
Abstract
The comparison of inter-population quantitative variation and neutral variation based on molecular markers (QST − FST) has been extensively used to infer the influence of different selection forces on potentially adaptive traits. Only recently have studies focused on two levels of inter-population genetic structuring: among regions (or groups) and among subpopulations within groups. This work aimed to compare quantitative and molecular variation within these two hierarchical levels for Hancornia speciosa Gomes, a fruit tree species that is native to the Brazilian Cerrado. Six quantitative traits related to initial plant growth were evaluated in a common garden environment using samples from 57 maternal families (treatments) derived from 29 subpopulations within four botanical varieties. The quantitative divergence among the botanical varieties (QGT) and among the subpopulations within varieties (QSG) for each trait were compared with the corresponding neutral variation (FGT and FSG) obtained based on six microsatellite loci using a parametric bootstrap procedure. The molecular results revealed a low degree of divergence among the botanical varieties and significant structuring among the subpopulations within varieties. The estimates of the quantitative divergence among the varieties (QGT) tended to be greater than the divergence among the subpopulations within varieties (QSG) for five out of the six quantitative traits. The comparison between the quantitative and molecular parameters suggests that divergent selection shaped the genetic structure among the botanical varieties for some traits, while the variation among the subpopulations within varieties was influenced by genetic drift and uniform selection.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Knowledge of the structure of genetic variation in natural populations is crucial for designing strategies for in situ or ex situ conservation. Natural plant populations, particularly when dispersed over a large geographical area, may exhibit different levels of structure among subpopulations (local populations). The effective size of a sample of individuals obtained from a natural population is highly dependent upon the degree of differentiation among the subpopulations, which can be determined using the expression \( {N}_e=\frac{1}{2{D}_1} \), where \( {D}_1={F}_{ST}\left[\frac{1+{C}^2}{S}\left(\frac{S^{\ast }}{S^{\ast }-1}\right)-\frac{1}{S^{\ast }-1}-\frac{1}{n}\right]+\frac{1+{F}_{IT}}{2n} \) (Vencovsky and Crossa 2003). In this formula, FST and FIT are Wright’s statistics and represent the allelic differentiation among subpopulations and the total fixation index for individuals in relation to the entire population, respectively (Wright 1951); S is the number of sampled subpopulations; S* is the number of subpopulations that are predicted to exist in nature; and C is the coefficient of variation of the number of individuals in each subpopulation. If the total number of sampled individuals (n) is relatively high, the value of the effective size is little influenced by FIT. Based on the assumption of the existence of a great number of subpopulations in nature (S∗ → ∞) and a small difference in the number of sampled individuals in each subpopulation (C → 0), the upper limit of the effective population size is \( {N}_e=\frac{S}{2{F}_{ST}} \) (Vencovsky and Crossa 2003). Therefore, even when FST is small and most of the genetic variation is within subpopulations, a large number of subpopulations are required to ensure that there is an adequate representativeness of samples for the purposes of genetic conservation for both ex situ and in situ strategies.
During the last few decades, molecular markers have been used extensively to evaluate the genetic structures of natural plant populations. The estimation of FST and/or analog parameters based on molecular data is widely used to infer the degree of genetic differentiation and gene flow among subpopulations. Despite its utility for studies of population genetics, the use of FST as a single measure of subpopulation differentiation may be insufficient in some instances. One of the situations that deserve special attention occurs when natural selection affects the differentiation of adaptive traits among subpopulations. The derivation of FST statistics based on mutation-drift equilibrium assumes selective neutrality; therefore, inferences of whole genome differentiation using estimates of FST based on neutral markers must be made with caution.
The effect of inbreeding due to the isolation of subpopulations on the structure of the variances in quantitative traits was demonstrated earlier by Wright (1951). Assuming the occurrence of random mating within subpopulations and additive effects of genes, he demonstrated that the total variance is \( {\sigma}_{T(F)}^2=\left(1+{F}_{ST}\right){\sigma}_{T(0)}^2 \), where \( {\sigma}_{T(0)}^2 \) represents the variance within the reference panmictic population. This total variance can be partitioned into the variance among subpopulations, which is represented by the equation \( {\sigma}_{ms}^2=2{F}_{ST}{\sigma}_{T(0)}^2 \), and the average variance within subpopulations, which can be represented by the equation \( {\sigma}_{S(0)}^2=\left(1-{F}_{ST}\right){\sigma}_{T(0)}^2 \). Based on these expressions, it follows that \( {F}_{ST}=\frac{\sigma_{ms}^2}{\sigma_{ms}^2+2{\sigma}_{S(0)}^2} \). This equation can be used to determine a parameter known as QST, which is an analog of FST and related to the components of variance of quantitative traits (Spitze 1993).
When subpopulations present themselves the average inbreeding coefficient (FIS) due to deviations from panmixia, the parameter can be estimated using the equation \( {\hat{Q}}_{ST}=\frac{\left(1+{F}_{IS}\right){\hat{\sigma}}_B^2}{\left(1+{F}_{IS}\right){\hat{\sigma}}_B^2+2{\sigma}_{AW}^2} \) (Bonnin et al. 1996). In this formula, the components of variance among and within subpopulations are represented by \( {\hat{\sigma}}_B^2 \) (total genetic variance among subpopulations instead of \( {\sigma}_{ms}^2 \) of the Wright’s formula) and \( {\sigma}_{AW}^2 \) (additive genetic variance within subpopulations instead of \( {\sigma}_{S(0)}^2 \) of the original formula). The intra-population fixation index FIS can be determined based on the mating system: FIS = 0 in the presence of alogamy and FIS = 1 in the presence of autogamy. If the species reproduces using a mixed system, FIS should be estimated separately using molecular markers or indirectly using the outcrossing rate (s), which will result in \( {F}_{IS}=\frac{s}{2-s} \), assuming Wright’s equilibrium (Vencovsky and Crossa 2003).
