
Abstract
Cedrela fissilis is an endangered timber species associated with seasonal forests throughout South America. We investigated a population of C. fissilis (PAN) located toward central Brazil to uncover insights on how an ecotone may have shaped the evolutionary history of this species at the local scale. PAN consisted of 18 mother trees and their 283 offspring (18 families), which were genotyped with ten microsatellite loci. We supplemented our dataset with equivalent microsatellite data from 175 specimens representing the east and west lineages of C. fissilis. An array of complementary methods assessed PAN for genetic diversity, population structure, and mating system. In PAN, the gene pool of the east lineage combined with a third (previously unidentified) lineage to form an admixture population. PAN is under inbreeding (Ho = 0.80 and 0.74, uHe = 0.85 and 0.82, Ap = 1.1 and 7.1, F = 0.06 and 0.10, for mother trees and offspring, respectively). Mother trees were predominantly outcrossing (tm = 0.95), with some selfing (1 − tm = 0.05), and crossing between related individuals (tm–ts = 0.07); they received pollen from few donors (Nep = 9). Restricted gene flow within PAN gave rise to a strong population structure, which split the 18 families into six groups. Some mother trees were reproductively isolated. Conservation perspectives are discussed.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Seasonal forests occur widely within the Neotropics. They occur from the Pacific coast of Mexico to the Caribbean; across northern Colombia and Venezuela; the inter-Andean valleys in Ecuador, Peru, and Bolivia; northwestern Argentina; and reach the Atlantic coast of Brazil (Prado and Gibbs 1993; Pennington et al. 2004). A fascinating feature of the Neotropical seasonal forests is their disjunct distribution. Where they occur, seasonal forests are found as scattered blocks within larger patches of other communities, such as moist forests, savannas, and shrublands (Pennington et al. 2004; Oliveira-Filho et al. 2006). There are two major blocks of continuous seasonal forests in eastern South America: the Mata Atântica dry forests (along the east coast of Brazil) and the Chiquitano dry forests (in lowland Bolivia and across the border with Brazil). The savanna-like vegetation of the Cerrado occupies most of the intervenient space between the two major blocks of continuous seasonal forests (Pennington et al. 2004). Scattered, small-size areas that hold a suitable combination of wetter conditions and favorable soil types allow for the endurance of blocks of seasonal forests within the drier vegetation of the Cerrado in central Brazil (Oliveira-Filho et al. 2006) and the Caatinga in the semiarid areas of northeastern Brazil (Queiroz 2006). At the present moment, the riverine vegetations that permeate the Cerrado—or the Caatinga—may provide the locked blocks of seasonal forests with some degree of genetic connectivity to the major blocks of continuous seasonal forests. Owing to the riverine vegetations, populations of species typically associated with seasonal forests may be able to overcome an otherwise impassable geographic barrier and maintain gene flow with other populations located within geographically disconnected blocks of seasonal forests, the extent of which the small, scattered blocks maintained gene flow with other larger, distant blocks of seasonal forests which continues to be unexplored.
Past climate changes resulted in major vegetation shifts in the Neotropics. In South America, recurrent vegetation shifts took place over broad geographic scales. These shifts were probably triggered when cycles of cooling/warming and dry/wet climates operated during the last two million years of the Pleistocene (Whitmore and Prance 1987), or even earlier (Mayle 2004; Pennington et al. 2004). Episodes of climate amelioration over time allowed for the recurrent expansion of forests at the expense of dry vegetations, such the Cerrado and Caatinga; but during drier periods, those dry vegetations likely expanded over closed-canopy forests (Whitmore and Prance 1987; Mayle 2004; Pennington et al. 2004).
In case geographically isolated populations endured genetic isolation within a block of seasonal forest, novel genetic variation had the chance to accumulate through genetic drift. Genetic differentiation that accumulated over time could set apart populations that evolved in isolation within those isolated blocks from populations of larger areas that continued to be interconnected. In such a scenario, levels of differentiation would vary depending upon the time the population remained in genetic isolation. Within central Brazil, areas that detained a more stable climate throughout the Late Pleistocene may have played an important role as paleorefuges for many plant species; those areas may congregate greater diversity and ancestral genotypes due to their greater long-term persistence and population structure (Carnaval et al. 2009).
Located toward central Brazil, the northern Minas Gerais State (NMG) comprises an ecotone with a unique mixture of three ecosystems. In NMG, the Caatinga finds its southernmost distribution and meets together with the Cerrado (advancing from the west) and the Mata Atlântica dry forests (advancing from the east). Small tributaries of the larger São Francisco River, the Peruaçu River, and the Pandeiros River (Nunes et al. 2009; Sales et al. 2009; Bethonico 2010) run through the area and allow for gallery forests and wetlands within an arid ecotone (Lopes et al. 2010). Differences in the flood regime and soil types along the rivers resulted in distinct floristic composition upstream compared to downstream (Rodrigues et al. 2009). It is plausible that the changes in ecological conditions that lead to contractions and expansions of forests in eastern South America (Mayle 2004; Pennington et al. 2004) also included NMG.
Cedrela P. Browne (Meliaceae) comprises 18 species; the genus is distributed from northern Mexico to northwestern Argentina (Styles 1981; Pennington and Muellner 2010). Several features suggest the dry seasonal forests as the likely origin of Cedrela: deciduous leaves; scale-protect gems; dry, capsular fruits; and winged seeds that are wind-dispersed and can survive the dry season (Pennington and Muellner 2010). While most of the congeners are narrow-ranged species, Cedrela fissilis and Cedrela odorata are broadly distributed in both Mesoamerica and South America (Pennington and Muellner 2010). They are associated with closed-canopy forests—seasonal forests and moist forests—and ecotones that are adjacent to seasonal forests (Styles 1981; Pennington and Muellner 2010).
Cedrela fissilis exhibits an extended juvenile phase and is a low-density species, with a natural density of about one to three trees per hectare (Carvalho 1994). Because it is a noble wood, these trees have experienced intense harvesting. Cedrela fissilis is currently classified as VU (vulnerable species) on the official list of threatened flora in Brazil (Ministério do Meio Ambiente [MMA] 2014) and as “Endangered A1acd+2cd” species on the IUCN Red List (International Union for Conservation of Nature [IUCN] 2017) primarily by habitat loss and overexploitation of subpopulations.
In eastern South America, C. fissilis contains two phylogenetic lineages: the east and west lineages, respectively (Garcia et al. 2011). Microsatellite markers (Mangaravite et al. 2016) largely agree with the phylogenetic analyses (Garcia et al. 2011) and corroborate the idea that C. fissilis splits into distinct gene pools. The east lineage occupies the Atlantic range, which occurs at the eastern side of the Cerrado, along the Atlantic coast of southeastern Brazil and includes both seasonal and moist forests. The west lineage inhabits the Chiquitano range at the western side of the Cerrado, further inland in the Chiquitano dry forests and the Madeira-Tapajós moist forests (in lowland Bolivia and neighboring lands of western Brazil) (Garcia et al. 2011; Mangaravite et al. 2016). In central Brazil, the Cerrado seems to be unsuitable to Cedrela. Likely, the Cerrado has been a long-standing geographic barrier that restrained gene exchange between the east and west lineages of C. fissilis, thus constrained them to evolve strong genetic differentiation under allopatry (Garcia et al. 2011; Mangaravite et al. 2016). Previous studies suggested that populations of Cedrela fissilis near ecotonal zones, such as those of NMG, may have undergone admixture extensively and may hold biogeographic clues about how the dispersion waves of seasonal forest elements progressed toward central Brazil over time (Mangaravite et al. 2016).
