
Abstract
Kersting’s groundnut (Macrotyloma geocarpum (Harms) Maréchal & Baudet) is an important source of protein and essential nutrients that contribute to food security in West Africa. However, the crop is still underexploited by the populations and under-researched by the scientific community. This study aimed to investigate the genetic diversity and population structure of 217 Kersting’s groundnut accessions from five origins using 886 DArTseq markers. Gene diversity was low and ranged from 0.049 to 0.064. The number of private alleles greatly varied among populations (42–192) and morphotypes (40–339). Moderate to very high levels of selfing and inbreeding were observed among populations (s = 56–85%, FIS = 0.389–0.736) and morphotypes (s = 57–82%, FIS = 0.400–0.691). Moreover, little to very high genetic differentiations were observed among populations (0.006 ≤ FIS ≤ 0.371) and morphotypes (0.029 ≤ FIS ≤ 0.307). Analysis of molecular variance partitioned 38.5% of the genetic variation among and 48.7% within populations (P < 0.001). Significant isolation by distance was detected between populations (R2 = 0.612, P = 0.011) and accessions (R2 = 0.499, P < 0.001). Discriminant analysis of principal components and neighbour joining consistently distinguished eight distinct clusters. These data provide a global picture of the existing genetic diversity for Kersting’s groundnut and will guide the choice of breeding strategies to increase production.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Kersting’s groundnut (Macrotyloma geocarpum (Harms) Maréchal & Baudet) is a geocarpic orphan legume crop grown in the semi-arid regions of West Africa. It is a diploid (2n = 22) species and likely originated from an area extending between central Benin and northern Togo (Achigan Dako and Vodouhè 2006), though a putative wild form M. geocarpum var. tisserantii (2n = 20) was found in northern Cameroon (Hepper 1963). Kersting’s groundnut is mainly grown for its grains which contain significant amounts of protein, essential amino acids, minerals such as calcium (183.25 mg/100 g), magnesium (1.46 mg/100 g), zinc (25.22 mg/100 g), phosphorus (345.45 mg/100 g) (Chikwendu 2015), sodium (23.38 mg/100 g) and potassium (520.93 mg/100 g) (Echendu et al. 2009). The grains also contain different vitamins including vitamin A (29.60 mg/100 g), B1 (2.86 mg/100 g) and B2 (0.22 mg/100 g), and are an excellent source of carbohydrate (Chikwendu 2015). Besides, the crop has the potential to restore soil fertility through atmospheric N2-fixation in root nodules (Jaiswal et al. 2019). This ranks Kersting’s groundnut among the crops that improve productivity in agroecosystems (Varshney et al. 2009). In Benin, Kersting’s groundnut is ranked as the highest priority and the most valuable orphan legume crop due to its substantial contribution to food security and income for resource-poor rural households (Dansi et al. 2012; Agoyi et al. 2019a). Despite these attributes, cultivation of Kersting’s groundnut is gradually being abandoned in the growing countries (Amujoyegbe et al. 2007; Adu-Gyamfi et al. 2011; Tamini 1997; Bampuori 2007; Mergeai 1993), especially in Benin (Assogba et al. 2015). Low yield, small grain size and lack of improved cultivars are among the commonly reported factors responsible for its reduced cultivation (Akohoue et al. 2018; Assogba et al. 2015; Kafoutchoni et al. 2021) and its anticipated extinction (Jaiswal et al. 2019; Adu-Gyamfi et al. 2011). Thus, it is urgent to develop new Kersting’s groundnut varieties with improved agronomic performances for boosted production.
Successful development of improved cultivars requires variations within germplasm (Govindaraj et al. 2015; Ajala et al. 2019). Assessment of genetic diversity and relatedness among accessions and populations is therefore fundamental to the success of any breeding program. Knowledge of this is crucial in the selection of parental lines and the identification of genotypes that harbour valuable alleles for breeding targets (Chen et al. 2017). Moreover, characterisation of genetic diversity is important for effective management of germplasm and development of core collection of genotypes representing maximum diversity in a minimum number of accessions (Kumar et al. 2016). However, genetic diversity in Kersting’s groundnut is poorly documented.
To date, most of the investigations on genetic diversity in Kersting’s groundnut were based on agromorphological traits (Akohoue et al. 2019; Mensah et al. 2016; Adu-Gyamfi et al. 2012; Assogba et al. 2015; Bayorbor et al. 2010). Although germplasm characterisation using agromorphological traits is frequently used by breeders in sub-Saharan Africa (Ajala et al. 2019), this approach also has many limitations. It is time consuming and prone to environmental effects compared to molecular characterisation (Singh et al. 2018). The first attempt to investigate the genetic diversity in Kersting’s groundnut using molecular markers used only 19 isozymes and was based on 20 accessions (Pasquet et al. 2002), thus limiting its power to depict the actual diversity in the crop. Isozyme markers are few in number, detect less polymorphism, have limited reproducibility and are known to be affected by the extraction method, plant tissues and plant growth stages, and can therefore produce unreliable diversity estimates (Govindaraj et al. 2015; Mondini et al. 2009). To understand the real genetic variation within the crop, a marker system that produces a lot more markers should be used in a large panel of accessions. The high throughput genotyping approach Genotyping-by-Sequencing (GBS), that reduces complexity in the genome to partially sequence the crop genome, has shown significant success in many annual legume crops including soybean (Agoyi et al. 2019b; Iquira et al. 2015; Jarquín et al. 2014), cowpea (Xiong et al. 2016; Fatokun et al. 2018) and Common beans (Schröder et al. 2016; Campa et al. 2018; Cortés and Blair 2018). DArTseq technology (Diversity Array Technology), is based on the GBS principle to generate thousands of SNP markers. It works well with many crops including neglected crops without reference genome (Kilian et al. 2012). DArTseq based markers have been successfully used to unfold genetic diversity patterns in various crops including watermelon (Yang et al. 2016), garlic (Egea et al. 2017), rice (Ndjiondjop et al. 2017), wheat (Robbana et al. 2019), and Bambara groundnut (Redjeki et al. 2020). Recently, Akohoue et al. (2020) used 493 DArTseq SNP markers on a collection of 281 Kersting’s groundnut accessions and found low genetic diversity influenced by the geographic origins of the accessions. Nevertheless, the study did not include accessions from all known geographical origins of the crop, which may affect the level of reported genetic diversity. The present study was undertaken to investigate the level of genetic diversity among a regional collection of 227 Kersting’s groundnut accessions from five west African countries and to determine population structure within the germplasm.
Materials and methods
Plant materials
A total of 227 Kersting’s groundnut accessions from five West African countries were used in this study (Table S1). Among them, 112 were from Benin, 48 from Burkina Faso, 36 from Ghana, 12 from Nigeria and 19 from Togo (Fig. 1). The accessions were all landraces collected directly from farmers or provided by the national agricultural research centre in each country. Details of the origin, characteristics and donor of each accession can be found on supplementary Table S1. The accessions were composed of 47% (102) cream seed, 22.5% (49) black seed, 13.8% (30) cream seed with black eye, 10.6% (23) brown mottled seed, and 6% (13) brown seed (Table S1). Hereafter, each seed colour was considered as a morphotype.