Since QST = FST is expected to be true under selective neutrality, the comparison of these two parameters for the same metapopulation constitutes a tool that can be used to test the effects of natural selection on quantitative traits using the value of FST estimated based on neutral markers as a null hypothesis (e.g., Spitze 1993; Bonnin et al. 1996; Goudet and Buchi 2006; Whitlock 2008; Leinonen et al. 2013; Boaventura-Novaes et al. 2018). When divergent selection favors local adaptations, the differentiation among subpopulations will be larger than expected based on neutrality, which will result in QST > FST; if all subpopulations are adapted to the same local optimum, uniform selection will result in QST < FST. In the absence of selection, no difference between the two parameters is expected.
In a review of comparative studies on population differentiation in terms of quantitative traits and neutral markers, Leinonen et al. (2008) performed a meta-analysis that included 50 species, among them 27 plant species. The results showed that in general, positive estimates of contrast tend to predominate, which suggests that divergence due to natural selection and local adaptation appears to be the norm in published studies. This confirms the results of earlier meta-analysis studies, with a larger number of species (Merilä and Crnokrak 2001; McKay and Latta 2002). Several other applications of QST − FST comparison in plants can be found in the literature (see Leinonen et al. 2013 for a review).
The Brazilian Cerrado is a biome located at the central region of Brazil that occupies an area of approximately 2.2 million km2 in which natural savannah-like vegetation predominates. This biome is very rich in terms of plant diversity due to presence of nearly 12,000 native species, most of which are endemic (Mendonça et al. 2008). This region has been affected greatly by agricultural activities during the last five decades that have reduced the original vegetation cover to fragments with different degrees of isolation. This has led to the inclusion of the Brazilian Cerrado in a list of 25 worldwide hotspots that are a priority for biodiversity conservation (Myers et al. 2000). Because of this, knowledge regarding the genetic structure of species is crucial for the design of adequate conservation strategies to be used within this biome to address habitat loss and future climatic changes (Diniz-Filho et al. 2018).
Hancornia speciosa Gomes (Apocinaceae) is a fruit tree that is native to the tropical regions of Brazil. Its fruit (“mangaba”) is highly regarded by the local human population and used in the form of fresh fruit, juice, jelly, or ice creams. Six botanical varieties have been described for the species based on morphological descriptors of leaves and flowers (Monachino 1945). According to this study, all six varieties occur in the Brazilian Cerrado. Only one variety (H. speciosa var. speciosa) occurs also outside of the Cerrado in coastal areas in the northeast and northern regions of Brazil (Silva-Junior and Lédo 2006). Currently, almost all fruit production is the result of direct collection from wild plant populations; research regarding agricultural and domestication techniques that can be used to cultivate the species is in the initial stages (Almeida et al. 2019). Some studies of genetic diversity have already been performed on the species based on molecular markers, morphological traits and chemical components in both ex situ and in situ conditions (Ganga et al. 2009; Ganga et al. 2010; Jimenez et al. 2015; Collevatti et al. 2016; Costa et al. 2017; Santos et al. 2017; Collevatti et al. 2018; Flores et al. 2018; Almeida et al. 2019). Collevatti et al. (2018) discuss the use of molecular data in support of the botanical varieties recognized based on morphological traits.
Models with two or more levels of population structure have been used extensively in studies that utilize F statistics. Only more recent theoretical studies have focused on the use of Q statistics in two hierarchical levels (Whitlock and Gilbert 2012; Cubry et al. 2017), and the results based on experimental data are also scarce (Volis et al. 2005; Boaventura-Novaes et al. 2018). The objective of this study was to evaluate the genetic structures among the botanical varieties and among the subpopulations within varieties of H. speciosa using microsatellite markers and quantitative traits to infer the effects of natural selection at both levels of population structure.
Materials and methods
Quantitative data
In October 2004, the prospection of local populations and the collection of fruits from H. speciosa were performed in different localities of the Brazilian Cerrado with the objective of sampling the majority of the genetic diversity of the species occurring within this biome. Fruits were collected from 35 localities and from three to six mother plants in each locality. After discarding the inadequate fruits, the seeds from 109 mother plants representing 35 subpopulations were sown in a nursery in Goiânia, GO, Brazil (latitude 16° 35′ 44″ S, longitude 49° 16′ 51″ W, altitude 717 m). Progenies that accounted for at least four well-grown seedlings were evaluated in the nursery in terms of plant height (NPH in cm) and stem diameter (NSD in mm) 12 months after they were sown. In December 2005, four seedlings from each progeny, the same ones evaluated for NPH and NSD, were transplanted into an experimental field in Goiânia, GO, Brazil (latitude 16° 35′ 38″ S, longitude 49° 17′ 27″ W, altitude 725 m), to produce an ex situ germplasm collection. It was planted using a randomized complete block design that included 57 treatments (maternal families) from 29 subpopulations, four replicates, and one plant per plot spaced at 6 m × 5 m. No artificial fertilization was performed and the cultural treatment was limited to the control of weeds and leaf-cutting ants. For more details of experimental conduction, see Ganga et al. (2009). In the field, the plant height and stem diameter were measured on a monthly basis from January 2006 to August 2007. The growth rates in terms of height (GRH in cm/month) and stem diameter (GRD in mm/month) were measured by the coefficient of linear regression estimated for each variable (Y) using the measurement date as independent variable (X). Additionally, the last measurements of plant height (FPH in cm) and stem diameter (FSD in mm) were used in this study, totaling six variables related to juvenile plant growth. The same variables were explored by Ganga et al. (2009) for agronomic purposes.