The mating system and the reproductive biology of C. fissilis remain poorly understood. The trees produce actinomorphic, unisex flowers, of 5 to 10 mm in length reunited in axillary thyrses. The flowers are pale green to red pink. The flowering season is from August to January. The maturity of the fruit is 8–10 months later, in the dry season when the trees shed their leaves. The fruits are dehiscent woody capsules from 3 to 11 cm in length, enclosing 30 to 300 winged, wind-dispersed seeds (Pennington and Muellner 2010). There are some suggestions that the pollination of C. fissilis may resemble that of Cupania guatemalensis, in which Trigona stingless bees play an important role (Bawa 1977), or may be similar to that of C. odorata, which exhibits reported monoecious reproduction with pollination carried out by nocturnal butterflies (Bawa et al. 1985).
Monoecy, protogyny, cross-fertilization, long-life cycles, and wind-dispersed seeds are traits of C. fissilis (Pennington and Muellner 2010) that may favor high levels of gene flow within the population and low differentiation among populations (Loveless and Hamrick 1984; Hensen and Oberprieler 2005). In C. fissilis, however, the low density in which the trees naturally occur together with the short-distance pollen dispersers travel may result in a scenario in which the genetic diversity is scattered in small assemblages of subpopulations within the larger population. When visiting a given flower, a pollinator may deposit several pollen grains transported from the nearest donor tree; thus, the fruit likely will yield a progeny of full-sibs (Muona et al. 1991). Progenies with low genetic diversity may be due to pollen coming from few male donors or when mating takes place between closely related neighbors (Surles et al. 1990).
In this study, we explored the potential role of the unique blend of ecosystems in NMG in shaping the evolutionary history of a population of C. fissilis. Firstly, we used microsatellite data from representative populations of either the Atlantic range or the Chiquitano range to determine the genealogical placement of a natural population of C. fissilis sampled from NMG. Subsequently, we obtained microsatellite data from mother trees and their offspring to investigate the mating system, genetic diversity, and genetic structure within this population. For the populations of C. fissilis within the ecotonal areas of NMG, this study proposes to uncover (1) the genealogical placement (within either the east lineage or the west lineage), (2) the level of lineage admixture (if any), (3) the levels of genetic diversity and genetic structure, and (4) the estimators of the mating system. We also present the implications of our results for the genetic conservation of C. fissilis and for the debate about the origins of seasonal forest distributions in eastern South America.
Materials and methods
Study site and sample collection
The sampling sites were located within three neighboring conservation units within an ecotone in NMG, toward central Brazil: Pandeiros River Basin Environmental Preservation Area, Peruaçu Caves National Park, and Mata Seca State Park. We used the Terrestrial Ecoregions of the World database from the World Wildlife Fund (Olson et al. 2001) to define the associated vegetation formations that surrounded the sampling sites. A total of 18 fruit-bearing trees of C. fissilis were located during the fruit season 2016–2017; we recorded their location using a global positioning system (GPS) receiver. Hereafter, each of those 18 fruit-bearing trees will be referred to as a “mother tree,” and collectively, they comprise the population “Pandeiros” (PAN). Vouchers were deposited at the Federal University of Viçosa (VIC) herbarium. Representative images of the study population are shown (Fig. 1).

Representative images of specimens of the population Pandeiros (PAN) of Cedrela fissilis. a Flowering branch. b Thyrses of flowers. c Capsules organized in an infrutescence. d Tree trunk. e Habit
In PAN, we sampled leaf tissue from each of the 18 mother trees for DNA analyses. Leaf samples were dried immediately using silica gel and kept at room temperature until subsequent use. During the fruit season, we sampled semi-open fruits from distinct branches around each mother tree and labeled them according to the mother tree. Fruits were placed on paper bags, transported to the laboratory, and kept at room temperature until the opening and releasing of the seeds. The seeds had their surface disinfected using the following procedure: the seeds were soaked into a solution of sodium hypochlorite (25% active chlorine) and Tween-20 (4 drops/100 mL) for 20 min, rinsed three times in sterile water, and then left in sterile water for 24 h. Subsequently, the seeds were washed in a solution of 70% ethanol for 1 min, disinfected again with the disinfection solution for 10 min, and rinsed three times in sterile water. Subsequently, the disinfected seeds were sown in Murashige-Skoog (MS20) liquid media (saccharose 20 g/L, myo-inositol 100 mg/L, MS salts 4.33 g/L, MS vitamins 10 mL/L, pH 5.8) (Murashige and Skoog 1962) and grown for 25–30 days. When the plantlets developed at least two leaflets, leaf tissue was taken and stored on silica gel at room temperature until subsequent use. For each mother tree, we obtained a set of plantlets. Hereafter, we will refer to each of those sets of plantlets as either an “offspring” (when the plantlet is considered individually) or a “family” (when the set of offspring from a given mother tree is considered collectively).
DNA extraction and microsatellite marker analyses
DNA extractions were carried out according to the protocol described previously (Cota-Sánchez et al. 2006) with modifications (Riahi et al. 2009). We genotyped each sample of PAN (both mother trees and offspring) using 11 nuclear microsatellite loci (Table 1). Eight markers (Ced2, Ced18, Ced41, Ced44, Ced54, Ced65, Ced95, and Ced131) were obtained for C. odorata (Hernández et al. 2008) and two markers (CF26, CF66) were for C. fissilis (Gandara 2009). The primer pair CF66 amplified two loci—CF66A and CF66B, respectively (Gandara et al. 2014). Distinct ranges of allele sizes (113–175 bp for CF66A; 199–253 bp for CF66B) allow for a clear differentiation between the genotypes of each of the two loci (Fig. 2).

Representative chromatograms of the ten microsatellite loci used in this study to genotype Cedrela fissilis. The chromatograms were obtained using fluorescent-labeled primers (6-FAM, HEX, or NED) in three different PCR systems: simplex (primers Ced66 and Ced44), duplex (primers Ced26 + Ced18), multiplex (M1: Ced54 + Ced41 + Ced95; M2: primers Ced2 + Ced65 + Ced131). Primer Ced66 amplified two loci; distinct allele ranges allowed for the differentiation between loci
Following a previously developed strategy (Mangaravite et al. 2016), the polymerase chain reaction (PCR) was performed using two multiplex (M1: Ced54-Ced41-Ced95; M2: Ced2-Ced65-Ced131), a duplex (D1: CF26-Ced18), and a simplex (S1: CF66 and S2: Ced44) systems (Fig. 2). We used a final volume of 12 μL containing 15 ng of DNA, 1X buffer (10 mM Tris-HCl, pH 8.4, 50 mM KCl, 1% Triton X-100), 0.2–0.3 mM of each primer (forward and reverse), 2.75 mM MgCl2, 0.25 mg/mL BSA (bovine serum albumin Invitrogen), 0.2 mM dNTPs, and 1 U Taq polymerase (Phytoneutria 7 Biotechnology). We used the following PCR program: 96 °C for 2 min, 30 cycles of 94 °C for 1 min, annealing temperature 55 °C for 1 min and 72 °C for 1 min; and 72 °C for 20 min. The forward primers were labeled with the fluorescence 6-FAM, HEX (MWG-Biotech), and NED (Applied Biosystems). Fragments were separated on a 96 capillary sequencer ABI PRISM 3730xl DNA Analyzer (Applied Biosystems), and their sizes were measured using GS500LIZ size standard (Thermo Fisher Scientific). The fragments were scored using GENEMAPPER 4.0 (Applied Biosystems) (Supplementary Table S1).
