
Abstract
Most commercially important rootstocks for peach [Prunus persica (L.) Batsch] had been selected for resistance to one or more of the root-knot nematode (RKN) species: Meloidogyne incognita, M. arenaria, and M. javanica. The peach root-knot nematode, M. floridensis (MF), is a relatively newly discovered threat to peach and is not controlled by resistance genes in “Nemared,” “Nemaguard,” and “Okinawa.” The “Flordaguard” peach seedling rootstock, conventionally bred to provide resistance to MF, has solely been used for low-chill peach production in Florida for over 20 years and has already shown signs of resistance breakdown. A source of high resistance to the pathogenic MF isolate (“MFGnv14”) was identified from wild peach Prunus kansuensis Rehder (Kansu peach), thereby suggesting the potential for broadening spectrum and increasing durability of resistance in peach rootstocks through interspecific hybridization with P. kansuensis. Using 12 F2 and BC1F1 populations derived from crosses between Okinawa or Flordaguard peach and P. kansuensis populations, we examined the genetic control for MF resistance by identifying associated microsatellite markers and determining genomic location of the resistance locus. One microsatellite marker (UDP98-025) showed strong and consistent association with resistance based on root-galling index. The resistance locus was mapped on the subtelomeric region of linkage group 2, co-localizing with other previously reported RKN resistance genes in Prunus. Segregation of gall-index-based resistance observed in F2 and BC1F1 populations is compatible with the involvement of a multiallelic locus wherein a dominant (Mf1) or recessive (mf3) resistance allele is inherited from P. kansuensis, and susceptibility alleles (mf2) from peach.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Development of peach rootstocks in the USA has historically been directed at improving nematode resistance in well-adapted varieties. The old nematode-resistant rootstocks such as “Shalil,” “Yunnan,” and “Bokhara” were resistant to Meloidogyne incognita but susceptible to Meloidogyne javanica (Hansen et al. 1956; Sharpe 1967). Continued efforts to broaden resistances in peach rootstocks while improving horticultural features have led to the development of rootstocks “Nemaguard,” “Nemared,” “Guardian,” and “Flordaguard” that were also effective against M. javanica (Okie et al. 1994; Ramming and Tanner 1983; Reighard and Loreti 2008; Sherman et al. 1991). The rootstock cultivar Flordaguard, with “Okinawa” and Prunus davidiana in its lineage, has been effective as a rootstock for peach production under Florida’s subtropical climate due to its low-chilling requirement, adaptability to non-alkaline soils, and ability to withstand infection by the endemic peach root-knot nematode (RKN), Meloidogyne floridensis (MF), which is not controlled by the RKN resistance (R) genes in Nemared, Nemaguard, and Okinawa rootstocks (Handoo et al. 2004; Olmstead et al. 2015).
Breeding for improved RKN resistance becomes increasingly important, as soil fumigants or nematicides are being phased out and even more so for production areas where the highly adaptive meiotic RKN species such as MF (Castagnone-Sereno 2006; Handoo et al. 2004) becomes a problem. Segregation analyses in F2 and BC1F1 interspecific populations from crosses of Okinawa or Flordaguard with Prunus kansuensis (PK) have established PK as a source of new alleles conferring high level of resistance against MF and revealed single-locus inheritance patterns (Maquilan 2017) congruent with what has been observed for other RKN R genes identified in Prunus (Claverie et al. 2004; Esmenjaud et al. 1996; Kochba and Spiegel-Roy 1975; Lu et al. 2000; Rubio-Cabetas et al. 1998; Van Ghelder et al. 2010; Yamamoto et al. 2001). These results suggest that interspecific hybridization with PK is a promising approach for broadening the genetic base of RKN resistance in existing rootstocks, mostly having been derived from Prunus persica and P. davidiana. Designing an efficient breeding strategy for the introgression of MF resistance from PK into locally adapted cultivars requires an understanding of the genetic control of resistance and an informed selection of parental allele combinations to increase the frequency of resistance alleles in the progeny.
Use of molecular markers tightly linked to the trait can accelerate the identification of desirable genotypes (Charcosset and Moreau 2004; Hospital 2005) especially those exhibiting nematode resistance, for which screening can be tedious and time-consuming. In this study, we used simple sequence repeat (SSR) microsatellite markers to identify genomic regions associated with MF resistance and utilize marker-trait linkage information to track the transmission of resistance alleles from PK into backcross progenies. Microsatellite markers are ideal for genetic mapping studies because of their abundance in the genome, codominance, high polymorphism, and transferability across species (Aranzana et al. 2003a; Aranzana et al. 2002; Claverie et al. 2004; Mnejja et al. 2010; Tautz 1989). Additionally, microsatellite markers have shown promise in marker-assisted selection of RKN resistance in several agronomic crops including rice, soybean, cotton, and peanut (Chu et al. 2011; Concibido et al. 2004; Jenkins et al. 2012; Miah et al. 2013). The Prunus reference map constructed from “Texas” almond × “Earlygold” peach F2 population is comprised of 562 markers including 185 SSRs (Aranzana et al. 2003b; Dirlewanger et al. 2004b; Joobeur et al. 1998). These markers have been used in initial mapping studies to locate underlying RKN R genes in several Prunus intra- and interspecific progenies (Cao et al. 2011; Claverie et al. 2004; Dirlewanger et al. 2004a). Genetic mapping studies have located Prunus RKN R genes Ma, RMja and Rjap in LG 7 at ≈ 2 cM from SSR marker pchgms6 (Claverie et al. 2004; Van Ghelder et al. 2010), as well as RMia, and PkMi in the subtelomeric region of LG 2 at ≈ 10 cM from the SSR marker UDP98-025 (Cao et al. 2011; Claverie et al. 2004).
Putative resistance genes, with the majority belonging to the toll/interleukin1 receptor (TIR) nucleotide-binding site (NBS) leucine-rich repeat (LRR) class, have been found to contain SSRs. In the peach genome, 60% of the disease-resistance-associated TIR-NBS-LRR genes with post-LRR regions contain SSR sequences in the intron between LRR and post-LRR exons, suggesting the potential utility of polymorphic SSRs for detecting and directly monitoring the introgression of disease resistance genes in breeding programs (Claverie et al. 2011; Lalli et al. 2005; Van Ghelder and Esmenjaud 2016). Multiple reported linkages for RKN resistance in the upper part of LG 2 and LG 7 indicate either clustering of genes or high allelic variability in these regions and therefore represent useful targets for generating rootstocks with broader spectrum and more durable resistance against RKN, as demonstrated for the three-way plum × (almond × peach) progeny containing completely dominant Ma, RMja, and RMia genes (Khallouk et al. 2013).