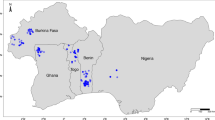
The analysis was performed based on field data from 29 subpopulations that represented 27 geographical localities (Fig. 1, Table S1) and nursery data from 27 subpopulations that represented 25 geographical localities. The subpopulations represented four botanical varieties (H. speciosa var. pubescens, H. speciosa var. gardneri, H. speciosa var. speciosa and H. speciosa var. cuyabensis); these will be referred to using only the variety name hereafter for the sake of simplicity. The varieties were identified according to the description of Monachino (1945), based on the morphological traits (see Ganga et al. 2009, for an illustration of the differences among varieties). Three subpopulations were excluded from the analysis due to uncertainty in the allocation to a specific botanical variety. The difference in numbers of subpopulations and localities of collection occurred because in two localities plants from the gardneri and pubescens varieties, which were considered to represent two different subpopulations for analysis purposes, occurred together. The coordinates of each collection locality and number of progenies per subpopulation and botanical variety can be seen in Ganga et al. (2009).

Map showing the 29 subpopulations (27 localities) within the four botanical varieties of Hancornia speciosa Gomes in the Brazilian Cerrado that are represented in the UFG germplasm collection in Goiânia, Goiás, Brazil
Molecular data
A sample consisting of two plants from each of the 57 families within the H. speciosa germplasm collection (first and second blocks) was genetically characterized using microsatellite markers (single sequence repeats - SSRs). When available, other progenies from the same subpopulation that were not included in the field experiment due to insufficient plant numbers were genotyped to improve the precision of the estimates, which resulted in a total of 116 genotyped plants. Six polymorphic SSR primers (HS 01, HS 05, HS 24, HS 26, HS 27, and HS 30, Table S2) that were initially developed for this species were used (Rodrigues et al. 2015). For each plant, genomic DNA was extracted from expanded leaves following the 2% CTAB protocol (Doyle and Doyle 1990). SSR amplifications were performed using a PTC-100 thermal cycler (MJ Research Inc.) and the amplified products were separated on 6% polyacrylamide gels stained with silver nitrate (Creste et al. 2001). More details about the methods used for DNA extraction and genotyping, as well the characterization of the primers, including new primers characterized later, can be found in Rodrigues et al. (2015).
Data analysis
The quantitative data from the nursery and field environments were submitted to analysis of variance according to the random nested model Yijkl = μ + vi + sj(i) + fk(ij) + el(ijk), where Yijkl represents the phenotypic value of the plant l from family k from subpopulation j from variety i; μ represents the general mean; vi represents the effect of variety i; sj(i) represents the effect of subpopulation j from variety i; fk(ij) represents the effect of family k within population j from the variety i; and el(ijk) is the effect of plant l from family k within the subpopulation j from variety i. The corresponding scheme of the analysis of variance is presented in Table 1.
The components of variance were estimated equating the mean squares to their expected values. The heritability coefficient at the family within population level was estimated by \( {\hat{h}}_{FS}^2=\frac{{\hat{\sigma}}_{FS}^2}{{\hat{\sigma}}_{FS}^2+\frac{{\hat{\sigma}}_{IF}^2}{k_1}}=\frac{MS_3-{MS}_4}{MS_3} \).
The formula for estimating the quantitative divergence index (QST) was adapted for populations with a two-level structure replacing (1 − FST) by (1 − FSG)(1 − FGT) in the basic formula that describes the variance structure of subdivided populations (Wright 1969), which resulted in the estimators \( {\hat{Q}}_{SG}=\frac{{\hat{\sigma}}_{SG}^2}{{\hat{\sigma}}_{SG}^2+\frac{2}{1+{F}_{IS}}{\hat{\sigma}}_{AW}^2} \), which measures the differentiation among the subpopulations within groups (botanical varieties, in this case), and \( {\hat{Q}}_{GT}=\frac{{\hat{\sigma}}_{GT}^2}{{\hat{\sigma}}_{GT}^2+{\hat{\sigma}}_{SG}^2+\frac{2}{1+{F}_{IS}}{\hat{\sigma}}_{AW}^2} \), which represents the differentiation among the groups. These formulas are equivalent to those developed by Whitlock and Gilbert (2012), except that we included the parameter FIS in order to account for deviation from panmixia within the subpopulation. A generalization of the formula that can be used for any levels of structure can be found in Cubry et al. (2017). The additive genetic variance within subpopulations (\( {\sigma}_{AW}^2 \)) was estimated from the component \( {\sigma}_{FS}^2 \), which represents the genetic variance among families within subpopulations. Because H. speciosa is an alogamous self-incompatible species (Darrault and Schlindwein 2005; Collevatti et al. 2016), it was assumed that progenies correspond to half-sib families, resulting in \( {\hat{\sigma}}_{AW}^2=4{\hat{\sigma}}_{FS}^2 \).