Genealogical placement of PAN
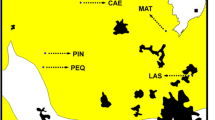
Raw microsatellite data from this study were combined with raw data obtained from 71 samples from four populations of the east lineage and 77 samples from five populations of the west lineage of C. fissilis that had been obtained in a previous study (Mangaravite et al. 2016). We also added raw data of 17 samples of PEU (a population sampled in the Uaimií State Park) and ten samples of PSB (from the Serra do Brigadeiro State Park) (Huamán-Mera, unpubl. data). Both PEU and PSB were populations located within the limits of the Atlantic range of C. fissilis. Thus, we obtained a single microsatellite dataset (193 samples). The populations were chosen to represent the two genealogical lineages of C. fissilis; together they covered most of the geographical range of the species in Brazil (Fig. 3).

Geographic locations of Cedrela fissilis populations in Brazil and their association with main vegetation types: Pandeiros (PAN), the six populations of the east lineage (DIA, CAP, CAM, BLU, PEU, and PSB), and five populations of the west lineage (TOC, ALT, FIG, POC, and ITA)
Two distinct approaches examined the likely placement of PAN relative to the two lineages of C. fissilis. Firstly, we carried out a principal coordinate analysis (PCoA) using the chord distance (Cavalli-Sforza and Edwards 1967) at the population level. The software POPULATIONS (Langella 1999) produced the pairwise distance matrix; the PCoA was carried out in the software GenAlEx v6.5 (Peakall and Smouse 2012). Custom-made scripts using the RGL package in the R programming language version 3.3.3 (R Development Core Team 2014) allowed for the visualization of the first three principal coordinates.
Secondly, the Bayesian model-based approach using the “Clustering of individuals” module, as implemented in the software BAPS v6 (Corander et al. 2008), inferred hidden genetic structure within the dataset, at the sample level. In BAPS, the number of clusters is treated as an unknown parameter (Corander et al. 2003). We input the following models independently for the “population mixture analysis”: K = 2, K = 3, and K = 4, with five replicates for each model. Then, BAPS performed a “population admixture analysis” for each of the three sets of K. This time, we selected the “admixture based on mixture clustering” option; BAPS was not provided with the information about the population of origin of the sample. For the admixture analysis, the minimum size of the population and the number of reference individuals were five, with 100 overall iterations and ten iterations for reference individuals.
Genetic diversity
Initially, the software MICROCHECKER (Van Oosterhout et al. 2004) estimated the frequency of scoring errors and the presence of null alleles in the microsatellite data of mother trees and offspring. The method ENA (excluding null alleles), as implemented in the software FREENA (Chapuis and Estoup 2007), estimated FST-ENA, a version of FST that avoids the positive presence of null alleles induces on FST (Weir 1996). To estimate the frequency of null alleles, FREENA used the EM algorithm (Dempster et al. 1977). Subsequently, FREENA made estimations to produce global and pairwise FST and FST-ENA, with a total of 10,000 replicates to calculate the bootstrap 95% confidence interval (95% CI).
We defined the following three arrays: (1) mother trees, (2) offspring, and (3) families. The software GenAlEx v6.5 (Peakall and Smouse 2012) inferred the average number of alleles per locus (A), number of effective alleles (NE), number of private alleles (Ap), unbiased expected heterozygosity (uHe), observed heterozygosity (Ho), coefficient of inbreeding (F), fixation indexes (FIS and FIT) (Hartl and Clark 1997), and the estimators of genetic differentiation (GST and GIS) (Nei 1973). Statistical analyses were carried out using the software JAMOVI v0.8.1.17 (0.Jamovi Project 2018). The Mann-Whitney U test and the Kruskal-Wallis test carri0ed out comparison between medians from genetic parameters.
The software POPGENE v4.7.0 (Rousset 2008) estimated the Fisher’s exact test to assess deviations from the Hardy-Weinberg equilibrium (HWE). The Holm-Bonferroni correction method (Holm 1979) was applied during estimates of linkage disequilibrium (LD). For locus across groups, we applied the multiple sample score U test for heterozygote deficiency and heterozygote excess (Raymond and Rousset 1995). Additionally, we performed the exact test for genotypic disequilibrium between all pairs of loci for offspring, with the following settings: 10,000 MCMC (Markov chain Monte Carlo), 100 batches, and 5000 iterations per batch (Rousset 2008).
The software INEST v2.2 (Chybicki and Burczyk 2009) obtained the average inbreeding coefficient with the null allele corrections (Fnull) for the offspring, therefore uncovering the effect of null alleles over inbreeding. The estimations used a Bayesian approach, with 500,000 MCMC iterations, and the thinning parameter was set to 5000 and burning to 50,000 cycles. We analyzed the full model (nfb), which includes estimations for the presence null alleles (n), inbreeding (f), and genotyping failures (b). We measured the robustness of the model by comparing the analysis using a random mating model (F = 0), determining which model fit the data better by choosing the model with the lowest DIC (deviance information criterion) value (Chybicki and Burczyk 2009).
Genetic structure
The software POPULATIONS produced the pairwise distance matrix for the 18 families using the chord distance (Cavalli-Sforza and Edwards 1967), and PCoA was carried out in the software GenAlEx v6.5 (Peakall and Smouse 2012). Custom-made scripts using the RGL package in the R allowed for the visualization of the first three principal coordinates. The package NbClust (Charrad et al. 2014) used a total of 23 methods to search the PCoA eigenvectors for determining the best number of clusters. Then, NbClust used the majority rule to suggest the number of cluster that fit best the PCoA dataset. A Bayesian model-based approach used the “Clustering on groups of individuals,” as implemented in BAPS. The best number of cluster obtained from NbClust was taken as the upper bound of the number models (K) we tested in BAPS. Then, we input the models independently for the “population mixture analysis,” with five replicates for each model, and selected “admixture based on mixture clustering.” BAPS was not provided with the information about which mother tree gave rise to which offspring. For the admixture analysis, the minimum size of the population and the number of reference samples were five, with the number of iterations of 100 and the number of iterations for reference samples of ten. Additionally, we estimated the gene flow among the inferred Bayesian groups from the “admixture analysis” using the option “Plot Gene-Flow” (Tang et al. 2009), as implemented in BAPS.
Mating system
The mating system was inferred using data from the 283 offspring obtained from the 18 mother trees of PAN. The analyses used both the mixed mating model (Ritland and Jain 1981) and the correlated mating model (Ritland 1989), applying the maximum expectation method (EM) as implemented in the software MLTR v3.4 (Ritland 2002). The standard error of the parameters was calculated from 1000 bootstraps; family was used as the resample unit. The following statistics were estimated: multilocus population outcrossing rate (tm), single locus population outcrossing rate (ts), mating among relatives rate (tm–ts), multilocus correlation of paternity (rp(m)), single locus correlation of paternity (rp(s)), effective number of pollen donors (Nep = 1/r(pm)), selfing sibs proportion (SSP), full-sibs proportion (FSP), and half-sibs proportion (HSP). The average coefficient of coancestry (Θxy) evaluated the genetic structure of the offspring within individuals between families (Sebbenn 2002; Carneiro et al. 2011). The variance effective population size (Ne(V)) and the number of seed trees for seed collection (m(150)) required to achieve a reference effective population size of 150 trees were calculated in accordance with a previous suggestion (Carneiro et al. 2011).