Map showing collection sites of Kersting’s groundnut accessions used in this study
Methods
DNA extraction
Each of the 227 accessions was planted in the field in single row plots of 5 m length. Young leaves were collected from 3-week-old plants into 96 deep well sample collection kits and sent to Biosciences Eastern and Central Africa (BecA-ILRI) Hub in Nairobi for Genotyping. DNA was isolated and purified using the NucleoMag® 96 Plant genomic DNA extraction kit (Macherey–Nagel, Düren, Germany), following the manufacturer’s instructions. Concentration of the extracted DNA was normalised within the range of 50–100 ng/µl. The quality and quantity of the DNA samples were checked on 0.8% agarose.
Library construction and genotyping by sequencing
Libraries were constructed following the protocol described in (Kilian et al. 2012). Two DArTseq™ complexity reduction methods were tested using the rare cutting restriction endonuclease enzyme PstI (5′-CTGCA|G-3′) in combination with two different frequently cutting restriction enzymes HpaII (5′-C|CGG-3′) and MseI (5′-T|TAA-3′). The PstI/MseI combination was selected as the best performing method. For each sample, 2 µL of DNA was digested with the PstI/MseI restriction enzyme combination. Digestion products were ligated to barcoded adapters pair annealed to the two restriction enzyme overhangs. The PstI-compatible adapter included the partial attachment sequence for the ‘Read 1 End’ of the Illumina flow cell, a barcode of variable length (4–8 bp) and the PstI-compatible overhang sequence. The reverse adapter included the partial sequence for the ‘Read 2 End’ of the Illumina flow cell and MseI-compatible overhang sequence. The adapter-ligated fragments were amplified in a Polymerase Chain Reaction (PCR) using optimized settings. After PCR, equimolar amounts of the amplified products from each sample were pooled together using a TECAN robotic liquid handler (TECAN, Männedorf, Switzerland) and loaded on the cBot (Illumina, Inc., San Diego, CA, USA) for clustering on a flow cell. Libraries were sequenced in the Illumina Hiseq 2500 using Single Read sequencing runs for 77 bases.
The IGSS platform uses a high-throughput genotyping-by-sequencing (GBS) DArTseq technology (DArT Pty Ltd, Canberra, Australia). DArTseq markers scoring was achieved using DArTsoft14 a proprietary marker scoring pipeline based on algorithms. SNP markers were scored as “0” (reference allele homozygote), “1” (alternate allele homozygote), “2” (heterozygote) and “-” (fragment missing in representation). As Kersting’s groundnut had no available reference genome, SNP markers were aligned to the reference genomes of Phaseolus vulgarius (ftp://ftp.ensemblgenomes.org/pub/plants/release-44/fasta/phaseolus_vulgaris/dna/), Vigna angularis (ftp://ftp.ensemblgenomes.org/pub/plants/release-44/fasta/vigna_angularis/dna/), Vigna radiata (ftp://ftp.ensemblgenomes.org/pub/plants/release-44/fasta/vigna_radiata/dna/) and Vigna subterranea (http://gigadb.org/dataset/view/id/101055) which are related species (Wojciechowski et al. 2004).
Quality filtering, imputation and markers diversity
SNP markers were imputed using the Diversity Arrays Technology’s KDCompute Optimal Imputation plugin (https://kdcompute.igss-africa.org/kdcompute/login). Seven imputation methods (MissForest, Random, Nipals, singular value decomposition (SVD), Expectation–Maximization (EM), k-nearest neighbour (kNN), and probabilistic principal component analysis (PPCA) were tested based on simple matching coefficient (SMC), calculated by excluding an additional 10% of missing values and evaluating the correlation between introduced missing values and the original dataset. Missing calls were imputed using the most accurate imputation method for which the highest SMC was recorded.
The pre-imputation SNP dataset was filtered for various quality control metrics, and markers that passed thresholds were retained for analysis. Various iterative filtering schemes were applied as recommended by O’Leary et al. (O'Leary et al. 2018) and the one that resulted in retaining more loci and individuals without compromising the markers quality was retained (data not shown). Consequently, only loci with < 50% missing data, > 5 read depth, > 75% call rate, > 95% reproducibility and > 0 polymorphic information content (PIC) were retained. In addition, accessions with > 50% missing SNPs were removed from downstream analyses. This narrowed down to 217 accessions that were retained for further analyses. SNP filtering was carried out in R 4.0.0 (R Core Team 2020).
Summary statistics (minimum, maximum and mean) were calculated for the remaining SNP markers for the following quality parameters: call rate, read depth, PIC, heterozygosity, minor allele frequency and reproducibility. The number of markers successfully aligned were graphed per reference genome.
Analysis of genetic variation and population structure.
The average number of alleles (Na), number of effective alleles (Ne), observed heterozygosity (Ho), expected heterozygosity (He), unbiased expected heterozygosity (uHe), the inbreeding coefficient (FIS) and the percentage of polymorphic loci (%P) were calculated by geographic population and by morphotype, using GenAlEx version 6.51b2 (Peakall and Smouse 2012). For this purpose, the 112 accessions from Benin were assigned to three putative populations (Atacora, Collines, Zou) while those from the other countries were considered belonging to a single population each. Average allelic richness (Ar), and the number of private alleles (Np) were calculated in the ‘PopGenReport’ (Adamack and Gruber 2014) R package. Moreover, selfing rate (s) was determined using the equation described in Ritland (1990): s = 2FIS/(1 + FIS). Nei’s pairwise FST was computed between all pairs of populations and morphotypes in the ‘hierfstat’ R package (Goudet and Jombart 2015). The FST statistics measures the overall level of genetic divergence among subpopulations. According to Wright (Wright 1978), a FST < 0.05 indicates little genetic differentiation, values comprised between 0.05 and 0.15 indicate moderate genetic differentiation, while FST values between 0.15 and 0.25 denotes great differentiation. FST > 0.25 indicates very great genetic differentiation (Wright 1978). Furthermore, analysis of molecular variance (AMOVA) was performed to determine the level of partitioning of diversity within and among populations and morphotypes. AMOVA were performed in the ‘poppr’ R package (Kamvar et al. 2014) using 999 permutations, while significance levels were estimated for variance components based on 9,999 permutations using the randtest function in the ‘ade4′ R package (Dray and Dufour 2007).
Population structure was inferred using a Discriminant Analysis of Principal Components (DAPC) performed in the ‘adegenet’ R package (Jombart 2008). The most probable number of clusters in the data was inferred using the find.clusters function from ‘adegenet’, running successive K-means and with the Bayesian Information Criterion (BIC) used as the statistical measure of goodness of fit. The number of principal components (PCs) to retain in the DAPC analysis was determined using a cross-validation procedure implemented with the xvalDapc function available in the ‘adegenet’ package, using the default parameters. Furthermore, an unrooted neighbour-joining tree was constructed in the package ‘poppr’ based on Edward’s genetic distance. Node strength in the dendrograms was tested using 1,000 bootstrap analyses in ‘poppr’ package.