This study aims to identify SSR markers linked with MF resistance, determine the genomic location of the resistance locus, and track resistance introgression based on genetic linkage with the SSR marker in early-generation (F2 and BC1F1) interspecific populations derived from crosses between peach and PK while analyzing recombination patterns across the genome. Results from this study will provide a useful basis for further genetic studies and marker-assisted introgression of novel sources of resistance alleles from PK into well-adapted peach rootstocks.
Materials and methods
Plant material
Twelve F2 and BC1F1 populations developed from interspecific crosses between Prunus persica (L.) Batsch (peach) and P. kansuensis Rehder (Kansu peach) were evaluated for genetic segregation at select microsatellite marker loci. The P. persica cultivars Flordaguard (FG) and Okinawa (OK) are utilized as genetic material at the University of Florida (UF) stone fruit breeding program to breed for low-chill adaptation. Okinawa is a domesticated peach seedling rootstock originating from Japan (Sharpe 1957). Flordaguard is the standard rootstock used for subtropical peach production and has both Okinawa peach and P. davidiana in its parentage (Sherman et al. 1991). “UFSharp” (SH), a low-chill-adapted peach scion cultivar, was used as one of the backcross female parents because it blooms profusely (Chaparro and Sherman 2006). The P. persica genotypes OK, FG, and SH are homozygous susceptible for “MFGnv14,” a resistance-breaking isolate of M. floridensis (MF), and the observed differences in segregation patterns among populations derived from the P. persica × P. kansuensis (PK) cross suggested that the PK accession is heterozygous at the resistance locus (Maquilan 2017). Seven F2 populations were developed from selfing five OK × PK hybrid selections and two FG × PK hybrid selections. Five of these interspecific hybrids were backcrossed to either FG or SH. A total of 478 individuals from 12 populations, each consisting of 23–43 individuals, were initially genotyped and evaluated for resistance to MF. All progenies were grown and evaluated for MF resistance in the greenhouse.
Phenotyping
The response of individual genotypes to MF was assessed by subjecting 4-month-old seedlings to a high inoculum pressure of 10,000 eggs or second-stage juveniles from a pure population of the MFGnv14 isolate. After 4 months from inoculation, root systems were visually rated for the number of galls. Each genotype was assigned a root galling index (GI) of 0 = no galls, 1 = one to two galls, 2 = three to 10, 3 = 11 to 30, 4 = 31 to 100, or 5 = more than 100 (Taylor and Sasser 1978). Root GI was the criterion previously established to separate resistant and susceptible phenotypes, where in values above 2 indicate host susceptibility (Maquilan 2017).
Genotyping
DNA extraction and amplification
Genomic DNA was extracted from the progenies and parents of the crosses following the modified CTAB procedure described by Chavez and Chaparro (2011). Genomic DNA was diluted to a final concentration of 20 ng μL−1 in TE buffer for PCR amplification of SSR loci. The PCR reactions were conducted in a 16 μL volume; a total of 10 μL master mix including 2.25 μL of 10× Thermopol® reaction buffer (New England BioLabs), 1 μL of 2.5 mM dNTP, 0.2 U of Taq DNA polymerase (New England BioLabs), and 6.5 μL of DNA-grade water, was combined with 2 μL each of 5 μM fluorescent-labeled forward primer and 5 μM reverse primer, and 2 μL of 20 ng μL−1 of genomic DNA. The PCR amplification proceeded using the following temperature profile: 94 °C for 3 min, then 40 cycles of (94 °C for 45 s, specific primer annealing temperature for 30 s, 72 °C for 1 min) finishing with 72 °C for 7 min.
SSR analysis
The linkage map developed from an interspecific cross of Texas almond with Earlygold peach (TxE) was used as a reference (Genomic Database for Rosaceae http://www.bioinfo.wsu.edu/gdr/) for selecting the SSR marker loci. Initially, 81 SSR markers spanning eight linkage groups (LG) were screened for stable amplification, polymorphism between P. persica and P. kansuensis parental genotypes, and transmission of alleles in selected hybrids. Of the 59 that showed polymorphisms, only 39 were selected for genotyping the mapping panel consisting of seven F2 and five BC1F1 families based on their location on the Prunus TxE reference map (Supplementary data, Table S1 and Fig. S2). Three SSR loci were selected from the linkage maps of “Contender” peach × “Fla.92-2C” peach (Fan et al. 2010) and “Ferjalou Jalousia” peach × “Fantasia” peach (Dirlewanger et al. 2007) F2 populations to extend coverage in LG 4 and LG 5. Details of the location, primer sequences, and annealing temperature of the selected markers are provided as supplementary data (Table S3). The selected SSR markers were spread out over the eight linkage groups with average spacing ranging from 7 centimorgans (cM) in LG 7 to 25 cM in LG 6. The number of selected markers on each linkage group ranged from three for LG 3 to eight for LG 1. Such a marker density would be sufficient to screen each chromosome for quantitative trait loci (QTL) in early generation populations such as F2 and backcrosses where a large linkage disequilibrium between loci is expected (Bus et al. 2009).
A total of 478 individuals from 12 interspecific peach × P. kansuensis populations were genotyped for 39 genome-wide SSR markers. The highly polymorphic SSR loci revealed 131 genotypes having off-type alleles at one or more loci (1–21 SSRs) and were consequently removed from the mapping files. The F2 seeds were produced by open-pollination and BC1F1 seeds through controlled pollination of flowers. The off-types could have resulted from cross-pollination of neighboring plants or from seed mixup, with the latter more likely to have occurred in BC1F1families; these were excluded and the final mapping data sets comprised of 18–40 individual genotypes from 12 populations, for a total of 345 genotypes.