Results
Genealogical placement of PAN
The results of the PCoA suggested that the 12 populations of C. fissilis were pulled together according to the range of origin, therefore forming two major groupings. The first group contained the six populations of the east lineage of C. fissilis that had been sampled at the Atlantic range (Fig. 4; depicted in blue). The second group brought together the five populations of the west lineage, from the Chiquitano range (Fig. 4; depicted in green). Distant from the west lineage, PAN was a population that occupied a placement near the east lineage (Fig. 4; depicted in purple). Together, the three principal coordinates explained about 48% of the total variation.

Plot of the first three principal coordinates for 12 populations of Cedrela fissilis, based on the Cavalli-Sforza and Edwards distance measure. Color code according to origin: blue, six populations of the east lineage (at the Atlantic range); green, five populations of the west lineage (at the Chiquitano range); red, PAN. Eigenvalues for each of the principal coordinate are shown in parenthesis
Admixture inferences shed further light into the genealogical placement of PAN within the east lineage and revealed hidden aspects of the genetic structure of PAN. When the K = 2 model was used (Fig. 5; top), the BAPS analysis indicated that the smaller Bayesian group (12 out of 18 mother trees of PAN) was split away from the larger group (six mother trees of PAN, together with 98 specimens of the six populations of the east lineage and 77 specimens of the five populations of the west lineage). When the K = 3 model was used (Fig. 5; bottom), the BAPS analysis showed that the previous split of PAN into two distinct Bayesian groups (as seen in the K = 2 model) was statistically stable. Furthermore, the K = 3 model indicated that the larger group (as seen in the K = 2 model) gave rise to two new groups, containing each either the east or the west lineages of C. fissilis, with few exceptions. In the K = 3 model, few specimens of the east lineage came together within the group formed mostly with the specimens of the west lineage, and vice versa.

Plots from admixture inferences obtained with K = 2 and K = 3 in BAPS for 12 populations (193 specimens) of Cedrela fissilis: six populations of the east lineage (at the Atlantic range), five populations of the west lineage (at the Chiquitano range), and PAN. Along the x-axis, each vertical bar represents a specimen. Along the y-axis, membership coefficient of a sample represents the fraction of the sample’s genome that has ancestry in a given Bayesian group (color-coded as indicated)
Genetic diversity
Once we have established the genealogical placement of PAN near the east lineage of C. fissilis, we shifted the analytic tools. Instead of tools for analyses at a large geographic scale, we used statistical tools that allowed us to understand the local forces that shaped the genetic diversity and differentiation within PAN.
The frequencies of null alleles were below the threshold value of 0.19; thus, dataset from all ten loci of the microsatellite markers was considered suitable for the subsequent analyses.
Ten nuclear microsatellite loci allowed us to identify a total of 189 alleles in PAN: 119 for the set of 18 mother trees and 178 for the set of 283 offspring (Table 1). Therefore, there was an influx of 58 new alleles through pollen gene flow that originated from nonsampled donor trees. All ten loci were polymorphic, both in mother trees and offspring. Across markers within mother trees, values of A ranged from 4 (Ced54) to 18 (CF66B) (Table 2); and within the offspring, it ranged from 7 (Ced54) to 32 (Ced44) (Table 3). Across families, values of A ranged from 4.2 (family 1) to 6.9 (family 18) (Table 4). For the set of 18 mother trees, the overall mean of the observed heterozygosity (Ho = 0.80) was lower than the observed unbiased heterozygosity (uHe = 0.85); the overall mean number of private alleles (Ap = 1.1) was very low (Table 2). Compared to mother trees, the offspring showed lower mean values of Ho (0.74) and uHe (0.82) and a much higher value of Ap (7.1; Table 3). There was a significant difference (p < 0.01) between values of Ap of mother trees compared to offspring. For the mother trees, the coefficient of inbreeding (F) ranged from − 0.048 (Ced2) to 0.232 (CF66A) (Table 2). For the offspring, values of F ranged from − 0.001 (Ced44) to 0.228 (Ced131) (Table 3). The overall mean values of F for the mother trees (F = 0.06) and the offspring (F = 0.10) were significantly different from 0 (p < 0.05).
For the offspring, six out of ten loci showed a significant deviation from the HWE. There were five loci (Ced2, Ced41, Ced44, Ced54, and CF66A) that exhibited heterozygote excess, while one locus (CF66B) showed heterozygote deficit (Table 3). The Holm-Bonferroni correction method indicated (p < 0.05) that there was LD between pairs of loci in five families: 4 (Ced41-CF26, Ced41-Ced2, Ced41-Ced131), 6 (Ced2-Ced44), 14 (Ced2-Ced44), 15 (Ced54-CF66B, Ced54-Ced44), and 17 (Ced2-Ced44).
Within families, the overall mean Ho (0.732) was higher than uHe (0.689; Table 4) and the values were significantly different (Ho > uHe; p < 0.001). As a consequence; the fixation index was negative (F = − 0.061) and significantly different from 0 (p < 0.001), which suggested deviations from random mating (Table 5). Among families, there were significant differences (p < 0.05) for A, uHe, and the number of effective alleles (NE; Table 4).
The overall mean of the inbreeding coefficient across families with null allele corrections (Fnull = 0.020) was obtained using INEST. Overall mean Fnull was different from 0 (p < 0.001). The inbreeding component of the model (f) played an important role in explaining the departures of HWE or LD in a total of five out of 18 families (4, 6, 11, 17, and 18). A single family (17) exhibited Fnull that did not overlap 0 (Table 4).
Differentiation among the 18 families
There was a significant (p < 0.01) genetic divergence (FST = 0.174; FST-ENA = 0.169, and GST = 0.163) among the 18 families of PAN (Table 5). The presence of null alleles biased the estimation of the fixation index, with FST-ENA being significantly different (p < 0.01) from FST.
The hierarchical analysis of molecular variance (AMOVA) using FST as the molecular distance indicated that there was no significant divergence (FST = 0.004; p = 0.057) between mother trees and their offspring (Table 6). Heterozygote deficit (FIS = 0.100) reached 10% of the variability among individuals. When RST was as molecular distance, AMOVA showed that the genetic divergence was strong (RST = 0.317; p < 0.001). Thus, SMM explains about 32% of the differences between mother trees and their offspring (Table 6).
Among families, AMOVA was significant for both FST (0.167; p < 0.001) and RST (0.047; p < 0.001), which corresponded to about 16 and 5% of total variance, respectively (Table 6). Differences among individuals within families (FIS = − 0.066; p = 1.00, RIS = 0.652; p < 0.001) were small and nonsignificant for the first and large and significant for the second (Table 6).
The results of PCoA revealed hidden genetic structures among the 18 families (Fig. 6). According to the majority rule as implemented in NbClust, seven methods suggested that the best number of clusters was six (Fig. 6; depicted as A to F). The number of families within a given cluster ranged from one to eight (Table 4).

Plot of the first three principal coordinates for 18 families of a population Cedrela fissilis (PAN) based on the Cavalli-Sforza and Edwards distance measure. NbClust defined the number of clusters (A to F, as indicated). Eigenvalues for each of the principal coordinate are shown in parenthesis
During the mixture analysis in BAPS, we used six models to probe how clustering would take place among the 283 offspring. The models ranged from K = 2 up to K = 6 (as six was the best number of clusters according to the preceding NbClust analysis). The results from three models (K = 2, K = 4, and K = 6) were presented (Fig. 7), with the 283 offspring arranged according to their family of origin (1 to 18). In the K = 2 model, the offspring were split into either of two Bayesian groups. Across the subsequent analyses (K = 4 and K = 6), the groups recovered in K = 2 model underwent further subdivisions.