Finally, isolation by distance (IBD) was tested using a Mantel test correlating genetic distance [FST/(1−FST)] and geographic distances among populations, and morphotypes. The correlation between Nei’s genetic distance (Nei 1973) and geographic distance was also examined among accessions using Mantel test. The Mantel test was performed with 999,999 permutations using the function mantel.randtest available in ‘ade4′. The genetic versus geographic distances were plotted along with a point density of accessions to visualise whether IBD existed as a continuous gradient or resulted from patches of distant and divergent individuals.
Results
Marker quality
The DArTseq genotyping of the panel of 227 Kersting’s groundnut generated a total of 1938 Single Nucleotide Polymorphism (SNP) markers of which 391 (20%) were aligned with the reference genome of Common bean (Phaseolus vulgaris), 279 (14%) with Adzuki bean (Vigna angularis), 279 (14%) with Mung bean (Vigna radiata), and 349 (18%) with Bambara groundnut (Vigna subterranea). Initially, the markers contained 20.23% missing data. The percentage of missing data was reduced to 10.4% after filtering. The probabilistic principal components analysis (PPCA) imputation method that had the highest performance, with an accuracy of 0.97 SMC (Simple matching coefficient), was used to impute the missing SNPs. After applying the various filtering criteria, 1052 SNPs (54.28%) were filtered out and the remaining 886 markers were used for further analyses (Table S2). Ten accessions had over 50% missing SNPs and were removed from the final dataset. All the filtered markers had 100% call rate and an average of 99.8% reproducibility. Read depth ranged between 5 and 140 with a mean across loci of 21.71. The distribution of read depths showed that most markers had read depth between 5 and 15. The mean PIC of the reference alleles (0.064) was greater than that of the SNP alleles (0.054). The average PIC ranged from 0.004 to 0.499 with a mean value of 0.059. About 83% of the SNP markers (738) had PIC below 0.05. Only 16.7% (148 SNPs) scored greater than 5% PIC. The minor allele frequency (MAF) varied from 0.002 to 0.495 with a mean of 0.044. About 12% (123) of markers had minor allele frequency greater than 0.05. Furthermore, mean expected and observed heterozygosity were estimated at 0.06 (0.010–0.500) and 0.008 (0–0.793) respectively. The total gene diversity (HT) across SNPs ranged from 0.002 to 0.501, with a mean HT of 0.057 (Table S3).
Genetic diversity analysis based on geographical populations and morphotypes
The genetic diversity parameters estimated for each geographical population are summarised in Table 1. Number of different alleles (Na), number of effective alleles (Ne) and allelic richness (Ar) varied slightly among the populations. Na ranged from 1.19 (Nigeria) to 1.54 (Zou). The number of effective alleles was nearly the same for all populations. The mean Ne across populations (1.10) was lower than mean Na (1.30) suggesting that only a few alleles contributed to the variation. Ar ranged between 1.12 (Nigeria) and 1.20 (Ghana) with a mean value of 1.17. Private alleles were found in all populations with Zou possessing the highest number of private alleles (Np = 192) whereas Nigeria had the lowest number (Np = 42). The accessions with the greater number of private alleles were Gbo5, Lili*Sie, and Itk from Zou, Ghana and Collines populations, respectively (Table S4). In general, the observed heterozygosity (Ho), expected heterozygosity (He) and unbiased expected heterozygosity (uHe) significantly varied (P < 0.001) among populations. Ho values were approximately 6–7 times lower than He and uHe. Ho ranged between 0.69 × 10–2 and 1.02 × 10–2, with an average of 0.79 × 10–2 across populations. The more diverse populations were from Atacora (He = 6.12 × 10–2, uHe = 6.24 × 10–2) followed by Zou population (He = 6.07 × 10–2, uHe = 6.12 × 10–2), whereas Ghana’s population was the least diverse (He = 5.32 × 10–2, uHe = 5.40 × 10–2). The inbreeding coefficient within populations varied from moderate in Zou (FIS = 0.39) to very high in Collines (FIS = 0.74) and Nigeria (FIS = 0.74). The selfing rate greatly varied among populations. It ranged between s = 0.56 for Zou population and s = 0.85 for Collines and Nigeria populations. The across populations mean was s = 0.72, indicating a considerable level of selfing. Selfing rates was proportional to inbreeding coefficients and inversely proportional to polymorphism within populations (Table 1). Percentage of polymorphism ranged from 18.51% (Nigeria) to 53.50% (Zou) with an average of 39.39% indicating a moderate level of polymorphism and variation within geographical populations.
The estimated genetic diversity parameters also varied among morphotypes as shown in Table 2. The number of alleles (Na) varied between 1.17 in brown seed to 1.59 in cream seed, with an average of 1.35 across morphotypes. The average number of effective alleles across morphotypes was 1.09. The highest allelic richness (1.22) was found in the black morphotype. The number of private alleles varied greatly among morphotypes with cream seed possessing the highest number of private alleles (Np = 339) and brown seed the lowest (Np = 40). Genetic diversity was low but significantly (P < 0.001) varied among morphotypes (Table 2). The more diverse morphotype were cream seed with black eye (He = 6.29 × 10–2, uHe = 6.40 × 10–2) and black (He = 6.18 × 10–2, uHe = 6.25 × 10–2), whereas the least diverse one was brown seed (He = 4.69 × 10–2, uHe = 4.88 × 10–2). Consistent with that, high levels of inbreeding (FIS = 0.69) and selfing (s = 0.82) were observed within brown seed, while the lowest levels were found in cream with black eye seed (FIS = 0.40, s = 0.57). The average percentage of polymorphism was 35.40% suggesting a moderate level of variation within morphotypes. The percentage of polymorphism was lowest within brown seed (17.27%) and highest in cream seed (59.37%).
Genetic differentiation across geographic populations and morphotypes
Genetic differentiation of geographic populations (Table 3) and morphotypes (Table 4) were examined based on FST. The FST coefficients among populations varied from 0.006 to 0.371, indicating little to very great differentiation. The highest level of differentiation was observed between Burkina and Zou populations. The lowest FST was observed between Collines and Zou. A very low genetic differentiation was also found between Atacora and Zou (FST = 0.008), and between Atacora and Collines (FST = 0.017). Therefore, individuals between populations from Benin (Atacora, Collines, Zou) were closely related (FST < 0.05). Equally low genetic differentiation was also observed between populations from Benin and Nigeria (0.033 < FST < 0.065) (Table 3). Low to very high differentiation was also observed between morphotypes (Table 4). The lowest differentiation was found between cream and brown (FST = 0.029) while differentiation was the highest between brown mottled and brown morphotypes (FST = 0.307). A very high differentiation was also found between brown mottled and cream morphotypes (FST = 0.302). The genetic distance between cream and cream with black eye morphotypes was also relatively high (FST = 0.265).
