stadler K (2018) Cultevo: tools, measures and statistical tests for cultural evolution. R package version 1.0.2 https://kevinstadler.github.io/cultevo/
Straub SCK, Parks M, Weitemier K, Fishbein M, Cronn RC, Liston A (2012) Navigating the tip of the genomic iceberg: next-generation sequencing for plant systematics. Am J Bot 99:349–364
Strauss S, Hong Y-P, Hipkins V (1993) High levels of population differentiation for mitochondrial DNA haplotypes in Pinus radiata, muricata, and attenuata. Theor Appl Gen 86:605–611
Sullivan AR et al (2020) The mitogenome of Norway spruce and a reappraisal of mitochondrial recombination in plants. Genome Biol Evol 12:3586–3598. https://doi.org/10.1093/gbe/evz263
Syring J, Willyard A, Cronn R, Liston A (2005) Evolutionary relationships among pine (Pinaceae) subsections inferred from multiple low-copy nuclear loci. Am J Bot 92:2086–2100
Toor N, Hausner G, Zimmerly S (2001) Coevolution of group II intron RNA structures with their intron-encoded reverse transcriptases. RNA 7:1142–1152
Tsutsui K, Suwa A, Ki Sawada, Kato T, Ohsawa TA, Watano Y (2009) Incongruence among mitochondrial, chlorplast, and nuclear gene trees in Pinus subgenus Strobus (Pinaceae). J Plant Res 122(5):509–521
Vander Wall SB (1993) Cache site selection by chipmunks (Tamias spp.) and its influence on the effectiveness of seed dispersal in Jeffrey pine (Pinus jeffreyi). Oecologia 96:246–252
Vander Wall SB, Enders MS, Barga S, Moore C, Seaman A, Perea R (2012) Jeffrey pine seed dispersal in the Sierra San Pedo Mártir Baja California, Mexico Western North American. Naturalist 72:534–542. https://doi.org/10.3398/064.072.0409
Wang B, Climent J, Wang X-R (2015) Horizontal gene transfer from a flowering plant to the insular pine Pinus canariensis (Chr. Sm. Ex DC in Buch). Heredity 114:413–418
Wang B, Wang X-R (2014) Mitochondrial DNA capture and divergence in Pinus provide new insights into the evolution of the genus. Mol Phylogen Evol 80:20–30
Wang X-Q, Ran J-H (2014) Evolution and biogeography of gymnosperms. Mol Phylogen Evol 75:24–40
Weidman RH (1939) Evidences of racial influence in a 25-year test of ponderosa pine. J Agric Res 59:855–887
Wells OO (1964) Geographic varation in ponderosa pine I The ecotypes and their distribution. Silv Gen 13:89–103
Williams CG (2010) Long-distance pine pollen still germinates after meso-scale dispersal. Am J Bot 97:846–855
Willyard A, Syring J, Gernandt DS, Liston A, Cronn R (2007) Fossil calibration of molecular divergence infers a moderate mutation rate and recent radiations for Pinus. Mol Biol Evol 24:90–101
Willyard A, Cronn R, Liston A (2009) Reticulate evolution and incomplete lineage sorting among the ponderosa pines. Mol Phylogen Evol 52:498–511
Willyard A et al. (2021) Phylogenomics in the hard pines (Pinus subsection Ponderosae; Pinaceae) confirms paraphyly in Pinus ponderosa, and places Pinus jeffreyi with the California big cone pines Sys Bot 46:538–561
Willyard A et al (2017) Pinus ponderosa: a checkered past obscured four species. Am J Bot 104:161–181
Wofford AM, Finch K, Bigott A, Willyard A (2013) A set of plastid loci for use in multiplex fragment length genotyping for intraspecific variation in Pinus (Pinaceae). Appl Plant Sci 2:1–10
Wolfe KH, Li WH, Sharp PM (1987) Rates of nucleotide substitution vary greatly among plant mitochondrial, chloroplast, and nuclear DNAs. Proc Nat Acad Sci USA 84:9054–9058
Won H, Renner SS (2003) Horizontal gene transfer from flowering plants to Gnetum. Proc Nat Acad Sci USA 100:10824–10829
Acknowledgements