Plots from mixture inferences obtained with K = 2, K = 4, and K = 6 in BAPS for 283 offspring from 18 families (1 to 18, as indicated) of a population Cedrela fissilis (PAN). Along the x-axis, each vertical bar represents a specimen. Along the y-axis, membership coefficient of a sample represents the fraction of the sample’s genome that has ancestry in a given Bayesian group (color-coded as indicated)
The composition of the groups of NbClust showed no apparent correspondence with the composition of the Bayesian groups recovered in BAPS. Nevertheless, the composition of six groups (K = 6; in BAPS) exhibited some degree of association with the geographic distribution of the families (Fig. 8). For instance, eight families (7 to 12, and 18; depicted in purple) were each sampled from mother trees located in close proximity within a single sampling site at the Pandeiros River Basin. Three families (2, 14, and 17; depicted in green) were collected each from neighboring mother trees at the Mata Seca State Park. Another set of three families (3, 4, and 5; depicted in pink) were from a separate sampling site at the Pandeiros River Basin. However, there were instances in which two neighboring mother trees gave rise each to a family that belonged to a discrete Bayesian groups. For example, the mother trees that gave rise to families 6 and 15 were from a single site but the families belonged to distinct groups. The pink group (3, 4, and 5) excluded family 16. Indeed, the geographic distribution of the families across the sampling area seems to have contributed to decrease gene flow among the six groups, as the Bayesian analysis of gene flow had demonstrated (Fig. 9). Mother trees within each of the six clusters received the contribution from genetically related pollen donors, therefore decreasing the rate of admixture among distinct genetic groups.

Geographic distribution of the 18 mother trees of a population of Cedrela fissilis (PAN) and mixture inferences obtained with K = 6 in BAPS for the 283 offspring. Each pie diagram represents the mean of the membership coefficients with the family. The pie size is proportional to the sample size of the family. NbClust defined the number of groups (N = 6; color-coded as indicated)

Gene flow network among the six groups of families (obtained with K = 6 in BAPS) for a population of Cedrela fissilis (PAN). A self-looping arrow denotes pollination took place with the own genetic composition of a given cluster. The direct arrow emerging from each cluster denotes the contributions made by means of gene flow among groups
Mating system
The overall mean for the multilocus outcrossing rate (tm) was 0.95. Across families, it ranged from 0.466 (family 6) to 0.998 (family 15) (Table 7). The averaged proportion of full-sibs (FSP = 13%), the overall mean half-sibs proportion (HSP = 82%), and the self-sib proportion (SSP = 5%) were shown (Table 7). Mating among genetically related individuals (tm–ts) showed that 7% of all offspring resulted from crossing among related trees (Table 7).
The value for the averaged multilocus correlation of paternity (rp(m) = 0.17) indicated that the probability of distinct families sharing the same pollen donor was low. However, rp(m) varied across families. There were instances in which most of the offspring of a given family were sired from the same pollen donor, such as the offspring of families 2 (0.59), 4 (0.35), 14 (0.24), 15 (0.26), and 16 (0.28). Thus, within each of those five families, the offspring combined half-sibs with self-sibs or self-half-sibs mostly (Table 7). The overall value of the crossing between pollinated related trees (rp(s)–rp(m) = − 0.04) was low and not significant (p = 0.236); thus, as a general trend, a given mother tree received pollen from few, unrelated donors. The overall mean for coancestry coefficient within families (Θxy = 0.184; p < 0.001) was significant. Among families, the values of Θxy ranged from 0.154 (family 5) to 0.292 (family 6); therefore, families 5 and 6 were of highly contrasting origin. Whereas the offspring of family 5 consisted mostly of half-sibs (HSP = 0.83), the offspring of family 6 was mostly self-sibs (SSP = 0.53; Table 7). The variance effective size (Ne(v)) was 3.03 and the number of seed trees per seed collection (m(150)) was 50.7 (Table 7).
Discussion
A local gene pool located in central Brazil
This study investigated PAN, a population of C. fissilis that is located within ecotones in NMG, toward central Brazil. We addressed a previous suggestion, by which populations of C. fissilis from small blocks of seasonal forests within central Brazil would detain gene pools entirely derived from parental sources located in larger blocks of seasonal forests at either the eastern or western side of the Cerrado, with possible admixture taking place where the two lineages reconnected (Garcia et al. 2011; Mangaravite et al. 2016).
Unexpectedly, most mother trees of PAN (12 out 18 specimens) belonged neither to the east nor to the west lineages. Instead, they clustered together into a third Bayesian group, a finding that did not fit within what we had anticipated. Lineages that exhibited local occurrences were not apparent from previous large-scale surveys (Mangaravite et al. 2016). The genetic variation of the remaining six mother threes of PAN matched the pattern expected for the east lineage. Thus, the current gene pool of PAN likely detained some degree of admixture because it seems to combine genetic components of both the east lineage (from the Atlantic range) and the third local lineage we just uncovered in this study.
The likely origin of the local lineage remains enigmatic. When the geographically isolated populations of C. fissilis endured genetic isolation within NMG owing to climatic changes that occurred in the Neotropics during Pleistocene (Whitmore and Prance 1987; Mayle 2004; Pennington et al. 2004), novel genetic variation might have the chance to accumulate over time through genetic drift and could explain why PAN exhibited a high degree of differentiation from the west lineage and some degree of differentiation from the remaining populations of the Atlantic range. In case the genetic drift we observed in PAN occurred recurrently within other ecotones across central Brazil, it is likely that additional populations of C. fissilis will exhibit genetic differentiation from either the east or the west lineages, with levels of differentiation varying depending upon the time the population remained in genetic isolation. Such lineages exhibiting genetic differentiation at the local scale are yet to be identified. It is plausible to assume that such scenario of genetic differentiation included other plant species that were co-distributed with C. fissilis; those species may have developed similar patterns of local genetic differentiation when their populations remained trapped within geographic isolated blocks of seasonal forests within the Cerrado or Caatinga.
Genetic diversity scattered among small subpopulations
The set of ten microsatellite markers uncover an appreciable amount of genetic diversity and a number of alleles within the 18 families of PAN. The dataset allowed for the realization of parentage estimations with confidence. Initially, we suspected that the presence of null alleles could explain the significant deviations from random mating we observed within each family of PAN. Deviations from the Hardy-Weinberg equilibrium and linkage disequilibrium have been suggested as a result of the presence of null alleles (Goicoechea et al. 2015; Islam et al. 2015). In our study, however, the frequencies of null alleles remained below the threshold value of 0.19 (Chapuis et al. 2008); thus, the presence of null alleles was assumed to detain little or no effect on shaping diversity and mating system estimations in PAN.
At first, we anticipated that PAN exhibit a lack of population structure, owing to high levels of gene flow within the population. Cedrela fissilis is a wind-dispersed species, with winged seeds adapted to long-distance dispersals (Styles 1981). At first glance, PAN resided within a small area that lacked observable barriers that would restrict gene flow among sampling sites. The ecotone that holds PAN consisted of small blocks of seasonal forests scattered within a matrix of open vegetation—either Cerrado or Caatinga—with significant anthropogenic activities that converted the natural vegetation into grassland and agricultural areas (Nunes et al. 2009; Sales et al. 2009; Bethonico 2010). Unexpectedly, the split of the 18 families into six Bayesian groups suggested that PAN detained population structure at levels beyond what we anticipated. Within PAN, gene flow via pollen seems to be severely restricted. For example, families 6 and 15 came from mother trees that were only 1.4 km apart. Nevertheless, those two mother trees produced no offspring together Fig. 9), rather each one of them originated distinctive families. The lack of cross-pollination between the mother trees of families 3, 4, and 5 and the mother tree of family 16 is another example of limited pollen flow within PAN, because those two sets of mother trees were located about 2.8 km apart (Fig. 8).