The AMOVA of 217 accessions from seven populations revealed that 38.53% of the total variation was among populations (FST = 0.385) and 48.72% within populations (FIS = 0.793), while the rest of variation (12.74%) was within accessions (FIT = 0.873; Table 5). All values were statistically significant (P < 0.001). The two-level AMOVA across morphotypes showed significant levels of genetic variation within (49.13%) and among morphotypes (38.73%), with 12.14% of the variation retained within accessions (Table 6). The overall fixation (FST) value was equal to 0.387 among morphotypes. There was homozygote excess as revealed by the high overall inbreeding coefficient (FIS = 0.802). The three-level AMOVA (Table 6) partitioned 22.10% of the genetic variation among the seven geographic populations and about 25% of variation among morphotypes. In addition, 40.39% of the total variation was observed within morphotypes, whereas the remaining 12.72% was found within individuals. All the values were statistically significant (P < 0.001) except at population level (P = 0.097).
Population structure
A discriminant analysis of principal components (DAPC) was used to infer population structure of Kersting’s groundnut. The optimum number of clusters inferred using the find.clusters function was eight (Fig. 2a, c). DAPC analysis was performed using the inferred number of clusters. Based on DAPC cross-validation analysis, the 60 first PCs (95% of variance conserved) of PCA (Fig. 2b) and 7 discriminant functions were retained. Clusters 1 to 8 consisted of 10, 22, 13, 98, 22, 17, 24 and 11 accessions, respectively. Cluster 1 was predominantly composed of accessions from Burkina Faso, with one accession from Atacora. Cluster 2 contained the majority (52.6%) of accessions from Togo, about 7.5% from Zou, with a few accessions from Nigeria, Burkina Faso, Collines and one accession from Atacora. Almost all the remaining accessions from Atacora possessed 80% membership in Cluster 4 which also included the majority of accessions from Zou, Nigeria and Collines (80.3%, 83.3% and 84.6% respectively). Cluster 3 contained mainly accessions from Ghana and Burkina Faso, whereas Cluster 5 included one third of the accessions from Burkina Faso and about 15% from Ghana. Clusters 6 and 7 were predominantly composed of accessions from Burkina Faso and the majority (~ 60%) of the accessions from Ghana, respectively (Table 7). Cluster 8 contained almost 37% of the accessions from Togo, and the remaining accessions from Atacora and Zou.

Discriminant analysis of principal components (DAPC) for 217 Kersting’s groundnut accessions using 886 SNPs. a Statistical determination of the optimum number of clusters, b Number of retained DAPC principal components, c Scatter plot from the DAPC analysis
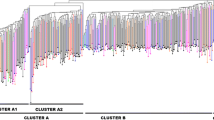
To obtain a more complete picture of the relationships among the 217 Kersting’s groundnut accessions a neighbour joining (NJ) tree was constructed based on Edward’s distance (Fig. 3). The NJ tree separated all inferred clusters from DAPC, in highly supported groups (> 75% bootstrap support (BS) value), though there was a branch of cluster 3 for which the BS value was slightly lower (66%; Fig. 3).

Unrooted neighbour joining tree based on Edward’s distance calculated for 217 accessions of Kersting’s groundnut, coloured by cluster calls DAPC. Numbers associated with branches indicate bootstrap values based on 999 replications
Isolation by distance
The Mantel test comparing genetic distance [FST/(1 − FST)] and geographic distance per population indicated the presence of a significant isolation by distance (IBD) (R2 = 0.612, P = 0.011; Fig. 4a). In contrast, no significant IBD was detected among morphotypes (R2 = 0.533, P = 0.083; Fig. 4b). Figure 4c showed that genetic distance between pairs of Kersting’s groundnut accessions increased significantly with geographic distance, with a Mantel test correlation of 0.499 and a probability of P < 0.001. Several densities of genetic relatedness with spatial discontinuities were found at accession level, indicating the presence of genetic patches (Fig. 4c).

Scatter plots illustrating the pairwise relationship between genetic distance (FST/(1−FST)) and geographic distances (Km) between populations (a) morphotypes (b) and samples (c). Warmer colors indicate higher densities of points
Discussion
Genetic diversity is particularly important to breeders as a source of novel alleles for improving diverse complex traits of agronomic interest, particularly with regards to exacerbated climate challenges and sophisticated end-user demands. In the present study, genetic diversity and population structure were assessed in a panel composed of 217 Kersting’s groundnut accessions from different origins, using 886 DArTseq-generated SNP markers. The information provided is useful to breeders and geneticists. It will help for efficient management and conservation of Kersting’s groundnut genetic resources, as the crop is almost on the verge of extinction in many growing countries (Pasquet et al. 2002; Adu-Gyamfi et al. 2011; Amujoyegbe et al. 2007).
SNP discovery and marker quality.
DArTseq has been successfully applied in many recent plant breeding studies for quantifying genetic diversity and deciphering population structure in major (Shaibu et al. 2020; Nemli et al. 2017; Mogga et al. 2018; Ndjiondjop et al. 2018) as well as minor or orphan crops (O’Connor et al. 2019; Redjeki et al. 2020; Robbana et al. 2019). The number of SNP markers detected in Kersting’s groundnut (1938 SNPs) is almost four-fold higher than that found by Akohoue et al. (2020) in the same crop (493 SNPs). This can be explained by the fact that the 281 accessions used by Akohoue et al. (2020) were all from Benin and Togo and may not have included important sources of genetic variation from the other Kersting’s groundnut growing countries.
Our results showed a relatively low alignment (14–20%) of markers with reference genomes of related crops such as common bean, mung bean, Bambara groundnut, and adzuki bean. This is in line with the findings of Akohoue et al. (2020), though we obtained better alignment here. It is therefore necessary to assemble a reference genome for Kersting’s groundnut to enhance the accuracy of SNP calling and thereby increase their use in the pre-breeding and future breeding pipelines of the crop.
The markers used in the study were highly reproducible (0.99) and scored 100% for call rate, denoting their high quality. However, they exhibited low polymorphism information contents (average PIC = 0.059) suggesting that most of them would not be very informative for linkage analysis (Alam et al. 2018). These low PIC values also deviate from the average reported for other underutilised legume crops including Bambara groundnut (PIC = 0.58) (Redjeki et al. 2020), mung bean (PIC = 0.25) (Islam and Blair 2018), pigeon pea (PIC = 0.25) (Zavinon et al. 2020), and cowpea (PIC = 0.23) (Fatokun et al. 2018). Kersting's groundnut is therefore not as polymorphic as other predominantly self-pollinated legume crops. Nevertheless, our results showed that SNPs with lower average PIC have greater density, which would increase their effectiveness in quantifying the genetic diversity and discriminating the Kersting’s groundnut accessions. Furthermore, the high sequencing depth observed in this study suggested that null alleles might be responsible for most missing data. In addition, low mean heterozygosity (Ho = 0.008) was found across markers. Low mean heterozygosity value (0.04) was also recorded in Kersting’s groundnut by Akohoue et al. (2020), indicating a low genetic variation within Kersting’s groundnut accessions.