Genotypic data were obtained by visual scoring of allele bands separated by agarose gel electrophoresis or by analyzing fluorescent peaks generated by capillary electrophoresis. The PCR products were separated via gel electrophoresis on 3–4% agarose gels stained with ethidium bromide (run for 3–4 h at 210 V) and markers segregating for alleles that differed by more than 5 base pairs (bp) were scored from the gel images. Bands that differed by less than 5 bp and those that were fuzzy or too faint to be reliably scored were resolved by the capillary electrophoresis sequencer (ABI 3730 DNA Analyzer, Applied Biosystems) at the UF Interdisciplinary Center for Biotechnology Research (Gainesville, Florida). Genotypes were determined from electropherograms generated in GeneMarker® software (v.1.5, Soft Genetics) using the Liz600 size standard to determine the size of sample peaks.
Marker scoring
Marker scoring followed the genotype coding conventions of JoinMap® (Van Ooijen 2006) for F2 and BC population types. For F2 populations, individuals were scored as “A” when they were homozygous for the P. persica allele, “B” when homozygous for the P. kansuensis allele, and “H” when heterozygous. For backcross populations, only SSR markers heterozygous in the F1 parent or both parents were used. Letters A-D or Ø (null) were designated to the allelic variants of each marker (Supplementary File S4) and were later grouped based on their segregation patterns. The polymorphic markers segregated according to AB × CD, EF × EG, and LM × LL. Alleles represented by the letters B, F, and M originated from P. kansuensis, while the letters A, C, D, E, G, and L were from P. persica. Considering the BC1F1 populations as species-level backcrosses, alleles from P. persica were lumped into a single A allele and genotypes with only P. persica alleles were treated as homozygotes, whereas genotypes with the combined alleles of P. kansuensis and P. persica were re-coded as H when converting genotypic data to the BC-type format. The phenotypic marker (resistance to M. floridensis) was scored as a dominant marker: “C” for resistant phenotypes and “D” for susceptible phenotypes.
Error checking
Some markers yielded ambiguous genotyping results because of faint or diffused bands on the gel, spurious peaks or overlapping stutter peaks on the electropherogram, and presence of null alleles. To resolve genotype ambiguities and to confirm the presence of null or off-type alleles, PCR amplification and electrophoresis were repeated once or twice. The electrophoretic band patterns and the allele peak patterns produced from GeneMarker® were compared. Markers were checked for Mendelian inconsistencies that may have resulted from genotyping errors by chi-square tests using JoinMap. Segregation distortion was found to commonly occur among selfed progenies, wherein P. kansuensis alleles were underrepresented at the distal marker locus in LG 6, a region known to be linked with gametophytic self-incompatibility locus in Prunus (Dirlewanger et al. 2004b). The electrophoresis output was re-examined when significant (P < 0.05) distortions were observed. Markers that could not be reliably scored were removed before performing linkage analysis. Furthermore, genotype errors or failed PCR reactions, usually due to poor-quality of DNA sample, were marked as missing data and individuals with more than 10% missing data were removed from the mapping files to avoid erroneous marker associations. Some markers revealed multiple stutter peaks that made it difficult to distinguish the true allele peaks. The true allele composition was determined by comparing segregation patterns across individuals within the population.
Map construction
Individual maps
Maps containing 34 to 36 SSR markers were constructed for each of the 12 F2 and backcross populations using the regression mapping algorithm in JoinMap. Marker data were subjected to chi-square tests to check for deviations from the 1:2:1 expected segregation in F2 or 1:1 in backcross population. The presence and magnitude of segregation distortion in mapping populations were assessed under the “locus genotypic frequency” command in JoinMap. Segregation-distorted markers were retained in the map if they did not dramatically affect mapping distances and marker order.
Markers were grouped by their independence LOD (logarithm [base 10] of odds) scores ranging from 2.0 to 10.0 with steps of 1 LOD unit. Marker groupings with the highest possible LOD scores were selected from the “groupings tree,” and any ungrouped markers, including segregation-distorted markers, were assigned into the existing linkage groups based on their known locations on the Prunus TxE reference map. Within each linkage group, markers were ordered starting with the most informative pair and the remaining markers were positioned one by one in subsequent rounds using the default settings (minimum LOD threshold of 1.0; maximum recombination frequency of 0.40; goodness-of-fit jump threshold for removal of loci = 5.0) and a ripple performed after each marker addition. Marker order was established usually after one round of fitting. The LOD threshold was successively reduced (0.5, 0.1, and 0.01) when a set of markers could not be mapped due to insufficient linkage. In some datasets where only two informative markers were available for LG 3, parameters were adjusted to the lowest stringency (recombination frequency = 0.5, LOD > 0.01) to place the two distant markers. Recombination frequencies were converted to map distances in centimorgans using Kosambi’s mapping function.
The marker order was checked for consistency with that of the Prunus TxE reference map. Conflicts in the order of the markers were resolved using the “start order” and “fixed order” map building functions in JoinMap. The tightly linked markers on LG 7 often generated negative map distances that could not be resolved using the fixed order command. Redundant markers (where no recombination occurred between two adjacent markers) were eliminated if they caused conflicts in marker order that could not be resolved due to insufficient resolution of the mapping population.
Combined maps
Combined maps were constructed using individual maps from populations with common parental allele combinations. Multiple maps for each linkage group were integrated using the “combine groups for map integration” command in JoinMap. Marker pairs were examined for heterogeneity of recombination frequencies under the “heterogeneity test” tab sheet. Where significant linkage heterogeneity (P < 0.05) occurred, marker data were re-examined for genotyping errors or inconsistencies in marker order. The marker pairs with deviant recombination frequencies causing map inflation were excluded from map construction. In LG 7, linkage heterogeneity was usually caused by clustering of markers at the same position in some mapping populations, particularly between CPPCT039 and UDP98-405 (2–4 cM interval on the Prunus TxE reference map) and between CPPCT017 and EPDCU3117 (3 cM interval on the TxE reference map). Linkage heterogeneity also occurred in LG 5 where only 2–3 markers were mapped. Linkage map calculations were based on mean recombination frequencies and combined LOD values of common markers mapped in more than one population. The regression mapping approach was used for determining marker order, and similar parameters used for individual map construction were considered for the combined maps. Marker pairs failing to meet set criteria were excluded from the map construction. Linkage maps were drawn and aligned with MapChart version 2.3 (Voorrips 2002).
Data analyses
Statistical analyses were performed using the SAS software (version 9.4, SAS Institute Inc., Cary, North Carolina, USA). The distribution of phenotypic data (root galling index) was assessed visually with histograms and checked for normality using the Proc Univariate procedure of SAS. The 12 F2 and backcross families were grouped based on common segregation patterns or parental allele combinations, and pooled data sets were checked for homogeneity of variances using the Levene’s test, which is robust to non-normality. Chi-square goodness-of-fit tests were performed on phenotype segregation data to determine their agreement with the expected Mendelian ratios using the Proc Freq procedure in SAS. All statistical tests used a P value threshold of 0.05 unless otherwise noted.