The SSR data from the same progenies that were evaluated in the field for quantitative traits were submitted to descriptive analysis in order to estimate the genetic parameters: number of alleles per locus (A), observed heterozygosity (HO), expected heterozygosity under Hardy–Weinberg equilibrium (He) and maximum expected heterozygosity supposing equal frequencies of alleles per locus (Hm). To infer the genetic structure among varieties and among subpopulations, it was performed a Bayesian clustering simulation to assess the number of discrete genetic clusters using software STRUCTURE 2.3.1 (Pritchard et al. 2000). The number of subpopulations (K) was estimated with ten replicates each for K = 1 to K = 35 using 100,000 iterations of Markov Chain after 100,000 burn-in period iterations using the admixture model. The K value was used to detect the most likely number of clusters (Evanno et al. 2005) using the STRUCTURE HARVESTER program (Earl and VonHold 2012).
It was also performed the analysis of variance of SSR-allele frequencies according to the method described by Weir (1996) in order to estimate the Wright’s F statistics. First, an analysis of the complete set of data was performed using the nested model with two hierarchical levels of population structure and five hierarchical sources of variation (botanical varieties, subpopulations within varieties, families within subpopulations, individuals within families and alleles within individuals). Based on this analysis of variance, the parameters FSG and FGT were estimated in correspondence with parameters QSG and QGT. Additionally, pair-wise analyses of the varieties were performed disregarding the population structures within varieties due to the small number of populations and/or progenies within some subpopulations in some varieties. In these cases, the parameter of interest was the one level \( {F}_{GT}^{\ast } \) among the pairs of varieties to be compared with the corresponding \( {Q}_{GT}^{\ast } \) value. A superscript (*) was used to differentiate these parameters from FGT and QGT because of the pooled nature of the within-variety component of variance in this case. Confidence intervals (95%) for each of the parameters were obtained using a bootstrap procedure across loci with 10,000 replicates. The analyses were performed using GDA software, version 1.1 (Lewis and Zaykin 2001). The parameter FGT corresponds to θP in the output of the GDA analysis. The parameter FSG was obtained using the equation \( {F}_{SG}=\frac{\theta_S-{\theta}_P}{1-{\theta}_P} \) for the sampling estimate and for each replicate of the bootstrap procedure, where θP is a measure of the coancestry at the population level (varieties in this study) and θS is a measure of the coancestry at the subpopulation level (Lewis and Zaykin 2001).
The contrasts QGT − FGT and QSG − FSG were tested for each quantitative trait using a parametric bootstrap procedure that was adapted for two hierarchical levels of structure and for the experimental design used here, from that described by Whitlock and Guillaume (2009) and Gilbert and Whitlock (2015). The simulated values of the components of variance among the varieties and subpopulations within varieties, assuming neutrality of traits, were obtained using the equations \( {\sigma}_{GT(neutral)}^2=\frac{F_{GT}}{1-{F}_{GT}}\left({\sigma}_{SG}^2+\frac{2{\sigma}_{AW}^2}{1+{F}_{IS}}\right) \) and \( {\sigma}_{SG(neutral)}^2=\frac{2{F}_{SG}{\sigma}_{AW}^2}{\left(1-{F}_{SG}\right)\left(1+{F}_{IS}\right)} \). The analysis was performed in Microsoft Excel™ by associating random values from a χ2 distribution with the values of neutral components of variance and the mean squares of the analysis of variance and re-estimating QGT and QSG, assuming selective neutrality. The 10,000 resampled values for each quantitative trait were randomly paired with 10,000 resampled values of FGT and FSG obtained from molecular data using bootstrap over loci. The bootstrap replicates of the F statistics were obtained using GDA software, version 1.1 (Lewis and Zaykin 2001). The values of \( {\ddot{Q}}_{GT}-{\ddot{F}}_{GT} \) and \( {\ddot{Q}}_{SG}-{\ddot{F}}_{SG} \) were used to simulate the null distributions for the contrasts, where ‘¨’ indicates the estimates obtained from each replicate during the bootstrap procedure.
Results
The number of alleles per SSR locus varied from 7 (HS 26) to 37 (HS 27), with an average of 19.1 allele per locus and a total of 115 alleles. The average expected heterozygosity (He) and observed heterozygosity (HO) per locus was equal to 0.852 and 0.559, respectively (Table S2). The observed heterozygosity (HO) was lower than He in all the varieties, which indicates a tendency toward inbreeding due to subdivision and deviations from panmixia, with similar values of FIG among varieties (Table 2). Bayesian clustering showed and optimum of three genetic groups (K = 3, Fig. 2).

Bayesian clustering of individuals and botanical varieties from the Hancornia speciosa Gomes germplasm collection using six microsatellite loci based on STRUCTURE software
The estimates of the F statistics based on the model that assumed two levels of genetic structure (botanical varieties and subpopulations within varieties) revealed a low amount of differentiation among the botanical varieties and a non-significant value for the intergroup fixation index (\( {\hat{F}}_{\mathrm{GT}}=0.0327 \); Table 3). The variation among subpopulations within varieties was highly significant (\( {\hat{F}}_{\mathrm{SG}}={0.2293}^{\ast \ast } \)) and reflected a genetic structuring at this hierarchical level. The mean intra-population fixation index was positive and significant (\( {\hat{F}}_{\mathrm{IS}}={0.1442}^{\ast } \)), which revealed deviation from panmixia within the subpopulations.
When the population structures within the varieties were ignored, the global value of \( {\hat{F}}_{\mathrm{GT}}^{\ast } \) (0.0726*) was higher than that of \( {\hat{F}}_{\mathrm{GT}} \) and differed significantly from zero. The pairwise estimates of \( {\hat{F}}_{\mathrm{GT}}^{\ast } \) ranged from 0.0310 (gardneri vs. speciosa) to 0.1289 (pubescens vs. cuyabensis) and were significant at the 5% probability level for all contrasts except pubescens vs. speciosa and gardneri vs. speciosa (Table 3).