Genetic diversity across populations and morphotypes
The overall genetic variability measured as expected heterozygosity He (Nei’s gene diversity) and unbiased expected heterozygosity uHe across geographical populations (He = 0.058; uHe = 0.059) and morphotypes (He = 0.057; uHe = 0.058) of Kersting’s groundnut is lower compared to the value of 0.22 reported by Akohoue et al. (2020). The seemingly higher average heterozygosity found by these authors could, however, be misleading regarding the real level of genetic diversity in the crop as it was estimated from 113 markers with minor allele frequency MAF ≥ 0.05. As demonstrated by O'Leary et al. (2018), the common practice to assume that loci with a MAF < 0.05 at population level are not useful and to exclude them from datasets may, unfortunately, lead to excluding true rare alleles from the datasets, especially in orphan crops with little or no genomic resources. However, consistently with Akohoue et al. (2020), we found genetic variation in Kersting’s groundnut, which is incongruent with Pasquet et al. (2002) who reported a total absence of genetic variation in the crop. The total absence of diversity reported by Pasquet et al. (2002) is due to the marker system (allozyme) used and the relatively low number (20) of accessions analysed. This further is an indication that genotyping-by-sequencing (GBS) has power to depict diversity in species with very narrow genetic base.
In any case, the present study showed that genetic variability within Kersting’s groundnut is undeniably limited. A possible reason for this limited genetic diversity could be the selfing nature of the crop which resulted in long generations of inbreeding. Indeed, from our results, selfing rates were estimated as 79% across populations, with values as high as 85% in some populations (Table 1). Similarly, very high selfing rates were observed across morphotypes with values reaching up to 82% within brown seed morphotypes. Moreover, there was a high predominance of homozygous individuals indicating that Kersting’s groundnut is primarily a selfing crop just like horsegram (Macrotyloma uniflorum) its closely related species (Priyanka et al. 2019; Morris 2008). In fact, Kersting’s groundnut bears ‘pre-anthesis cleistogamous’ flowers in which pollination and fertilisation occur in closed flower buds (Kafoutchoni et al. 2021), leading to very high levels of homozygosity as reflected here by the low observed heterozygosity among populations and morphotypes (Tables 1 and 2). This should enable the easy development of pure lines which can then be crossed to develop segregating populations whereby superior phenotypes can then be selected to develop new cultivars. Nonetheless, it is worth noting that all the attempts to cross Kersting’s groundnut remained unsuccessful so far. Hence, the development of new cultivars largely depends on the success of hybridization. Meanwhile, induced mutagenesis seems to be the most promising way to broaden the genetic base of Kersting’s groundnut.
The higher genetic diversity found in Atacora and in Cream seeds with black eye can be favourably used in Kersting’s groundnut genetic improvement. However, the greatest numbers of private alleles found respectively in Zou and in Cream seed morphotypes suggest that there are potential sources of novel alleles and traits of qualitative or agronomic importance. The first three accessions with greater number of private alleles were Gbo5, Lili*Sie and Itk and possessed respectively 18, 16 and 12 private alleles (Table S4). These accessions can thus be good candidate parents for the development of improved Kersting’s groundnut cultivars with desirable traits for farmers and consumers.
Genetic differentiation and partitioning of genetic variation across populations and morphotypes
The ranges of FST values among populations (Table 4) and among morphotypes (Table 5) of Kersting’s groundnut revealed substantial genetic differentiation. As expected, little differentiation is observed between the three populations from Benin. This suggests a high level of gene flow in the form of seed exchange among farmers, especially between Zou and Collines, two neighbouring districts. In general, considerable genetic differentiation was observed between populations from different countries, indicating some degree of genetic isolation among countries. Indeed, a significant correlation was detected between genetic and geographic distances between individuals and populations suggesting that seed exchange between very distant localities may not be common. These are in agreement with many previous studies that reported cultural, social, and natural barriers may strengthen reproductive isolation, thereby limiting or promoting gene flow among cultivars (Pusadee et al. 2009; Sharma et al. 2013).
The results of AMOVA indicated that majority of genetic variation (~ 49%) was intra-population, while 38.5% of genetic variation was inter-population, and only ~ 13% of variation was apportioned within accessions. Partitioning of genetic variation in plant populations can be affected by many factors including the breeding system (e.g. selfing or outcrossing rates) of the species (Duminil et al. 2007; Hamrick and Godt 1996; Szczecińska et al. 2016). Evidence showed that inbreeding or selfing annual plants predominantly maintain more genetic variation among populations than within populations contrary to long-lived perennial and/or outcrossing species (Yang et al. 2010; Hamrick et al. 1991). Our finding is not consistent with this theory. Higher within population variation is also reported in other annual crops such as wheat (Eltaher et al. 2018; Kabbaj et al. 2017), safflower (Hassani et al. 2020) and rice (Yelome et al. 2018). Besides, greater genetic variation (49%) was also partitioned within morphotypes while the variation among morphotypes was moderate (about 39%) but significant. Therefore, intensive collection of accessions of various morphotypes from different populations/countries would substantially increase the genetic diversity within Benin Kersting’s groundnut germplasm.
Population structure
DAPC divided the accessions into eight well-defined clusters which were further confirmed by neighbour joining (NJ) tree. The high number of detected clusters was expected as the accessions included in the study were all landraces from a wide geographical origin. The differences between DAPC and NJ in the assignment of accessions to clusters could be explained by the fact that the NJ method is an agglomerative hierarchical method of clustering, whereas DAPC is based on k-means which is a non-hierarchical clustering method (Campoy et al. 2016). It is well established that population structure may lead to spurious associations in Genome-wide association studies (GWAS) (Korte and Farlow 2013; Barabaschi et al. 2016) and should be considered in the development of training populations for Genomic selection (GS) (Isidro et al. 2015). Therefore, the clustering of accessions presented in this work provides useful data to inform decision-making for genetic improvement of Kersting’s groundnut using molecular breeding tools and techniques.
Conclusions
The present study was undertaken to investigate the level of genetic variation and determine population structure among a collection of 217 Kersting’s groundnut accessions from different origins using 886 high-quality DArTseq derived SNP markers. Low levels of genetic diversity were found overall. The highest genetic diversity was found in Atacora population while the highest number of private alleles was observed in Zou population. Similarly, the more diverse morphotype was cream seed with black eye whereas the highest number of private alleles was found in cream seed. Furthermore, very high genetic differentiations were observed among populations and morphotypes, with maximal differentiation between populations from different countries. The available level of genetic diversity could be efficiently exploited to develop improved Kersting’s groundnut cultivars with desirable characteristics such as high yield and quality traits. However, the use of induced mutagenesis is recommended for fast broadening of the genetic base of the crop. Substantial genetic differentiation was observed among populations and among morphotypes. Likewise, considerable genetic variations were contained within populations as well as within morphotypes. The population structure analysis identified eight clusters containing accessions from different geographic origins. The genomic resource developed in this study can significantly contribute to integrating modern breeding techniques, including Genome-wide association studies and Genomic selection into the Kersting’s groundnut breeding pipeline in Benin and West Africa.
Availability of data and material
The datasets supporting the conclusions of this article are included within the article.