In Cedrela, the inflorescences display thyrses with a proportion of female to male flowers of about 1♀:2♂ (Gouvêa et al. 2008). Several genera within Swietenioideae, including Cedrela, share an inflorescence arrangement in which the central flower of a cyme or a three-flowered cymule is female, while the lateral flowers are male (Styles 1972). On certain occasions, however, cymules may display only male flowers; more rarely, the flowers may become all functional females. Thus, the number of male flowers may exceed the number of functional female flowers (Gouvêa et al. 2008). Nutrition and other environmental factors may influence the tendency toward either maleness or femaleness; moreover, the proportion of female to male flowers can also vary during a given flowering season (Styles 1972). In PAN, it is likely that differences in the availability of flowers, different sex proportions, protogyny, phenological differences, and availability of suitable pollinators contributed to gene flow to become restricted among neighboring mother trees and allowed for high genetic differentiation among families, such as families 6 and 15, and 3–4–5, and 16. Moreover, in a scenario that included maleness, mother trees with a high proportion of male to female flowers would become less receptive to incoming gene flow; thus, such scenario would explain why families in the groups orange (families 1 and 16), cyan (15), and yellow (6) had their origin owing to self-pollination exclusively Fig. 9). On the other hand, femaleness or a lower proportion of male flowers would account for the higher than average receptivity to incoming gene flow from external sources into family groups purple (families 7 to 13, and 18), pink (3, 4, and 5), and green (2, 14, 17) (Fig. 9).
Several statistics (Ho, uHe, Ap, and F) showed significant differences between mother trees and offspring, thus suggesting that PAN is under inbreeding. Although nonsampled donor trees were sources of new alleles, the origin of some families was highly dependent upon mating among relatives. We found significant differences between the fixation indexes FST and FST-ENA. Such bias (about 3%) likely took place when true heterozygotes were falsely considered as (null) homozygotes during data acquisition prior to analyses with FREENA. The strong differentiation among families confirmed that barriers at the local scale interrupted pollen flow among subpopulations. Within the landscape, for example, the subpopulation of eight mother trees that gave rise to families that came together as group A (Fig. 6)—and came together as the purple Bayesian group (Fig. 4)—was neighbors within a grassland area of about 5.6 ha; this subpopulation was about 9 km from any other mother tree sampled for this study. Moreover, such subpopulation likely comprised descendents from a recent ancestor; thus, the probability of correlated mating became high within the site and may explain the origin of group A—and the purple Bayesian group.
High levels of kinship within families
With an average value of tm ~ 1.0 for most families of PAN, C. fissilis behaved predominantly as an outcrossing species. However, at least some of the seeds within each family resulted from selfing or biparental inbreeding. The exceptionally low values of tm suggested that families 1 (tm = 0.82) and 6 (tm = 0.47) displayed a mixed mating system (0.2 < tm ≤ 0.8; Goodwillie et al. 2005). Within families 1 and 6, levels of selfing and mating between related parents were higher than the average value of PAN. Family 1 comes from a mother tree that was apparently an isolated tree located within a landscape with no observable pollen donors nearby. Family 6 comes from a mother tree that had three mature trees at close proximity (less than 50 m), but those trees did not provide pollen as they did not set flowers during the study season. Families 1 and 6 fit well within the suggestion that outcrossing rates and distances among reproductive individuals are negatively associated, with the tendency of geographically isolated trees to become reproductive isolated trees (Rymer et al. 2013; Vinson et al. 2015).
The average value of correlated mating within families (rp(m) = 0.17; Table 7) indicated that parents held some degree of relatedness. The low value of the effective number of pollen donors (Nep = 8.95; Table 7) indicated that the majority of the offspring within a given family resulted from related crosses, with only two to ten pollen donors per family. The coancestry coefficient within families (Θxy = 0.184; Table 7) was closer to the value expected for half-sibs families than full-sibs families (Θxy = 0.125 and Θxy = 0.250, respectively; Sebbenn 2006). In PAN, therefore, Θxy was about 47% higher than the value expected for a panmictic population (Sebbenn 2006). Mating among relatives, such as that we uncovered in PAN, can arise when pollinators visit persistently related neighbors. Nocturnal bees, butterflies, and thrips (Thysanoptera) are able to carry out pollination in several species of Meliaceae (Patino-Valera 1997). In the tropical tree Bagassa guianensis, wind-borne trips arrive in great numbers as they visit receptive flowers and distribute pollen among trees (Silva et al. 2008). In PAN, trips may carry out pollination among nearby, possibly related trees, as those insects are short flyers (Silva et al. 2008).
Given that the value of the half-sibs proportion (HSP = 0.82) was higher than the full-sibs proportion (FSP = 0.13) and that sharing of pollen donors was an uncommon feature (rp(m) = 0.17), we concluded that PAN is under a nonrandom mating pattern. Flowering asynchrony, small population size, and the foraging behavior of pollinators systematically visiting near neighbor trees (Sebbenn 2006) can be factors that account for the correlated mating we observed in PAN. Moreover, in a low-density species, such as C. fissilis (Carvalho 1994), pollen availability is naturally less diverse. In contrast, high-density species may rely on multiple pollen sources and pollen donors are shared with high frequency (Murawski and Hamrick 1991).
Conservation perspectives
Our study population (PAN) occurred within isolated blocks of seasonal forests near the westernmost border of the Atlantic range, toward central Brazil. The ecotone in NMG is the meeting point of three major vegetation covering: patches of seasonal forests are intermingled with either the Cerrado or the Caatinga. Our results suggested that PAN detained unique blend of alleles. Thus, the gene pools of either west or east lineages of C. fissilis cannot serve as a replacement for the gene pool of PAN.
Although PAN is secured within three neighboring conservation units—Pandeiros River Basin Environmental Preservation Area, Peruaçu Caves National Park, and Mata Seca State Park—the almost complete absence of both seedlings and juvenile trees across the sampling sites (Dias-Soto, unpubl. data) is worrisome and suggests that tree replacement across generations may become a severe threat to C. fissilis in NMG in the near future. Thus, the genetic diversity of PAN deserves immediate attention and requires conservation management plans.
Although the average number of pollen donors was about 9 (Nep = 8.95; Table 7), PAN detained some mother trees with exceptionally low number of pollen donors and little pollen diversity. This situation promotes selfing and correlated mating and can lead to isolated trees on grassland or forest patches to produce offspring with low genetic diversity. In PAN, the genetic diversity was unevenly distributed across families, which imply that not all mother trees were equally suitable as seed sources for a germplasm bank that aims the reintroduction or the enrichment of C. fissilis in NMG. We recommend measures to be taken to protect seed and pollen donors with a significant role in maintaining the remaining genetic diversity of PAN.
In PAN, the variance effective size (Ne(v) = 3.0) was lower than expected under the random mating expectations (Ne(v) = 4.0; Sebbenn 2006). Therefore, seed collection for conservation genetics, progeny tests, and reforestation must be taken from about 51 mother trees (m(150) = 50.7) to retain an effective population size of 150 trees. In case PAN had been a panmictic population, seed collection would require 38 mother trees (m(150) = 37.4; Sebbenn 2006). During the study season (2016–2017), most of the mature trees of PAN set neither flower nor seeds. We located only 18 fruit-bearing trees, among a total of 104 mature trees (Dias-Soto, unpubl. data). Local environmental factors, such as the accessibility to suitable levels of water and nutrient and availability of pollinator agents, can play a crucial role in determining the success of flowering and fruit production within a given season; most likely, those factors determined the low success in fruit production we observed. Based on our field experience, we anticipate that the identification of 51 mother trees for seed collection would be challenging and time-consuming in PAN. Our results strongly suggest that seed collection for conservation purpose should not be carried out on few, easily accessible donor trees. Alternatively, collecting few seeds from as many donor trees as possible would represent a more efficient strategy for the genetic conservation of C. fissilis.