Data sets from similarly grouped crosses were first separately analyzed for the presence of QTLs and then combined and analyzed together to increase the statistical power of detecting QTLs. Because the data did not follow a normal distribution and the data could not be transformed to fit the normality assumption for parametric tests, the non-parametric mapping function in MapQTL® (Van Ooijen 2009) based on Kruskal-Wallis (KW) test was used to explore individual associations between SSR markers and resistance based on root GI. A threshold level of P = 0.005 for genome-wide significance (Van Ooijen 2006) was considered for identifying marker(s) closely associated with the GI trait in both individual and combined families. Exact P values for the significant marker were obtained by a Kruskal-Wallis test in SAS (Proc Npar1way). The proportion of phenotypic variation (R2) accounted for by the significant marker locus was determined by a simple linear regression model for each family and combined families of a particular cross combination. In addition, a two-way ANOVA (Proc GLM) was performed to investigate main and interaction effects between marker genotype and family. When no significant family effects and marker × family interaction effects were observed, the data sets were pooled to provide improved estimates of the phenotypic effect of shared QTL alleles. Following a significant KW test, the effects of the genotypes at the associated marker locus were further examined with post hoc pairwise comparisons using the Wilcoxon rank-sum test run with α = 0.005 for two groups (BC genotypes segregating 1:1 at a locus) and the KW test for three groups (F2 genotypes segregating 1:2:1) with Bonferroni-adjusted critical P value (ά = 0.0017) to account for multiple comparisons. Ranks were assigned for GI values, ordered from lowest to highest, and the mean ranks of genotypes were compared. The genotype effects were expressed as mean GI values ± standard deviation (SD) followed by different letters denoting significantly different mean ranks among genotypes.
Results
Resistance segregation
A total of 345 individuals from 12 F2 and BC1F1 families derived from the cross P. persica × P. kansuensis were tested for resistance to the pathogenic isolate of M. floridensis (MfGnv14). Two different patterns of segregation for root galling index (GI) were observed among the seven F2 families, of which six showed 3:1 resistant (R) to susceptible (S) ratio (Pcombined = 0.894) and one showed a 1R:3S ratio (P = 0.494). The segregation patterns suggest that a major resistance locus is involved with resistance determined by a dominant allele in the families segregating 3R:1S, and a recessive allele in one family segregating 1R:3S. When backcrossed to a homozygous-susceptible P. persica genotype (Flordaguard [FG] or UFSharp [SH]), the BC1F1 progenies segregated 1R:1S (Pcombined = 0.916) as expected if the dominant resistance allele was inherited, and 0R:1S if the recessive resistance allele was inherited in the progeny save for three resistant individuals that might have escaped infection. The segregation ratios in each population and mean GI values for the R and S phenotypes are presented in Table 1.
The variation in segregation patterns among the F2 progenies indicate the heterozygous allelic status of the wild peach parent, P. kansuensis (PK), at the locus conferring resistance to M. floridensis (MF) and the segregation of two different PK alleles into the F1 hybrids. Through backcrosses to a homozygous-susceptible P. persica (PP) parent, we have confirmed that PK is the progenitor of MF resistance, for which we assigned the symbols “Mf1” and “mf3” to represent dominant and recessive resistance alleles, respectively. The symbol “mf2” was used to represent susceptible allelic forms of P. persica.
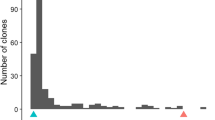
Based on common segregation patterns, the different crosses could be classified according to the different allele combinations in the F1 parent as follows: Mf1 mf2 × Mf1 mf2 and mf2 mf3 × mf2 mf3 for selfed F1 hybrids; and mf2 mf2 × Mf1 mf2 and mf2 mf2 × mf2 mf3 for backcrosses to P. persica. The phenotypic distributions of the different cross combinations for the GI trait are presented in Fig. 1. The GI trait displayed non-normal, strongly skewed distributions with the Mf1 mf2 × Mf1 mf2 crosses (N = 169) heavily skewed towards resistance and the mf2 mf3 × mf2 mf3 cross (N = 35) heavily skewed towards susceptibility. The mf2 mf2 × Mf1 mf2 crosses (N = 91) displayed the expected bimodal distribution, while the mf2 mf2 × mf2 mf3 crosses (N = 50) consisted almost entirely of susceptible types. The histograms further show that resistance to MF is controlled by a major locus with dominant and recessive resistance alleles. Combined information from populations having the same phenotypic distributions corresponding to similar allelic constitutions at the MF locus could be used to provide better estimates of allele effects and increase the power to detect QTLs due to the increased sample size (Mackay et al. 2009).

Histograms of the resistance response to a pathogenic isolate of Meloidogyne floridensis (“MFGnv14”) among 12 interspecific peach × Prunus kansuensis progenies grouped according to the parental cross. The populations segregate for dominant (Mf1) or recessive (mf3) resistant alleles from P. kansuensis and recessive susceptible (mf2) alleles from peach. The top and bottom panels show the distributions of root galling index among seven F2 progenies and five BC1F1 progenies, respectively
Polymorphisms and genotype segregation at microsatellite loci
Among the 39 SSR markers selected for mapping, 29 consistently showed informative polymorphisms across 12 peach × P. kansuensis populations, while the other markers were informative only in certain progenies. Markers CPPCT026, EPPISF032, BPPCT038, and UDP98-405 were not informative in some F2 progenies and backcrosses because the F1 parents inherited the common alleles of P. persica (Okinawa [OK] or Flordaguard [FG]) and P. kansuensis (PK), and similarly when the F1 parents were backcrossed to another P. persica genotype (UFSharp [SH]). Markers UDP96-013 and CPPCT005 displayed polymorphisms in the F2, but the PK alleles could not be traced in the backcross progenies due to shared alleles with the backcross parent (SH or FG). Marker CPDCT025 was monomorphic between OK and PK, while CPSCT034 did not show segregation in some F2 and backcross progenies.