References
Achigan Dako EG, Vodouhè SR (2006) Macrotyloma geocarpum (Harms) Maréchal and Baudet. Brink M, Belay G (Editeurs) PROTA 1
Adamack AT, Gruber B (2014) PopGenReport: simplifying basic population genetic analyses in R. Methods Ecol Evol 5(4):384–387
Adu-Gyamfi R, Dzomeku IK, Lardi J (2012) Evaluation of growth and yield potential of genotypes of Kersting’s groundnut (macrotyloma geocarpum harms) in Northern Ghana. Int Res J Agric Sci Soil Sci 2(12):509–515
Adu-Gyamfi R, Fearon J, Bayorbor T, Dzomeku I, Avornyo V (2011) The Status of Kersting’s Groundnut (Macrotyloma Geocarpum [Harms] Marechal and Baudet) An Underexploited Legume in Northern Ghana. Outlook Agric 40(3):259–262
Agoyi EE, N’danikou S, Kafoutchoni M, Ayena M, Sodedji FA, Agbahoungba S, Sossou HS, Vodouhe R, Assogbadjo AE (2019a) Kersting’s Groundnut [Macrotyloma geocarpum (Harms) Maréchal & Baudet] crop attracts more field pests and diseases than reported before. Agric Res Technol Open Access J 21(5)
Agoyi EE, Tumuhairwe JB, Chigeza G, Tukamuhabwa P, Diers BW (2019b) First attempt to identify and map QTLs associated with promiscuous nodulation In Soybean. bioRxiv:688028
Ajala SO, Olayiwola MO, Ilesanmi OJ, Gedil M, Job AO, Olaniyan AB (2019) Assessment of genetic diversity among low-nitrogen-tolerant early generation maize inbred lines using SNP markers. South Afr J Plant Soil 36(3):181–188
Akohoue F, Achigan-Dako EG, Coulibaly M, Sibiya J (2019) Correlations, path coefficient analysis and phenotypic diversity of a West African germplasm of Kersting’s groundnut [Macrotyloma geocarpum (Harms) Maréchal & Baudet]. Genet Resour Crop Evol 66(8):1825–1842
Akohoue F, Achigan-Dako EG, Sneller C, Van Deynze A, Sibiya J (2020) Genetic diversity, SNP-trait associations and genomic selection accuracy in a west African collection of Kersting’s groundnut [Macrotyloma geocarpum (Harms) Maréchal & Baudet]. PLoS ONE 15(6):e0234769
Akohoue F, Sibiya J, Achigan-Dako EG (2018) On-farm practices, mapping, and uses of genetic resources of Kersting’s groundnut [Macrotyloma geocarpum (Harms) Maréchal et Baudet] across ecological zones in Benin and Togo. Genet Resour Crop Evol 66(1):195–214
Alam M, Neal J, O’Connor K, Kilian A, Topp B (2018) Ultra-high-throughput DArTseq-based silicoDArT and SNP markers for genomic studies in macadamia. PLoS ONE 13(8):e0203465
Amujoyegbe B, Obisesan I, Ajayi A, Aderanti F (2007) Disappearance of Kersting's groundnut (Macrotyloma geocarpum (Harms) Marechal and Baudet) in south-western Nigeria: an indicator of genetic erosion. Plant Genetic Resources Newsletter (Bioversity International/FAO)
Assogba P, Ewedje EB, Dansi A, Loko Y, Adjatin A, Dansi M, Sanni A (2015) Indigenous knowledge and agro-morphological evaluation of the minor crop Kersting’s groundnut (Macrotyloma geocarpum (Harms) Maréchal et Baudet) cultivars of Benin. Genet Resour Crop Evol 63(3):513–529
Bampuori AH (2007) Effect of Traditional Farming Practices on the Yield of Indigenous Kersting’s Groundnut (Macrotyloma geocarpum Harms) Crop in the Upper West Region of Ghana. J Dev Sustain Agric 2(2):128–144
Barabaschi D, Tondelli A, Desiderio F, Volante A, Vaccino P, Vale G, Cattivelli L (2016) Next generation breeding. Plant Sci 242:3–13. https://doi.org/10.1016/j.plantsci.2015.07.010
Bayorbor TB, Dzomeku IK, Avornyo VK, Opoku-Agyeman MO (2010) Morphological variation in Kersting’s groundnut (Kerstigiella geocarpa Harms) landraces from northern Ghana. Agric Biol J N Am 1(3):290–295
Campa A, Murube E, Ferreira JJ (2018) Genetic diversity, population structure, and linkage disequilibrium in a spanish common bean diversity panel revealed through genotyping-by-sequencing. Genes 9(11):518
Campoy JA, Lerigoleur-Balsemin E, Christmann H, Beauvieux R, Girollet N, Quero-García J, Dirlewanger E, Barreneche T (2016) Genetic diversity, linkage disequilibrium, population structure and construction of a core collection of Prunus avium L. landraces and bred cultivars. BMC Plant Biol 16(1):1–15
Chen W, Hou L, Zhang Z, Pang X, Li Y (2017) Genetic diversity, population structure, and linkage disequilibrium of a core collection of Ziziphus jujuba assessed with genome-wide SNPs developed by genotyping-by-sequencing and SSR markers. Front Plant Sci 8:575
Chikwendu JN (2015) Comparative evaluation of chemical composition of fermented ground bean flour (Kerstingella geocarpa), cowpea flour (Vigna unguiculata) and commercial wheat flour (Triticum spp.). Pak J Nutr 14:218–224
Cortés AJ, Blair MW (2018) Genotyping by sequencing and genome-environment associations in wild common bean predict widespread divergent adaptation to drought. Front Plant Sci. https://doi.org/10.3389/fpls.2018.00128
Dansi A, Vodouhe R, Azokpota P, Yedomonhan H, Assogba P, Adjatin A, Loko YL, Dossou-Aminon I, Akpagana K (2012) Diversity of the neglected and underutilized crop species of importance in Benin. ScientificWorldJournal 2012:932947. https://doi.org/10.1100/2012/932947
Dray S, Dufour A-B (2007) The ade4 package: implementing the duality diagram for ecologists. J Stat Softw 22(4):1–20
Duminil J, Fineschi S, Hampe A, Jordano P, Salvini D, Vendramin GG, Petit RJ (2007) Can population genetic structure be predicted from life-history traits? Am Nat 169(5):662–672
Echendu A, Obizoba I, Ngwu E, Anyika J (2009) Chemical composition of ground bean based cocoyam, yam and plantain pottage dishes and roasted ground bean. Pak J Nutr 8(11):1786–1790
Egea LA, Mérida-García R, Kilian A, Hernandez P, Dorado G (2017) Assessment of genetic diversity and structure of large garlic (Allium sativum) germplasm bank, by diversity arrays technology “genotyping-by-sequencing” platform (DArTseq). Front Genet 8:98
Eltaher S, Sallam A, Belamkar V, Emara HA, Nower AA, Salem KF, Poland J, Baenziger PS (2018) Genetic diversity and population structure of F3: 6 Nebraska winter wheat genotypes using genotyping-by-sequencing. Front Genet 9:76
Fatokun C, Girma G, Abberton M, Gedil M, Unachukwu N, Oyatomi O, Yusuf M, Rabbi I, Boukar O (2018) Genetic diversity and population structure of a mini-core subset from the world cowpea (Vigna unguiculata (L.) Walp.) germplasm collection. Sci Rep 8(1):1–10