We thank Jean Bousquet, David Charlet, George Ferguson, Valerie Hipkins, Stephen K. Langer, Robert Latta, Sergio Hernández León, Aaron Liston, Diana Ramos Dorantes, Charles S. Rand, and anonymous reviewers for their comments on earlier versions of the manuscript. Hendrix College students Adam Bigott, Hassan Karemera, Nicole Segear, Mason Sifford, and Austin Wofford contributed to the laboratory and analytical work on these mitochondrial data as part of their undergraduate research projects. Other Hendrix students helped with field collections and DNA isolations: Blake Cooper, Connor Douglas, Kristen Finch, Jack Finney, Payton Lea, Julia Lefler, Brandon Linz, Samuel Lockhart, Trang Nguyen, Dakota Pouncey, Brian Schumacher, Mason Sifford, Joshua Smith, Kevin Spatz, and Pete Wills. Land managers who protect these populations and permitted our collections are gratefully acknowledged: Bighorn, Black Hills, Cleveland, Colville, Coronado, Deschutes, Eldorado, Fremont-Winema, Humboldt-Toiyabe, Inyo, Kaibab, Klamath, Los Padres, Mendocino, Modoc, Nez Perce, Ochoco, San Bernardino, San Juan, Santa Fe, Sequoia, Shasta-Trinity, Sierra, Six Rivers, Umpqua, and Wasatch National Forests; California State Parks (Henry Coe and Henry Cowell), Fred Lawrence Whipple Observatory, Guadalupe National Park, Hualapai Mountain Park (Mohave County, AZ), Joint Base Lewis-McChord, North Sierra Tree Improvement Association, Sierra Pacific Industries, Quail Hollow Park (Santa Cruz County, CA), the Nature Conservancy, University of California Santa Cruz Arboretum, and University of California Reserves (Landel-Hills Big Creek and James San Jacinto).
Funding
A. W. and her students received support from Arkansas SURF, Arkansas Academy of Science Undergraduate Research Fund, and the Hendrix College Odyssey program; K. M. P. was funded with Cost Share Agreement 18-CS-11330110–026 between the U.S. Department of Agriculture, Forest Service, Southern Research Station, and North Carolina State University; D. S. G. was funded from Programa de Apoyo a Proyectos de Investigación e Innovación Tecnológica (DGAPA PAPIIT-IN228209).
Author information
Authors and Affiliations
Contributions
A. W. designed the experiment, collected plant samples, supervised student lab work, aligned the nucleotide sequences, and wrote the manuscript; D. S. G. collected plant samples, supervised lab work, and contributed to the manuscript; A. L. R. collected plant samples, sequenced nucleotides, and contributed to the manuscript; K. M. P. advised experimental design and contributed to the manuscript.
Corresponding author
Ethics declarations
Ethics approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Data archiving statement
Nucleotide sequences were submitted to GenBank (MK766513–MK766718). Three Supplemental Data files were included with this article. Online Resource 3 lists the geographic location and observed haplotypes for 206 individuals of 22 taxa from 123 locations in the USA, Mexico, and Guatemala. The 11 GenBank accessions that were used are included in the table without geographic location. Online Resource 4 is the alignment of 217 samples, with motif numbers 1–8 (Fig. 2) inserted before each motif to aid alignment and Online Resource 5 is the alignment without motif columns. The first characters of each sample name in the alignments give the haplotype. Online Resource 6 is the binary matrix based on the presence/absence of each motif repeat that was used to create the median joining network.
Additional information
Responsible Editor: G.G. Vendramin
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Willyard, A., Gernandt, D.S., López-Reyes, A. et al. Mitochondrial phylogeography of the ponderosa pines: widespread gene capture, interspecific sharing, and two unique lineages. Tree Genetics & Genomes 17, 47 (2021). https://doi.org/10.1007/s11295-021-01529-4
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11295-021-01529-4