According to the quantitative analysis of variance, there were significant differences among the botanical varieties for all traits, with the exception of plant height in nursery (NPH, Table 4), which demonstrated that the variation in the growth traits in juvenile plants was present at this hierarchical level. The effects of the subpopulations within the botanical varieties tended to be non-significant, with the exception of plant height in nursery conditions. The variances among the families (progenies) within subpopulations and, consequently, the additive genetic variance were significant for nursery stem diameter (NSD), nursery plant height (NPH) and diameter grow rate (GRD) and were not significant for stem diameter in the field (FSD), plant height in the field (FPH) and height grow rate (GRH). The coefficients of heritability at the progeny-within-subpopulations level ranged from 30.7 to 67.1%, and the residual coefficients of variation ranged from 29.6 to 40.1% (Table 4). The varieties cuyabensis and gardneri showed greater values for the means for most traits. The pubescens variety was intermediate in terms of growth, while speciosa exhibited lower growth (Table 4).
The contrasts QGT − FGT were not significant for the NSD and NPH traits in the nursery. In terms of the traits in the field, the contrast QGT − FGT was significant at the 5% level for FSD only. At a relaxed level of significance of 10%, the three other field traits (FPH, GRD, and GRH) exhibited a quantitative divergence among the varieties that was greater than the molecular divergence (Table 4). In the nursery, the values of \( {\hat{Q}}_{\mathrm{GT}} \) were lower for the NSD and NPH traits than in the field, which demonstrated an increase in differentiation among varieties with plant growth. The pair-wise \( {\hat{Q}}_{\mathrm{GT}}^{\ast } \) values showed no apparent correlation with the respective \( {\hat{F}}_{\mathrm{GT}}^{\ast } \) values. The speciosa variety exhibited a higher quantitative differentiation in comparison with the other varieties, while the cuyabensis and gardneri varieties exhibited low differentiation between them (Table 5).
All of the values for the difference \( {\hat{Q}}_{\mathrm{SG}}-{\hat{F}}_{\mathrm{SG}} \) were negative; however, the contrast QSG − FSG was significant at the 5% probability level for only one trait (NSD). If we allowed for a relaxed level of probability (10%), the contrast value for one more trait (GRD) was found to be significant.
Discussion
SSR analysis
The six SSR loci showed high polymorphism with 115 alleles and a mean of 19.1 alleles per locus. This value is higher than those reported by Rodrigues et al. (2015) (A = 8.1) who characterized 34 SSR loci using 35 individuals of H. speciosa and Collevatti et al. (2018) (A = 9.6), who evaluated 777 individuals from 28 subpopulations using seven SSR loci. The high levels of the expected heterozygosity (He) observed per locus (Table S2) and per variety (Table 2) indicate considerable molecular diversity conserved in the germplasm collection. The variety gardneri presented the highest He (0.843) (Table 2), being the variety with the largest number of populations (Table S1). This value is higher than that found by Collevatti et al. (2018) (He = 0.70) for the same variety with a much larger number of individuals per population.
Although the Bayesian clustering analysis indicated the formation of three clusters, the allocation of individuals into each cluster was not clear. These results indicate historical gene flow between varieties of H. speciosa. However, clusters were distributed in all of the sampled areas, although in different proportions. The greater proportion of individuals occurred in the cluster purple, representing the H. speciosa var. gardneri (Fig. 2).
The low values of \( {\hat{F}}_{\mathrm{GT}} \) and high values of \( {\hat{F}}_{\mathrm{SG}} \) found for the molecular data initially appeared to be unexpected. They were not surprising, however, if we consider that each botanical variety can be considered to be a large population that is subdivided into smaller subpopulations. Therefore, the stochastic process of drift may greatly affect differentiation among local finite populations but cause little change in the gene frequencies of the entire variety, which in practice represents an infinite population in the absence of bottleneck events. If the gene flow among the subpopulations is restricted, the differences caused by drift may remain and thereby result in significant values for the inter-population fixation index.
The pair-wise estimates of \( {F}_{\mathrm{GT}}^{\ast } \) showed that the pubescens, gardneri and speciosa varieties tend to be more similar, while the cuyabensis variety was shown to be more differentiated from the others. This variety apparently occupies a more restricted geographical area within the western portion of the biome (Fig. 1) and, consequently, may be more isolated from the other three varieties that were studied. Another hypothesis for the higher degree of differentiation may be that a founder effect or bottleneck affected this variety more than the others. The contrasts for pubescens vs speciosa and gardneri vs speciosa varieties were non-significant at the 5% probability level. The pubescens and gardneri varieties occur in the central region of the biome and are sympatric in some areas, but gardneri occupies a more extensive area. They are botanically differentiated by a single discontinuous morphological trait: the presence of pubescence in leaves and young branches of the former variety (Monachino 1945). Some subpopulations of both varieties are neighbors of speciosa subpopulations. Botanical differentiation among the other three varieties is based on the continuously varied traits leaf size and leaf petiole length, in addition to some flower traits. This has led to uncertainty in the characterization of three subpopulations that were not included in the analysis, two of which are located at the border of the areas of distribution of the gardneri and cuyabensis varieties and one of which was collected at the border of areas occurring gardneri and speciosa varieties. The presence of these intermediate plants suggests the occurrence of gene flow among botanical varieties in overlapping areas.