For the four markers on LG 1 (CPSCT008, CPPCT029, BPPCT028, and CPPCT026) that showed unexpected homozygote frequencies, several independent rounds of PCR amplification and fragment analyses were conducted to compare the outputs and verify the scores. False homozygotes due to null alleles, allele dropout, and adjacent-allele heterozygotes were corrected. For CPSCT008, many genotypes tended to be misinterpreted as homozygous because the larger allele (234 bp) appears as a very low peak or a very faint band when the other allele (182 bp) is present. In the case of CPPCT029 and BPPCT028, the OK × PK and FG × PK F1 parents appeared to be homozygous, and there was no apparent segregation among the F2 progenies. However, the genotypes segregated 1:1:1:1 when backcrossed to SH, which is heterozygous for the marker; this could be explained by the F1 parent being heterozygous for the null allele. For BPPCT028, the heterozygous-null genotypes were distinguished from the homozygous genotypes by the extended range of their stuttering peaks. Segregating null alleles from marker CPPCT006 on LG 8 were also observed, but these could not be confirmed in most populations due to poor repeatability at the locus that is not attributable to poor-quality DNA. For CPPCT026, difficulty in differentiating stutters of homozygotes from adjacent-allele heterozygotes (191/189 bp) was frequently encountered in the segregating OK × PK F2 populations. The marker CPPCT019B on LG 8 presented a similar problem (for all 12 populations), which is a common occurrence for markers having a dinucleotide repeat motif (Clarke et al. 2001; DeWoody et al. 2006). Only the marker loci that gave consistent genotyping results were included in the mapping files.
Linkage maps
Linkage information from multiple populations was combined to improve the recombination frequency estimates and to construct maps with more accurate marker distances, as our common set of markers displayed varied degrees of polymorphism and recombination in different populations. The combined maps represented the four parental crosses: two for F2 populations (OK × PK and FG × PK) and two for backcrosses to P. persica (SH or FG × [OK × PK] and SH × [FG × PK]) (Supplementary data, Figs. S5–S8). The four combined maps consisted of 34 to 36 markers covering 59 to 64% of the TxE reference map. The marker coverage of the combined maps varied by linkage group. More markers were tested for LG 1, 2, 7, and 8 than for LG 3, 4, 5, and 6 (Supplementary data, Fig. S2). Several markers had to be tested in LG 1 and LG 8 because of the difficulty in identifying usable markers based on polymorphism. Because LG 1 is longer and more saturated with SSR markers than LG 8, more markers were used to map LG 1 (5 to 8 markers at 8.7 to 15.0 cM intervals) compared to LG 8 (two to three markers at 10.9 to 22.8 cM intervals). The region in LG 4 had the smallest coverage (28.6%) on the TxE map because two (EPPISF032 and CPDCT014) of the four markers were selected from different linkage maps. Greater coverage (> 70%) and sufficient marker density (5 to 8 markers with 6.2 to 14.1 cM intervals) were ensured for linkage regions in LG 2 and LG 7 where the root-knot nematode resistance genes for Prunus had been localized (Cao et al. 2011; Claverie et al. 2004; Van Ghelder et al. 2010).
The total map lengths ranged from 248.5 to 351.2 cM with the average marker spacing per linkage group ranging from 6.2 to 25.1 cM, excluding LG 3 in OK × PK and FG/SH [OK × PK] combined maps that were dramatically inflated by the two distant markers. Only three markers were used to map LG 3, being the smallest (48.4 cM) of the eight linkage groups in the TxE map, and one marker (CPDCT025 placed between BPPCT007 and CPDCT027) could not be used because it was monomorphic between OK and PK. Three loci (BPPCT008, BPPCT025, and CPPCT030) in LG 6 produced map lengths shorter than the TxE map by 53% in OK × PK and 56% in FG × PK combined maps. Comparison of total map lengths between F2 and backcross populations indicated increased recombination frequencies in the latter; the combined map lengths in backcross populations were longer by 28% in FG/SH [OK × PK]) and by 30% in SH × [FG × PK) than those of their respective F2 counterparts, OK × PK and FG × PK (Supplementary data, Tables S9–S12).
Marker-trait association
The Kruskal-Wallis (KW) test was performed to identify microsatellite regions in the genome that are significantly associated with the resistance to M. floridensis (MF) based on root GI values. A threshold of P < 0.005 was considered for genome-wide significance (Van Ooijen 2009). Because of the non-normal distribution of the GI trait, we used a non-parametric distribution-free test, which analyzes differences in medians for ordinal data. The KW test suggested the presence of QTL for MF resistance in LG 2 close to the UDP98-025 locus. Among the 34 to 36 SSR regions screened, no other marker locus showed a stronger association with the GI trait than UDP98-025 and there were no markers in other linkage groups that showed a genome-wide level of significance. Segregation distortion was not evident at the significant marker locus as determined by chi-square tests of 1:2:1 ratio in F2 progenies and 1:1 in backcrosses. The phenotypic variance explained (PVE) by the QTL ranged from 23.9 to 80.4% when resistance is dominantly conferred in the progeny and 8.6 to 22.4% when resistance is recessively conferred. The degree of association of UDP98-025 marker with the resistance response to MF infection, mean GI values for the genotypes, and the PVE (R2) in each progeny are presented in Table 2.
The F2 progenies were classified into two genotype cross combinations based on the assumption of a segregating dominant resistance allele (Mf1) or recessive resistance allele (mf3) inherited from PK. The F2 progenies were partitioned into their genotypes for the codominant SSR marker UDP98-025 (B, homozygous for PK allele; H, heterozygous; A, homozygous for PP allele) which, under the assumption of tight linkage, should correspond to their putative genotypes at the MF resistance locus (B = Mf1 Mf1 or mf3 mf3; H = Mf1 mf2 or mf2 mf3 depending on the F1 parent; A = mf2 mf2). In case of a multi-progeny cross type, the GI data were checked for non-significant interaction effects between progeny and marker genotype before combining datasets.