References
Bawa KS (1977) The reproductive biology of Cupania guatemalensis Radlk. (Sapindaceae). Evolution 31:52–63
Bawa KS, Perry DR, Beach JH (1985) Reproductive biology of tropical lowland rain forest trees. I. Sexual systems and incompatibility mechanisms. Am J Bot 72:331–345
Bethonico MBM (2010) Rio Pandeiros: território e história de uma área de proteção ambiental no norte de Minas Gerais. Acta Geogr 3:23–38
Carnaval AC, Hickerson MJ, Haddad CFB, Rodrigues MT, Moritz C (2009) Stability predicts genetic diversity in the Brazilian Atlantic forest hotspot. Science 323:785–789
Carneiro FS, Lacerda AEB, Lemes MR, Gribel R, Kanashiro M, Wadt LHO, Sebbenn AM (2011) Effects of selective logging on the mating system and pollen dispersal of Hymenaea courbaril L. (Leguminosae) in the eastern Brazilian Amazon as revealed by microsatellite analysis. Forest Ecol Manag 262:1758–1765
Carvalho PER (1994) Espécies florestais brasileiras: recomendações silviculturais, potencialidades e uso da madeira. EMBRAPA-CNPF/SPI, Brasilia
Cavalli-Sforza LL, Edwards AWF (1967) Phylogenetic analysis: models and estimation procedures. Evolution 21:550–570
Chapuis MP, Estoup A (2007) Microsatellite null alleles and estimation of population differentiation. Mol Biol Evol 24:621–631
Chapuis MP, Lecoq M, Michalakis Y, Loiseau A, Sword GA, Piry S, Estoup A (2008) Do outbreaks affect genetic population structure? A worldwide survey in Locusta migratoria, a pest plagued by microsatellite null alleles. Mol Ecol 17:3640–3653
Charrad M, Ghazzali G, Boiteau V, Niknafs A (2014) NbClust: an R package for determining the relevant number of clusters in a data set. J Stat Softw 61:1–36
Chybicki IJ, Burczyk J (2009) Simultaneous estimation of null alleles and inbreeding coefficients. J Hered 100:106–113
Corander J, Marttinen P, Sirén J, Tang J (2008) Enhanced Bayesian modelling in BAPS software for learning genetic structures of populations. BMC Bioinformatics 9:539
Corander J, Waldmann P, Sillanpää MJ (2003) Bayesian analysis of genetic differentiation between populations. Genetics 163:367–374
Cota-Sánchez JH, Remarchuk K, Ubayasena K (2006) Ready-to-use DNA extracted with a CTAB method adapted for herbarium specimens and mucilaginous plant tissue. Plant Mol Biol Rep 24:161–167
Dempster AP, Laird NM, Rubin DB (1977) Maximum likelihood from incomplete data via the EM algorithm. J R Stat Soc B 39:1–38
Gandara FB (2009) Diversidade genética de populações de Cedro (Cedrela fissilis Vell. (Meliaceae) no Centro-Sul do Brasil. D. Phil. thesis, Universidade de São Paulo
Gandara FB, Tambarussi EV, Sebbenn AM, Ferraz EM, Moreno MA, Ciampi AY, Vianello RP, Grattapaglia D, Kageyama PY (2014) Development and characterization of microsatellite loci for Cedrela fissilis Vell (Meliaceae), an endangered tropical tree species. Silvae Genet 63:240–243
Garcia MG, Silva RS, Carniello MA, Veldman JW, Rossi AAB, Oliveira LO (2011) Molecular evidence of cryptic speciation, historical range expansion, and recent intraspecific hybridization in the Neotropical seasonal forest tree Cedrela fissilis (Meliaceae). Mol Phylogenet Evol 61:639–649
Goicoechea PG, Herrán A, Durand J, Bodénès C, Plomion C, Kremer A (2015) A linkage disequilibrium perspective on the genetic mosaic of speciation in two hybridizing Mediterranean white oaks. Heredity 114:373–386
Goodwillie C, Kalisz S, Eckert CG (2005) The evolutionary enigma of mixed mating systems in plants: occurrence, theoretical explanations, and empirical evidence. Annu Rev Ecol Evol Syst 36:47–79
Gouvêa CF, Dornelas MC, Rodriguez APM (2008) Floral development in the tribe Cedreleae (Meliaceae, sub-family Swietenioideae): Cedrela and Toona. Ann Bot 101:39–48
Hartl DL, Clark AG (1997) Principles of population genetics. Sinauer Associates, Sunderland
Hensen I, Oberprieler C (2005) Effects of population size on genetic diversity and seed production in the rare Dictamnus albus (Rutaceae) in central Germany. Conserv Genet 6:63–73
Hernández G, Buonamici A, Walker K, Vendramin GG, Navarro C, Cavers S (2008) Isolation and characterization of microsatellite markers for Cedrela odorata L. (Meliaceae), a high value neotropical tree. Conserv Genet 9:457–459
Holm S (1979) A simple sequentially rejective multiple test procedure. Scand J Stat 6:65–70
International Union for Conservation of Nature [IUCN] (2017) The IUCN red list of threatened species. Version 2017-3. http://www.iucnredlist.org. Accessed 26 May 2018
Islam MS, Lian C, Kameyama N, Hogetsu T (2015) Analysis of the mating system, reproductive characteristics, and spatial genetic structure in a natural mangrove tree (Bruguiera gymnorrhiza) population at its northern biogeographic limit in the southern Japanese archipelago. J For Res 20:293–300
Jamovi Project (2018) Jamovi (Version v0.8.1.17). https://www.jamovi.org. Accessed 26 May 2018
Langella O (1999) POPULATIONS. Version 1.2.28. A population genetic software. CNRS, UPR9034. http://bioinformatics.org/;tryphon/populations. Accessed 26 May 2018
Lopes LE, Neto SD, Leite LO, Moraes LL, Capurucho JMG (2010) Birds from Rio Pandeiros, southeastern Brazil: a wetland in an arid ecotone. Rev Bras Ornitol 18:267–282
Loveless MD, Hamrick JL (1984) Ecological determinants of genetic structure in plant populations. Annu Rev Ecol Syst 15:65–95
Mangaravite E, Vinson CC, Rody HVS, Garcia MG, Carniello MA, Silva RS, Oliveira LO (2016) Contemporary patterns of genetic diversity of Cedrela fissilis offer insight into the shaping of seasonal forests in eastern South America. Am J Bot 103:307–316
Mayle FE (2004) Assessment of the Neotropical dry forest refugia hypothesis in the light of palaeoecological data and vegetation model simulations. J Quat Sci 19:713–720
Ministério do Meio Ambiente [MMA] (2014) Lista Nacional Oficial de Espécies da Flora Ameaçadas de Extinção. Portaria 443, December 17th, 2014. http://pesquisa.in.gov.br/imprensa/jsp/visualiza/index.jsp?data=18/12/2014andjornal=1andpagina=110andtotalArquivos=144. Accessed 26 May 2018
Muona O, Moran GF, Bell JC (1991) Hierarchical patterns of correlated mating in Acacia melanoxylon. Genetics 127:619–626
Murashige T, Skoog F (1962) A revised medium for rapid growth and bio assays with tobacco tissue cultures. Physiol Plant 15:473–497
Murawski DA, Hamrick JL (1991) The effect of the density of flowering individuals on the mating systems of nine tropical tree species. Heredity 67:167–174