The significance of the parameter FIS obtained from the analysis based on the two levels model suggests the occurrence of inbreeding due to deviations from panmixia within the subpopulations. Studies of the reproduction system in a population from a coastal area in the northeast region of Brazil (H. speciosa var. speciosa) revealed the occurrence of self-incompatibility (Darrault and Schlindwein 2005). A pollen dispersal study carried out in the same collection used in this work demonstrated that there was no reproductive barrier between the botanical varieties and that there was an absence of self-pollinated seedlings (Collevatti et al. 2016), which corroborated the existence of self-incompatibility at the specie level. Therefore, the occurrence of intra-population inbreeding suggests that non-random mating occurs in natural areas and leads to biparental inbreeding. Evidence of biparental inbreeding has been reported in other studies of H. speciosa (Collevatti et al. 2016; Costa et al. 2017).
Quantitative analysis
In contrast with the molecular analysis, the analysis of variance of the quantitative traits showed a clear genetic differentiation among the botanical varieties for five out the six quantitative traits and a low degree of differentiation among the subpopulations within varieties. This fact suggests that different evolutionary forces have shaped the actual structure of the variation of the quantitative traits. The only significant variation among the subpopulations within varieties, which was found for the plant height in the nursery (NPH), can be inflated by differences in seed vigor among populations within the same variety that can results in variations in seedling development. In this case, some of the differences likely occur due to maternal effects that tend to decrease with plant growth.
The high values for the residual coefficients of variation (29.6 to 40.1%) reflect great variation among plants within families, which was expected since the variance at this level is the result of the accumulation of the environmental variance among plots, 3/4 of the additive genetic variance and the total dominance variance within the subpopulations. The use of only one plant per plot makes it difficult to control this source of variation in the experiment. Since the germplasm collection can be used as a seed orchard in future, the use of single plant in each plot has the function of preventing the crossbreeding between plants of the same family.
In general, the cuyabensis and gardneri varieties exhibited higher means for the evaluated traits. From an agronomic point of view, these botanical varieties can be recommended as the most promising under the conditions of this experimental area (Ganga et al. 2009; Almeida et al. 2019).
Quantitative vs. molecular divergence
Based on the hypothesis that FGT measures the neutral variation among the botanical varieties, the high estimates of QGT observed here for most traits suggest that natural selection plays a role in molding the structural pattern of quantitative genetic variation among the varieties in terms of juvenile growth traits. The geographical distribution of the sampled subpopulations shows that the botanical varieties occur from west/southwest to northeast of the biome approximately in the following order: cuyabensis, gardneri, pubescens, and speciosa. During collection mission, we observed that cuyabensis occurs predominantly in latosols, which comprise a class of deep soils with fertility that is greater than average soils of the Cerrado biome, while gardneri is the most common variety in the southwest and central region of the biome and occurs in different classes of soils. Pubescens also occurs in the central region of the Cerrado, but at a low frequency, and occurs predominantly in plinthosols and cambisols, which are soil classes that are more limited in their fertility and water retention. The speciosa variety occurs predominantly in sandy soils at the northeast region of the biome. The rainfall intensity decreases from the west/southwest to the northeast, which affects the mean growth of the varieties in common garden conditions, which is consistent with the environment of origin of each variety.
The non-significance of the QGT − FGT contrast for the nursery variables (NSD and NPH) indicates that there is no evidence that selection forces shaped the divergence among the varieties in terms of seedling growth traits. Seedlings from trees from the Cerrado biome in general, and H. speciosa in particular, direct more energy to the development of the root system than that to aerial structures (Rosa et al. 2005). This is important for seedling establishment during the rainy season and survival during the next dry season, which is typical of the biome. Therefore, there is no apparent reason for the occurrence of divergent selection at this stage.
The positive and high magnitude differences between the QGT and FGT estimates for the field variables suggest that divergent selection is shaping the variation among the varieties. The use of the 10% level of significance in addition to the usual 5% level is justified by the low power of the statistic test used due to the intrinsic nature of the errors that is associated with the estimates. In this case, the components of variance used as numerator of the estimation formula, for both QGT and FGT, were estimated from the mean squares associated with three degrees of freedom only. For a more detailed description of the QST/FST comparison, see Whitlock (2008) and Whitlock and Guillaume (2009). Our results reinforce the need for caution when designing conservation strategies based only on the use of molecular neutral markers, particularly when the subpopulations exhibit low levels of differentiation. When the FST value is low, virtually any value can be obtained for QST estimate (Leinonen et al. 2008).
In contrast with the inter-variety level, the non-significance for the difference QSG − FSG for most traits reflects a pattern of variation that is compatible with differentiation caused by genetic drift and no evidence of divergent selection among the subpopulations within varieties. The negative estimates for the contrast QSG − FSG, which were shown to be significant at the 5% level for one trait and at 10% for an additional trait, suggest that the hypothesis of uniform selection within each variety for some traits is coherent. The estimates of the Q statistics are affected downward by dominance effects. Therefore, lower values of QSG must be considered with caution when making inferences about selection (Cubry et al. 2017).
Initial development is an important aspect of plant establishment in the field. Therefore, the presence of uniform selection within the more uniform areas of occurrence in each species appears to be congruent with expected for adaptive juvenile traits. Similar results for the comparison QST − FST have been verified for Eugenia dysenterica DC., which is another fruit tree that is native to the Brazilian Cerrado (Boaventura-Novaes et al. 2018).