Goudet J, Jombart T (2015) hierfstat: estimation and tests of hierarchical F-statistics. R package version 004-22 10
Govindaraj M, Vetriventhan M, Srinivasan M (2015) Importance of genetic diversity assessment in crop plants and its recent advances: an overview of its analytical perspectives. Genet Res Int 2015:431487. https://doi.org/10.1155/2015/431487
Hamrick J, Godt M, Murawski D, Loveless M (1991) Correlations between species traits and allozyme diversity: implications for conservation biology. Genet Conserv Rare Plants 3:30
Hamrick JL, Godt MW (1996) Effects of life history traits on genetic diversity in plant species. Philos Trans R Soc Lond B Biol Sci 351(1345):1291–1298
Hassani SMR, Talebi R, Pourdad SS, Naji AM, Fayaz F (2020) In-depth genome diversity, population structure and linkage disequilibrium analysis of worldwide diverse safflower (Carthamus tinctorius L.) accessions using NGS data generated by DArTseq technology. Mol Biol Rep 47(3):2123–2135
Hepper F (1963) Plants of the 1957–58 West African Expedition: II. The Bambara Groundnut (Voandzeia subterranea) and Kersting’s Groundnut (Kerstingiella geocarpa) Wild in West Africa. Kew Bull 16(3):395–407
Iquira E, Humira S, François B (2015) Association mapping of QTLs for sclerotinia stem rot resistance in a collection of soybean plant introductions using a genotyping by sequencing (GBS) approach. BMC Plant Biol 15(1):5
Isidro J, Jannink J-L, Akdemir D, Poland J, Heslot N, Sorrells ME (2015) Training set optimization under population structure in genomic selection. Theor Appl Genet 128(1):145–158
Islam AF, Blair MW (2018) Molecular characterization of Mung bean germplasm from the USDA core collection using newly developed KASP-based SNP markers. Crop Sci 58(4):1659–1670
Jaiswal SK, Mohammed M, Dakora FD (2019) Microbial community structure in the rhizosphere of the orphan legume Kersting’s groundnut [Macrotyloma geocarpum (Harms) Marechal & Baudet]. Mol Biol Rep 46(4):4471–4481
Jarquín D, Kocak K, Posadas L, Hyma K, Jedlicka J, Graef G, Lorenz A (2014) Genotyping by sequencing for genomic prediction in a soybean breeding population. BMC Genom 15(1):740
Jombart T (2008) adegenet: a R package for the multivariate analysis of genetic markers. Bioinformatics 24(11):1403–1405
Kabbaj H, Sall AT, Al-Abdallat A, Geleta M, Amri A, Filali-Maltouf A, Belkadi B, Ortiz R, Bassi FM (2017) Genetic diversity within a global panel of durum wheat (Triticum durum) landraces and modern germplasm reveals the history of alleles exchange. Front Plant Sci 8:1277
Kafoutchoni KM, Agoyi EE, Dassou GH, Sossou HS, Ayi S, Glèlè CA, Adomou AC, Yédomonhan H, Agbangla C, Assogbadjo AE (2021) Reproductive biology, phenology, pollen viability and germinability in Kersting’s groundnut (Macrotyloma geocarpum (Harms) Maréchal & Baudet, Fabaceae). S Afr J Bot 137:440–450
Kafoutchoni KM, Agoyi EE, Sossou HS, Agbahoungba S, Agbangla C, Assogbadjo AE (2021) Impacts of sociodemographic factors on the production system of the orphan legume Kersting’s groundnut (Macrotyloma geocarpum) and prioritisation of production constraints in Benin. Environ Dev Sustain (in press)
Kamvar ZN, Tabima JF, Grünwald NJ (2014) Poppr: an R package for genetic analysis of populations with clonal, partially clonal, and/or sexual reproduction. PeerJ 2:e281
Kilian A, Wenzl P, Huttner E, Carling J, Xia L, Blois H, Caig V, Heller-Uszynska K, Jaccoud D, Hopper C (2012) Diversity arrays technology: a generic genome profiling technology on open platforms. In: Data production and analysis in population genomics. Humana Press, Totowa, NJ, pp 67–89
Korte A, Farlow A (2013) The advantages and limitations of trait analysis with GWAS: a review. Plant Methods 9(1):29. https://doi.org/10.1186/1746-4811-9-29
Kumar S, Ambreen H, Variath MT, Rao AR, Agarwal M, Kumar A, Goel S, Jagannath A (2016) Utilization of molecular, phenotypic, and geographical diversity to develop compact composite core collection in the oilseed crop, safflower (Carthamus tinctorius L.) through maximization strategy. Front Plant Sci 7:1554
Mensah AG, Gyan AD, Yeboah AM (2016) An assessment on variation among genotypes of kersting’s groundnut in Northern Ghana. Adv Agric Agric Sci 2(7):149–155
Mergeai G (1993) Influence des facteurs sociologiques sur la conservation des ressources phytogénétiques. Le cas de la lentille de terre (Macrotyloma geocarpum (Harms) Marechal & Baudet) au Togo. Bull des Recherches Agronomiques de Gembloux 28(4):487–500
Mogga M, Sibiya J, Shimelis H, Lamo J, Yao N (2018) Diversity analysis and genome-wide association studies of grain shape and eating quality traits in rice (Oryza sativa L.) using DArT markers. PloS one 13(6):e0198012
Mondini L, Noorani A, Pagnotta MA (2009) Assessing plant genetic diversity by molecular tools. Diversity 1(1):19–35
Morris J (2008) Macrotyloma axillare and M. uniflorum: descriptor analysis, anthocyanin indexes, and potential uses. Genet Resour Crop Evol 55(1):5–8
Ndjiondjop M-N, Semagn K, Gouda AC, Kpeki SB, Dro Tia D, Sow M, Goungoulou A, Sie M, Perrier X, Ghesquiere A (2017) Genetic variation and population structure of Oryza glaberrima and development of a mini-core collection using DArTseq. Front Plant Sci 8:1748
Ndjiondjop MN, Semagn K, Sow M, Manneh B, Gouda AC, Kpeki SB, Pegalepo E, Wambugu P, Sié M, Warburton ML (2018) Assessment of genetic variation and population structure of diverse rice genotypes adapted to lowland and upland ecologies in Africa using SNPs. Front Plant Sci 9:446
Nei M (1973) Analysis of gene diversity in subdivided populations. Proc Natl Acad Sci 70(12):3321–3323
Nemli S, Aşçioğul TK, Ateş D, Eşiyok D, Tanyolac MB (2017) Diversity and genetic analysis through DArTseq in common bean (Phaseolus vulgaris L.) germplasm from Turkey. Turk J Agric For 41(5):389–404
O’Leary SJ, Puritz JB, Willis SC, Hollenbeck CM, Portnoy DS (2018) These aren’t the loci you’e looking for: principles of effective SNP filtering for molecular ecologists. Mol Ecol 27(16):3193–3206