Following a significant KW test (P < 0.005), non-parametric pairwise comparisons per cross type were made to determine differences among the genotypes with respect to the GI data. The analysis was conducted on the combined dataset to increase representation of resistant/susceptible individuals in corresponding genotypes at the marker locus and thereby increase the statistical power to detect significant genetic effects. Rank-based analyses revealed that, in the Mf1 mf2 × Mf1 mf2 cross, the PK-homozygotes (Mf1 Mf1) and heterozygotes (Mf1 mf2) were significantly different from the PP-homozygotes (mf2 mf2). The Mf1 Mf1 and Mf1 mf2 genotypes corresponded to low mean GI values of 0.18 ± 0.69 and 0.66 ± 1.55, respectively, whereas the mf2 mf2 genotypes corresponded to high mean GI values (4.00 ± 1.97). These results indicate the involvement of the Mf1 allele in reducing the amount of root galling, while mf2 showed no such contribution. The significant effect of the Mf1 allele on resistance could also be observed in the backcross progenies of the mf2 mf2 × Mf1 mf2 cross, in which the Mf1 mf2 genotypes corresponded to lower mean GI values (0.78 ± 1.67) compared to mf2 mf2 genotypes (4.37 ± 1.65). In the mf2 mf3 × mf2 mf3 cross, the difference in the levels of root galling between PK-homozygotes (3.27 ± 1.79) and PP-homozygotes (5.00 ± 0.00) was marginally significant (P = 0.006) but, when compared with the heterozygotes (4.33 ± 1.23), the PK-homozygotes did not show significantly (P = 0.454) reduced levels of root galling as would be expected if two copies of the recessive allele (mf3 mf3) confer resistance and not when only a single copy (mf2 mf3) is present. The recessive contribution of the mf3 resistance allele may have been underestimated because the mf3mf3 genotypes were underrepresented (only 4 resistant out of 11 individuals with the PK-homozygous marker genotype) due to recombination between the UDP98-025 marker and the QTL. Transmission of the recessive resistance allele to the backcross progenies of the mf2 mf2 × mf2 mf3 cross yielded predominantly susceptible phenotypes associated with the mf2 mf3 and mf2 mf2 genotypes.
The strong linkage of UDP98-025 with the MF resistance locus allowed us to follow the transmission of the resistant/susceptible alleles in the backcross progenies, and the results provide further support for the existence of three different alleles at the resistance locus: two resistance alleles from PK that are either dominant (Mf1) or recessive (mf3), and susceptible alleles (mf2) from PP. In the F2 and backcross progenies of F1 parents carrying the dominant resistance allele (Mf1), the UDP98-025 marker correctly predicted resistance among PK-homozygotes and heterozygotes (co-segregating with Mf1 Mf1 and Mf1 mf2 genotypes, respectively), as well as susceptibility among PP-homozygotes (co-segregating with mf2 mf2 genotypes) up to 100%. In [OK × PK]3-derived F2 and backcross progenies where the resistance trait was recessively inherited, the marker has a low prediction accuracy (36%) in PK-homozygotes due to the low frequency of resistant individuals but could still reliably predict susceptibility in heterozygotes and PP-homozygotes (co-segregating with the respective mf2 mf3 and mf2 mf2 genotypes) up to 100% (Table 3).
Localization of major resistance locus on LG 2
Using the regression mapping method in JoinMap, four to five polymorphic SSR markers were mapped on LG 2 in seven interspecific F2 progenies and three backcross progenies. The map lengths ranged from 13.5 to 66.8 cM in F2 and 45.2 to 48.9 cM in the backcrosses. The SH × [OK × PK]3 and FG × [OK × PK]3 backcross progenies showed no segregation for resistance, so these populations were not used to map the MF locus. Three markers (BPPCT013, UDP96-013, and BPPCT030) in LG 2 generally appeared to be tightly linked (and clustered more tightly in F2 maps than in BC1F1 maps). These results are not unexpected. Proximity of the three marker loci and separation of UDP98-025 from the three loci corresponded to their distance on the TxE reference linkage map; the three marker loci had close intervals of 3 to 10 cM, whereas UDP98-025 is positioned from the most proximal marker BPPCT013 by 15 cM on the reference map. The variability in the map lengths for LG 2 resulted mainly from differences in recombination between UDP98-025 and the three tightly linked loci.
The MF locus and UDP98-025 SSR locus exhibited different degrees of linkage across F2 populations that could have resulted from sampling variation. In F2 populations with dominantly inherited resistance, the linkage distances between UDP98-025 and MF locus ranged from 0.8 cM in [OK × PK]4 (n = 29) to 13.6 cM in [OK × PK]1 (n = 36). Combining linkage information from the six F2 populations, UDP98-025 was linked at 7.2 cM to the MF locus. In the [OK × PK]3 population, the MF locus was located at a greater distance (24.6 cM) from UDP98-025 as expected due to the recessively inherited trait. The BC1F1 populations generally showed greater map distances; the MF locus was positioned 8.5 cM from the UDP98-025 locus in SH × [FG × PK]6 (n = 40) to 20.6 cM in SH × [OK × PK]2 (n = 33). The combined BC1F1 linkage map (N = 90) showed the association of UDP98-025 with the MF locus at 14.5 cM, which is twofold greater than that of the combined F2 linkage map (7.2 cM, N = 169) (Fig. 2).

Comparison of marker distances for linkage group 2 and approximate position of Meloidogyne floridensis resistance locus from combined maps and component maps within each cross type. a Mf1 mf2 × Mf1 mf2: OP “Okinawa” × P. kansuensis, FP “Flordaguard” × P. kansuensis (letters followed by accession number of the F1 parent). b mf2 mf3 × mf2 mf3. c mf2 mf2 × Mf1 mf2: OP2S “UFSharp” × (“Okinawa” × P. kansuensis), FP1S “UFSharp” × (“Flordaguard” × P. kansuensis), FP6S “UFSharp” × (“Flordaguard” × P. kansuensis). Lines between linkage maps connect common markers
Discussion
The F2 peach × P. kansuensis interspecific progenies segregated in a ratio of 1:3 or 3:1 between resistance and susceptibility to MF consistent with Mendelian expectations of a single major gene controlling resistance, which was also validated in backcrosses segregating 0:1 or 1:1 between resistance and susceptibility. The F2 phenotypic distributions appeared strongly skewed, and those of the backcrosses followed a bimodal distribution confirming that a major locus is involved. To identify SSR markers associated with resistance to MF and to elucidate the genetic architecture of the trait, the interspecific-hybrid-derived populations, consisting of 23 to 43 individuals each, were genotyped with 34 to 36 SSR markers distributed at 6.2 to 25.1 cM intervals across eight linkage groups based on the Prunus TxE reference map. Based on the assumption of a single locus with major phenotypic effect, the population size and marker density for our mapping populations were deemed sufficient for initial QTL detection in the presence of large non-recombined linkage blocks characteristic of early-generation populations (Flint-Garcia et al. 2003; Mackay et al. 2009).