In nested models used to study natural populations, random effects are usually assumed to stem from infinite populations at each hierarchical level. In some instances, however, the number of groups or subpopulations would be finite. This is the case for the botanical varieties in the present study, which are clearly finite in nature. A general method for transforming the mean squares expectation from that used for infinite to that used for finite models was described by Searle and Fawcett (1970). In the case of a nested model, the variance component at each level is affected by the finiteness of the level nested immediately within it. Therefore, when only the higher level of the hierarchical model is finite, as in this case, the expectations of the mean squares are the same as those used in infinite models. This principle applies to the estimation of both FGT and QGT.
In conclusion, our results suggest that divergent selection is a factor that shapes differentiation among botanical varieties of H. speciosa for some juvenile growing traits, while differentiation among the subpopulations within varieties is shaped mostly by genetic drift or uniform selection.
References
Almeida GQ, Vieira MC, Ganga RMD, Chaves LJ (2019) Agronomic evaluation of a Hancornia speciosa Gomes germplasm collection from the Brazilian Cerrado. Crop Breed Appl Biotechnology:19 (In press)
Boaventura-Novaes CRD, Novaes E, Mota EES, Telles MPC, Coelho ASG, Chaves LJ (2018) Genetic drift and uniform selection shape evolution of most traits in Eugenia dysenterica DC. (Myrtaceae). Tree Genet Genomes 14:76. https://doi.org/10.1007/s11295-018-1289-2
Bonnin I, Prosperi J, Olivierit I (1996) Genetic markers and quantitative genetic variation in Medicago truncutula (Leguminosae): a comparative analysis of population structure. Genetics 143:1795–1805
Collevatti RG, Olivatti AM, Telles MPC, Chaves LJ (2016) Gene flow among Hancornia speciosa (Apocynaceae) varieties and hybrid fitness. Tree Genet Genomes 12:74–85. https://doi.org/10.1007/s11295-016-1031-x
Collevatti RG, Rodrigues EE, Vitorino LC, Lima-Ribeiro MS, Chaves LJ, Telles MPC (2018) Unravelling the genetic differentiation among varieties of the Neotropical savanna tree Hancornia speciosa Gomes. Ann Bot 122:973–984. https://doi.org/10.1093/aob/mcy060
Costa CF, Collevatti RG, Chaves LJ, Lima JS, Soares TN, Telles MPC (2017) Genetic diversity and fine-scale genetic structure in Hancornia speciosa Gomes (Apocynaceae). Biochem Syst Ecol 72:63–67. https://doi.org/10.1016/j.bse.2017.03.001
Creste S, Tulmann-Neto A, Figueira A (2001) Detection of single sequence repeat polymorphism in denaturing polyacrylamide sequencing gels by silver staining. Plant Mol Biol Rep 19:299–306
Cubry P, Scotti I, Oddou-Muratorio S, Lefevre F (2017) Generalization of the QST framework in hierarchically structured populations: impacts of inbreeding and dominance. Mol Ecol Resour 17:76–83. https://doi.org/10.1111/1755-0998.12693
Darrault RO, Schlindwein C (2005) Limited fruit production in Hancornia speciosa (Apocynaceae) and pollination by nocturnal and diurnal insects. Biotropica 37:381–388. https://doi.org/10.1111/j.1744-7429.2005.00050.x
Diniz-Filho JAF, Barbosa ACOF, Chaves LJ, Souza KS, Dobrovolski R, Rattis L, Terribile LC, Lima-Ribeiro MS, Oliveira G, Brum FT, Loyola R, Telles MPC (2018) Overcoming the worst of both worlds: integrating climate change and habitat loss into spatial conservation planning of genetic diversity in the Brazilian Cerrado. Biodivers Conserv 29:1–16. https://doi.org/10.1007/s10531-018-1667-y
Doyle JJ, Doyle JL (1990) Isolation of plant DNA from fresh tissue. Focus 12:13–15
Earl DA, VonHold BM (2012) STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv Genet Resour 4:359–361. https://doi.org/10.1007/s12686-011-9548-7
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14:2611–2620. https://doi.org/10.1111/j.1365-294X.2005.02553.x
Flores IS, Silva AK, Furquim LC, Castro CFS, Chaves LJ, Collevatti RG, Lião LM (2018) HR-MAS NMR Allied to chemometric on Hancornia speciosa varieties differentiation. J Braz Chem Soc 29:708–714. https://doi.org/10.21577/0103-5053.20170191
Ganga RMD, Chaves LJ, Naves RV (2009) Parâmetros genéticos em progênies de Hancornia speciosa Gomes do Cerrado. Sci For 37:395–404
Ganga RMD, Ferreira GA, Chaves LJ, Naves RV, Nascimento JL (2010) Caracterização de frutos e árvores de populações naturais de Hancornia speciosa Gomes do cerrado. Rev Bras Frutic 32:101–113. https://doi.org/10.1590/S0100-29452010005000019
Gilbert KJ, Whitlock MC (2015) QST - FST comparisons with unbalanced half-sib designs. Mol Ecol Resour 15:262–267. https://doi.org/10.1111/1755-0998.12303
Goudet J, Buchi L (2006) The effects of dominance, regular inbreeding and sampling design on QST, an estimator of population differentiation for quantitative traits. Genetics 172:1337–1347. https://doi.org/10.1534/genetics.105.050583
Jimenez HJ, Martins LSS, Montarroyos AVV, Silva-Junior JF, Alzate-Marin AL, Moraes-Filho RM (2015) Genetic diversity of the Neotropical tree Hancornia speciosa Gomes in natural populations in Northeastern Brazil. Genet Mol Res 14:17749–17757. https://doi.org/10.4238/2015.December.21.48