O’Connor K, Kilian A, Hayes B, Hardner C, Nock C, Baten A, Alam M, Topp B (2019) Population structure, genetic diversity and linkage disequilibrium in a macadamia breeding population using SNP and silicoDArT markers. Tree Genet Genomes 15(2):24
Pasquet RS, Mergeai G, Baudoin J-P (2002) Genetic diversity of the African geocarpic legume Kersting’s groundnut, Macrotyloma geocarpum (Tribe Phaseoleae: Fabaceae). Biochem Syst Ecol 30(10):943–952
Peakall R, Smouse PE (2012) GenAlEx 6.5: genetic analysis in Excel. Population genetic software for teaching and research–an update. Bioinformatics 28(19):2537
Priyanka S, Sudhagar R, Vanniarajan C, Ganesamurthy K, Souframanien J (2019) Combined mutagenic ability of gamma ray and EMS in horsegram (Macrotyloma uniflorum (Lam) Verdc.). Electron J Plant Breed 10(3):1086–1094
Pusadee T, Jamjod S, Chiang Y-C, Rerkasem B, Schaal BA (2009) Genetic structure and isolation by distance in a landrace of Thai rice. Proc Natl Acad Sci 106(33):13880–13885
R Core Team (2020) R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria
Redjeki ES, Ho WK, Shah N, Molosiwa OO, Ardiarini N, Kuswanto MS (2020) Understanding the genetic relationships between Indonesian bambara groundnut landraces and investigating their origins. Genome 999:1–9
Ritland K (1990) Inferences about inbreeding depression based on changes of the inbreeding coefficient. Evolution 44(5):1230–1241
Robbana C, Kehel Z, Naceur B, Sansaloni C, Bassi F, Amri A (2019) Genome-wide genetic diversity and population structure of tunisian durum wheat landraces based on DArTseq technology. Int J Mol Sci 20(6):1352
Schröder S, Mamidi S, Lee R, McKain MR, McClean PE, Osorno JM (2016) Optimization of genotyping by sequencing (GBS) data in common bean (Phaseolus vulgaris L.). Mol Breed 36(1):6
Shaibu AS, Sneller C, Motagi BN, Chepkoech J, Chepngetich M, Miko ZL, Isa AM, Ajeigbe HA, Mohammed SG (2020) Genome-wide detection of SNP markers associated with four physiological traits in groundnut (Arachis hypogaea L.) mini core collection. Agronomy 10(2):192
Sharma S, Upadhyaya H, Varshney R, Gowda C (2013) Pre-breeding for diversification of primary gene pool and genetic enhancement of grain legumes. Front Plant Sci. https://doi.org/10.3389/fpls.2013.00309
Singh S, Gupta S, Thudi M, Das RR, Vemula A, Garg V, Varshney R, Rathore A, Pahuja S, Yadav DV (2018) Genetic diversity patterns and heterosis prediction based on SSRs and SNPs in hybrid parents of pearl millet. Crop Sci 58(6):2379–2390
Szczecińska M, Sramko G, Wołosz K, Sawicki J (2016) Genetic diversity and population structure of the rare and endangered plant species Pulsatilla patens (L.) Mill in East Central Europe. PLoS ONE 11(3):e0151730
Tamini Z (1997) Etude ethnobotanique et analyses morphophysiologiques du développement de la lentille de terre (Macrotyloma geocarpum (Harms) Marechal et Baudet. Université de Ouagadougou, Burkina-Faso
Varshney RK, Close TJ, Singh NK, Hoisington DA, Cook DR (2009) Orphan legume crops enter the genomics era! Curr Opin Plant Biol 12(2):202–210. https://doi.org/10.1016/j.pbi.2008.12.004
Wojciechowski MF, Lavin M, Sanderson MJ (2004) A phylogeny of legumes (Leguminosae) based on analysis of the plastid matK gene resolves many well-supported subclades within the family. Am J Bot 91(11):1846–1862
Wright S (1978) Evolution and the genetics of populations, volume 4: variability within and among natural populations, 4th edn. University of Chicago Press, Chicago
Xiong H, Shi A, Mou B, Qin J, Motes D, Lu W, Ma J, Weng Y, Yang W, Wu D (2016) Genetic diversity and population structure of cowpea (Vigna unguiculata L. Walp). PLoS ONE 11(8):e0160941
Yang X, Ren R, Ray R, Xu J, Li P, Zhang M, Liu G, Yao X, Kilian A (2016) Genetic diversity and population structure of core watermelon (Citrullus lanatus) genotypes using DArTseq-based SNPs. Plant Genet Resour 14(3):226–233
Yang Y, Pan Y, Gong X, Fan M (2010) Genetic variation in the endangered Rutaceae species Citrus hongheensis based on ISSR fingerprinting. Genet Resour Crop Evol 57(8):1239–1248
Yelome OI, Audenaert K, Landschoot S, Dansi A, Vanhove W, Silue D, Van Damme P, Haesaert G (2018) Analysis of population structure and genetic diversity reveals gene flow and geographic patterns in cultivated rice (O. sativa and O. glaberrima) in West Africa. Euphytica 214(11):215
Zavinon F, Adoukonou-Sagbadja H, Keilwagen J, Lehnert H, Ordon F, Perovic D (2020) Genetic diversity and population structure in Beninese pigeon pea [Cajanus cajan (L.) Huth] landraces collection revealed by SSR and genome wide SNP markers. Genet Resour Crop Evol 67(1):191–208
Acknowledgements
We are grateful to the different institutions that provided the accessions used in this study: IITA-Nigeria, INERA-Burkina-Faso, SARI-Ghana. Finally, we thank Dr. Mustapha Mohammed (Tshwane University of Technology, Pretoria, South Africa) for providing additional accessions from Ghana.
Funding
This research was funded by the World Academy of Sciences (TWAS), Grant number 18-238 RG/BIO/AF/AC_G-FR3240303667. We thank Carnegie Cooperation of New York for funding part of the work through the Regional Universities Forum for Capacity Building in Agriculture (RUFORUM) Grant number RU/2018/TQA/38.
Author information
Authors and Affiliations
Contributions
Conceptualization, Konoutan M. Kafoutchoni and Eric E. Agoyi; Data curation, Konoutan M. Kafoutchoni; Formal analysis, Konoutan M. Kafoutchoni; Funding acquisition, Konoutan M. Kafoutchoni, Symphorien Agbahoungba and Achille E. Assogbadjo; Project administration, Eric E. Agoyi; Resources, Eric E. Agoyi, Achille E. Assogbadjo and Clément Agbangla; Supervision, Achille E. Assogbadjo and Clément Agbangla; Writing – original draft, Konoutan M. Kafoutchoni; Writing – review & editing, Konoutan M. Kafoutchoni, Eric E. Agoyi and Symphorien Agbahoungba.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Kafoutchoni, K.M., Agoyi, E.E., Agbahoungba, S. et al. Genetic diversity and population structure in a regional collection of Kersting’s groundnut (Macrotyloma geocarpum (Harms) Maréchal & Baudet). Genet Resour Crop Evol 68, 3285–3300 (2021). https://doi.org/10.1007/s10722-021-01187-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10722-021-01187-4