The QTL analysis in multiple independent populations detected one SSR locus associated with the resistance QTL corroborating our findings of single-locus inheritance from the progeny tests (Maquilan 2017). The QTL was localized in the same genomic region that harbored the genetic factors controlling M. incognita and M. arenaria in P. kansuensis, as well as in P. persica cultivars Shalil, Nemared, and “Juseitou” (Cao et al. 2011; Claverie et al. 2004; Yamamoto et al. 2005) and mapped close to the SSR marker UDP98-025 at the subtelomeric region of LG 2. The UDP98-025 marker amplified three different fragment sizes in PK and PP genotypes. The 115-bp allele in PK was associated with resistance, while the 111-bp (OK and FG) and 127-bp (SH) allele sizes in P. persica genotypes were associated with susceptibility. The use of such a codominant SSR marker, which can distinguish heterozygotes, in examining genotypic differences based on GI values enabled validation of the proposed single-locus dominant/recessive model from the segregation analyses. The QTL was confirmed to be completely dominant when levels of root galling in PK-homozygotes were similar to the heterozygotes and significantly lower than those of the PP-homozygotes at the linked marker locus. The QTL was confirmed to be recessive when levels of root galling in PK-homozygotes were significantly lower than those of the heterozygotes, whereas the heterozygotes had levels similar to those of PP-homozygotes. Although the effect of the recessive QTL was not apparent in the F2 due to the low frequency of resistant individuals and some recombination occurring between the QTL and the marker locus, the recessive effect was confirmed in the backcrosses, which showed non-significant differences between the heterozygous and PP-homozygous marker genotypes. The study also demonstrated the advantage of observing multiple F2 interspecific populations to capture the possible allelic combinations in segregating F1 hybrids (Chaparro et al. 1994). The OK × PK and FG × PK hybrids each inherited a resistance allele from PK (designated as Mf1 or mf3) and a susceptible allelic form from PP (designated as mf2); the inferred relationship of the three different alleles at the same locus and their contribution to MF resistance is shown in Table 4. Multiple alleles conferring high resistances to major RKN have also been identified in Myrobalan plum (P. cerasifera) by examining segregation in several intraspecific F1 and F2 progenies of crosses between heterozygous and homozygous accessions (Esmenjaud et al. 1996; Rubio-Cabetas et al. 1998).
The codominant SSR marker was useful for detecting the resistance alleles especially in the heterozygotes and for tracking the transmission of resistance alleles via linkage to the backcross progenies when the genetic model is known. However, its use as a diagnostic marker for MF resistance in a breeding program will be complicated if the PK accession is heterozygous at the resistance locus. If resistance is recessively inherited in the progeny, several individuals carrying the 115-bp marker allele may not have the resistance allele and therefore will be incorrectly classified as resistant. It would be necessary to identify other co-dominant markers within the region that can differentiate the two resistance alleles in PK. However, if a homozygous-dominant PK accession is identified and used to make crosses with homozygous-susceptible PP genotypes, the UDP98-025 marker can predict resistance with high accuracy (≈ 98%) among F2 individuals that are homozygous for the 115-bp allele.
In this study, the distance of the linked SSR marker locus UDP98-025 from the MF resistance locus spans a large chromosomal segment corresponding to an interval size of 7.2 cM in F2 and 14.5 cM in backcrosses. The same marker was placed ≈ 10 cM from the RMia gene on the TxE reference map (Dirlewanger et al. 2004b) and from the PkMi gene using a BC1F1 population derived from a cross between peach “Bailey” and P. kansuensis “Honggengansutao” (Cao et al. 2011). These RKN R genes located on LG 2, along with those located on the telomeric region of LG 7 (Ma, Rjap, and RMja) (Cao et al. 2011; Claverie et al. 2004; Van Ghelder et al. 2010; Yamamoto et al. 2005), have been characterized as a class of toll/interleukin1 receptor-(TIR) nucleotide-binding-(NB) leucine-rich repeat (LRR) genes (Cao et al. 2014; Claverie et al. 2011; Duval et al. 2014). The MF resistance gene in PK mapped in this study broadens the suite of R genes on the top region of chromosome 2, which has been found to contain a fivefold higher density of genes encoding disease-resistance-associated NB-LRR proteins than the peach genome average (Verde et al. 2013). Further studies require a denser set of markers in this QTL region. Higher throughput single nucleotide polymorphisms (SNP) from the 9 K peach SNP array (Verde et al. 2012) may prove useful for fine-mapping the QTL in peach × P. kansuensis populations. Other codominant markers such as sequence-tagged sites and SSRs (AMP117 and AMP116) flanking RMia or PkMi on LG 2 as well as resistance gene analogues have been used to examine polymorphisms among Prunus accessions (Cao et al. 2014; Duval et al. 2014; Lu et al. 1999; Yamamoto et al. 2005). The same markers could also be used to examine the level of allelic variation at the MF resistance locus among PK accessions and in Prunus rootstock breeding germplasm due to collinearity between Prunus genomes (Dirlewanger et al. 2004b). Whether the Mf, RMia, and PkMi are distinct genes that are tightly linked in a single locus or are part of a single, multi-allelic resistance locus is an important question that remains unresolved and a complexity not directly addressed in this study. Further investigations involving different RKN species and isolates are needed to establish the resistance specificities of the Mf alleles and to determine their relationship with the possibly linked or interacting R genes identified in LG 2.
The genetic maps constructed from the 12 interspecific peach × P. kansuensis populations showed variability in recombination rates across the genome that is inherent in the interspecific cross. The combined genetic maps provide information about the recombination landscape in the early-generation populations. For fine-mapping the QTL region, there is a need for additional markers where intermarker distances are large or greater number of individuals where there is tight SSR linkage, in order to capture more recombination events when analyzing interspecific-hybrid-derived F2 and backcross populations. The map lengths for backcross populations were generally longer across eight linkage groups than those of F2 indicating that more recombination has occurred in the former, which can be utilized further to achieve a higher-resolution linkage map particularly at the target QTL interval on the top region of LG 2 where recombination distance was twofold longer.