Leinonen T, McCairns RJS, O’Hara RB, Merilä J (2013) QST–FST comparisons: evolutionary and ecological insights from genomic heterogeneity. Nat Rev Genet 14:179–190. https://doi.org/10.1038/nrg3395
Leinonen T, O’Hara RB, Cano JM, Merilä J (2008) Comparative studies of quantitative trait and neutral marker divergence: a meta-analysis. J Evol Biol 21:1–17. https://doi.org/10.1111/j.1420-9101.2007.01445.x
Lewis PO, Zaykin D (2001) Genetic data analysis: computer program for the analysis of allelic data (software). http://lewis.eeb.uconn.edu/lewishome/software.html. Accessed 16 march 2019
McKay JK, Latta RG (2002) Adaptive population divergence: markers, QTL and traits. Trends Ecol Evol 17:285–291. https://doi.org/10.1016/S0169-5347(02)02478-3
Mendonça RC, Felfili JM, Walter BMT, Silva-Junior MC, Rezende AV, Filgueiras TS, Nogueira PE, Fagg CW (2008) Flora vascular do bioma Cerrado: Checklist com 12.356 spécies. In: Sano SM, Almeida SP, Ribeiro JF (eds) Cerrado: ecologia e flora. Embrapa, Brasília, pp 421–442
Merilä J, Crnokrak P (2001) Comparison of genetic differentiation at marker loci and quantitative traits. J Evol Biol 14:892–903. https://doi.org/10.1046/j.1420-9101.2001.00348.x
Monachino J (1945) A revision of Hancornia (Apocynaceae). Lilloa, Tucumán 11:19–48
Myers N, Mittermeier RA, Mittermeier CG, Fonseca GAB, Kent J (2000) Biodiversity hotspots for conservation priorities. Nature 403:853–858. https://doi.org/10.1038/35002501
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959. https://doi.org/10.1111/j.1471-8286.2007.01758.x
Rodrigues AJL, Yamaguishi AT, Chaves LJ, Coelho ASG, Lima JS, Telles MPC (2015) Development of microsatellite markers for Hancornia speciosa Gomes (Apocynaceae). Genet Mol Res 14:7274–7278. https://doi.org/10.4238/2015.July.3.2
Rosa MEC, Naves RV, Oliveira-Junior JP (2005) Substrates for production and growth of mangaba (Hancornia speciosa Gomes) seedlings. Pesqui Agropecuária Trop 35:65–70
Santos PS, Freitas LS, Santana JGS, Muniz EM, Rabbani ARC, Silva AVC (2017) Genetic diversity and the quality of Mangabeira tree fruits (Hancornia speciosa Gomes—Apocynaceae), a native species from Brazil. Sci Hortic (Amsterdam) 226:372–378. https://doi.org/10.1016/j.scienta.2017.09.008
Searle SR, Fawcett RF (1970) Expected mean squares in variance components models having finite populations. Biometrics 26:243–254
Silva-Junior JF, Lédo AS (2006) Botânica. In: Silva-Junior JF, Lédo AS (eds) A cultura da Mangaba. Embrapa, Brasília, pp 26–33
Spitze K (1993) Population structure in Daphnia obtusa: quantitative genetic and allozymic variation. Genetics 135:367–374
Vencovsky R, Crossa J (2003) Measurements of representativeness used in genetic resources conservation and plant breeding. Crop Sci 43:1912–1921. https://doi.org/10.2135/cropsci2003.1912
Volis S, Yakubov B, Shulgina I, Ward D, Mendlinger S (2005) Distinguishing adaptive from nonadaptive genetic differentiation: comparison of QST and FST at two spatial scales. Heredity (Edinb) 95:466–475. https://doi.org/10.1038/sj.hdy.6800745
Weir BS (1996) Genetic data analysis II. Methods for discrete population genetic data. Sinauer, Sunderland
Whitlock MC (2008) Evolutionary inference from QST. Mol Ecol 17:1885–1896. https://doi.org/10.1111/j.1365-294X.2008.03712.x
Whitlock MC, Gilbert KJ (2012) QST in a hierarchically structured population. Mol Ecol Resour 12:481–483. https://doi.org/10.1111/j.1755-0998.2012.03122.x
Whitlock MC, Guillaume F (2009) Testing for spatially divergent selection: comparing QST to FST. Genetics 183:1055–1063. https://doi.org/10.1534/genetics.108.099812
Wright S (1951) The genetical structure of populations. Ann Eugenics 15:323–354
Wright S (1969) Evolution and the genetics of population. In: The theory of gene frequencies, vol 2. University of Chicago Press, Chicago
Acknowledgments
This paper is dedicated to Professor Roland Vencovsky (in memoriam) who inspired and encouraged us in quantitative and population genetic studies of wild plant species.
Our research has been supported by the project CERGEN (PRONEX/FAPEG/CNPq, Proc. 201210267000802). L.J. Chaves has been continuously supported by productivity grants from the National Council for Scientific and Technological Development, CNPq, Brazil.
We are very grateful to two anonymous reviewers who have greatly contributed to the improvement of this paper.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no conflicts of interest.
Data archiving statement
All data provided as supplementary material are available in the online version of this article.
Additional information
Communicated by L.A. Meisel
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
ESM 1
(DOCX 35 kb)
Rights and permissions
About this article

Cite this article
Chaves, L.J., Ganga, R.M.D., Guimarães, R.A. et al. Quantitative and molecular genetic variation among botanical varieties and subpopulations of Hancornia speciosa Gomes (Apocynaceae). Tree Genetics & Genomes 16, 50 (2020). https://doi.org/10.1007/s11295-020-01444-0
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11295-020-01444-0