Nei M (1973) Analysis of gene diversity in subdivided populations. Proc Natl Acad Sci U S A 70:3321–3323
Nunes YRF, Azevedo IFP, WV VMDM, Souza RA, Fernandes GW (2009) Pandeiros: o Pantanal Mineiro. MGBiota 2:4–17
Oliveira-Filho AT, Jarenkow JA, Rodal MJN (2006) Floristic relationships of seasonally dry forests of eastern South America based on tree species distribution patterns. In: Pennington RT, Ratter JA, Lewis GP (eds) Neotropical savannas and seasonally dry forests: plant diversity, biogeography and conservation. CRC, Boca Raton, pp 159–192
Olson DM, Dinerstein E, Wikramanayake ED, Burgess ND, Powell GVN, Underwood EC, D’amico JA, Itoua I, Strand HE, Morrison JC, Loucks CJ, Allnutt TF, Ricketts TH, Kura Y, Lamoreux JF, Wettengel WW, Hedao P, Kassem KR (2001) Terrestrial ecoregions of the world: a new map of life on earth. BioScience 51:933–938
Patino-Valera F (1997) Recursos geneticos de Swietenia y Cedrela en los Neotropicos: propuestas para acciones coordinadas. FAO, Rome
Peakall R, Smouse PE (2012) GenAlEx 6.5: genetic analysis in Excel. Population genetic software for teaching and research—an update. Bioinformatics 28:2537–2539
Pennington RT, Lavin M, Prado DE, Pendry CA, Pell SK, Butterworth CA (2004) Historical climate change and speciation: neotropical seasonally dry forest plants show patterns of both Tertiary and Quaternary diversification. Philos Trans R Soc B 359:515–538
Pennington TD, Muellner AN (2010) A monograph of Cedrela (Meliaceae). DH Books, Sherborne
Prado DE, Gibbs PE (1993) Patterns of species distributions in the dry seasonal forests of South America. Ann Mo Bot Gard 80:902–927
Queiroz LP (2006) The Brazilian caatinga: phytogeographical patterns inferred from distribution data of the Leguminosae. In: Pennington RT, Ratter JA, Lewis GP (eds) Neotropical savannas and seasonally dry forests: plant diversity, biogeography and conservation. CRC, Boca Raton, pp 113–149
R Development Core Team (2014) R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna
Raymond M, Rousset F (1995) An exact test for population differentiation. Evolution 49:1280–1283
Riahi M, Zarre S, Maassoumi AA, Attar F, Kazempour Osaloo S (2009) An inexpensive and rapid method for extracting papilionoid genomic DNA from herbarium specimens. Genet Mol Res 9:1334–1342
Ritland K (1989) Correlated matings in the partial selfer Mimulus guttatus. Evolution 43:848–859
Ritland K (2002) Extensions of models for the estimation of mating systems using n independent loci. Heredity 88:221–228
Ritland K, Jain S (1981) A model for the estimation of outcrossing rate and gene frequencies using n independent loci. Heredity 47:35–52
Rodrigues PMS, Azevedo IFP, Veloso MDM, Santos RM, Menino GCO, Nunes YRF, Fernandes W (2009) Riqueza florística da vegetação ciliar do rio Pandeiros, norte de Minas Gerais. MGBiota 2:18–37
Rousset F (2008) Genepop'007: a complete reimplementation of the Genepop software for Windows and Linux. Mol Ecol Resour 8:103–106
Rymer PD, Sandiford M, Harris SA, Billingham MR, Boshier DH (2013) Remnant Pachira quinata pasture trees have greater opportunities to self and suffer reduced reproductive success due to inbreeding depression. Heredity 115:115–124
Sales RH, Souza SCA, Luz GR, Costa FM, Amaral VB, Santos RM, Veloso RM, Nunes YRF (2009) Flora arbórea de uma floresta estacional decidual na APA Estadual do Rio Pandeiros, Januária/MG. MGBiota 2:31–41
Sebbenn AM (2002) Número de árvores matrizes e conceitos genéticos na coleta de sementes para reflorestamentos com espécies nativas. Rev Inst Florestal 14:115–132
Sebbenn AM (2006) Sistemas de reprodução em espécies tropicais e suas implicações para a seleção de árvores matrizes para reflorestamentos ambientais. In: Higa AR, Duque SL (eds) Pomares de sementes de espécies florestais nativas. FUPEF, Curitiba, pp 93–138
Silva MB, Kanashiro M, Ciampi AY, Thompsom I, Sebbenn AM (2008) Genetic effects of selective logging and pollen gene flow in a low-density population of the dioecious tropical tree Bagassa guianensis in the Brazilian Amazon. For Ecol Manag 255:1548–1558
Styles BT (1972) The flower biology of the Meliaceae and its bearing on tree breeding. Silvae Genet 5:175–182
Styles BT (1981) Swietenioideae. In: Pennington TD, Styles BT, Taylor DAH (eds) Flora Neotropica monograph: Meliaceae. New York Botanical Gardens, Bronx
Surles SE, Arnold J, Schnabel A, Hamrick JL, Bongarten BC (1990) Genetic relatedness in open-pollinated families of two leguminous tree species, Robinia pseudoacacia L. and Gleditsia triacanthos L. Theor Appl Genet 80:49–56
Tang J, Hanage WP, Fraser C, Corander J (2009) Identifying currents in the gene pool for bacterial populations using an integrative approach. PLoS Comput Biol 5:e1000455
Van Oosterhout C, Hutchinson WF, Wills DPM, Shipley P (2004) Micro-checker: software for identifying and correcting genotyping errors in microsatellite data. Mol Ecol Notes 4:535–538
Vinson CC, Dal’Sasso TCS, Sudré CP, Mangaravite E, Oliveira LO (2015) Population genetics of the naturally rare tree Dimorphandra wilsonii (Caesalpinioideae) of the Brazilian Cerrado. Tree Genet Genomes 11:46
Weir BS (1996) Genetic data analysis II: methods for discrete population genetic data. Sinauer Associates, Sunderland
Whitmore TC, Prance GT (1987) Biogeography and Quaternary history in tropical America. Clarendon, Oxford
Funding
This work was supported by The Minas Gerais State Foundation of Research Aid – FAPEMIG (grant numbers PPM 00561-15 and APQ 03504-15) and by The National Council of Scientific and Technological Development – CNPq (fellowship number PQ 305827/2015-4) to LOO. JMDS received financial support from the Universidad de Sucre for his doctoral training. IBAMA and IEF-MG provided collecting permits.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Data archiving statement
Microsatellite genotype data are available in Supplementary Table S1.
Additional information
Communicated by L. A. Meisel
Electronic supplementary material
Table S1
Genotypic data (diploid individuals) for the population PAN (mother trees and offspring) of Cedrela fissilis obtained with ten microsatellite loci (XLS 84 kb)
Rights and permissions
About this article

Cite this article
Diaz-Soto, J.M., Huamán-Mera, A. & Oliveira, L.O. Population genetics of Cedrela fissilis (Meliaceae) from an ecotone in central Brazil. Tree Genetics & Genomes 14, 73 (2018). https://doi.org/10.1007/s11295-018-1287-4
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11295-018-1287-4