The genetic analyses have revealed the effects of the resistance alleles from PK as tracked by the linked, codominant SSR marker and are consistent with the suggested dominance/recessive model from the segregation analyses (Maquilan 2017). No evidence for distorted segregation was detected in LG 2 indicating that the introgression of MF resistance from PK into cultivated peach is predictable by single-gene-based Mendelian inheritance. Analyses of 12 interspecific peach × P. kansuensis progenies using microsatellite markers allowed us to infer the chromosomal location of the Mf locus, which confers resistance to M. floridensis. The Mf locus was found to be in the same region on linkage group 2 where other Prunus RKN resistance genes have been reported, e.g., RMia from peach (Shalil and Nemared), and PkMi from wild peach conferring resistance against M. incognita and M. arenaria. The findings about the mode of inheritance and the resistance locus would direct our breeding strategies towards developing new nematode-resistant rootstocks by effectively selecting resistant parents from diverse germplasm and keeping track of the resistance in the progeny. It must be noted, however, that the use of a single MF population to evaluate resistance among the interspecific progenies does not allow for definitive conclusions to be made about whether or not the same alleles confer resistance against other MF populations. There are possibly multiple alleles (or haplotypes) in the highly polymorphic resistance locus from the wild-related PK, and different interspecific peach × PK hybrids may carry different resistance alleles at the MF locus. In this context, it is important to understand the influence of different allele combinations on the level of MF resistance when considering interspecific hybridization and introgression of RKN resistances from PK into peach rootstocks. In Myrobalan plum (P. cerasifera), resistance variability to major RKN was observed among the different accessions, and diallel crosses of five accessions revealed different modes of inheritance for resistance when challenged with M. arenaria (Esmenjaud et al. 1996; Esmenjaud et al. 1994). Screening additional markers within the subtelomeric region of LG 2 should identify more closely linked markers that can distinguish the various polymorphisms associated with resistance to MF in PK and therefore facilitate the efficient introgression of the trait into existing peach rootstocks, particularly Okinawa and Flordaguard.
References
Aranzana MJ, Carbo J, Arus P (2003a) Microsatellite variability in peach [Prunus persica (L.) Batsch]: cultivar identification, marker mutation, pedigree inferences and population structure. Theor Appl Genet 106:1341–1352. https://doi.org/10.1007/s00122-002-1128-5
Aranzana MJ, Garcia-Mas J, Carbo J, Arus P (2002) Development and variability analysis of microsatellite markers in peach. Plant Breed 121:87–92. https://doi.org/10.1046/j.1439-0523.2002.00656.x
Aranzana MJ, Pineda A, Cosson P, Dirlewanger E, Ascasibar J, Cipriani G, Ryder CD, Testolin R, Abbott A, King GJ, Iezzoni AF, Arus P (2003b) A set of simple-sequence repeat (SSR) markers covering the Prunus genome. Theor Appl Genet 106:819–825. https://doi.org/10.1007/s00122-002-1094-y
Bus VGM, Esmenjaud D, Buck E, Laurens F (2009) Application of genetic markers in rosaceous crops. In: Folta KM, Gardiner SE (eds) Genetics and genomics of rosaceae. Springer, New York, pp 563–599
Cao K, Wang LR, Zhao P, Zhu GR, Fang WC, Chen CW, Wang XW (2014) Identification of a candidate gene for resistance to root-knot nematode in a wild peach and screening of its polymorphisms. Plant Breed 133:530–535. https://doi.org/10.1111/pbr.12174
Cao K, Wang LR, Zhu GR, Fang WC, Chen CW, Zhao P (2011) Construction of a linkage map and identification of resistance gene analog markers for root-knot nematodes in wild peach, Prunus kansuensis. J Am Soc Hortic Sci 136:190–197
Castagnone-Sereno P (2006) Genetic variability and adaptive evolution in parthenogenetic root-knot nematodes. Heredity 96:282–289. https://doi.org/10.1038/sj.hdy.6800794
Chaparro JX, Sherman WB (2006) ‘UFSharp’ peach. J Amer Pomolog Soc 60:95–96
Chaparro JX, Werner DJ, Omalley D, Sederoff RR (1994) Targeted mapping and linkage analysis of morphological isozyme, and RAPD markers in peach. Theor Appl Genet 87:805–815. https://doi.org/10.1007/bf00221132
Charcosset A, Moreau L (2004) Use of molecular markers for the development of new cultivars and the evaluation of genetic diversity. Euphytica 137:81–94. https://doi.org/10.1023/b:euph.0000040505.65040.75
Chavez DJ, Chaparro JX (2011) Identification of markers linked to seedlessness in Citrus kinokuni hort. ex Tanaka and its progeny using bulked segregant analysis. HortScience 46:693–697
Chu Y, Wu CL, Holbrook CC, Tillman BL, Person G, Ozias-Akins P (2011) Marker-assisted selection to pyramid nematode resistance and the high oleic trait in peanut. Plant Genome 4:110–117. https://doi.org/10.3835/plantgenome2011.01.0001
Clarke LA, Rebelo CS, Gonçalves J, Boavida MG, Jordan P (2001) PCR amplification introduces errors into mononucleotide and dinucleotide repeat sequences. J Clin Pathol: Mol Pathol 54:351–353. https://doi.org/10.1136/mp.54.5.351
Claverie M, Bosselut N, Lecouls AC, Voisin R, Lafargue B, Poizat C, Kleinhentz M, Laigret F, Dirlewanger E, Esmenjaud D (2004) Location of independent root-knot nematode resistance genes in plum and peach. Theor Appl Genet 108:765–773. https://doi.org/10.1007/s00122-003-1463-1
Claverie M, Dirlewanger E, Bosselut N, Van Ghelder C, Voisin R, Kleinhentz M, Lafargue B, Abad P, Rosso MN, Chalhoub B, Esmenjaud D (2011) The Ma gene for complete-spectrum resistance to Meloidogyne species in Prunus is a TNL with a huge repeated C-terminal post-LRR region. Plant Physiol 156:779–792. https://doi.org/10.1104/pp.111.176230
Concibido V, Diers B, Arelli R (2004) A decade of QTL mapping for cyst nematode resistance in soybean. Crop Sci 44:1121–1131
DeWoody JA, Nason JD, Hipkins VD (2006) Mitigating scoring errors in microsatellite data from wild populations. Mol Ecol Notes 6:951–957. https://doi.org/10.1111/j.1471-8286.2006.01449.x
Dirlewanger E, Cosson P, Boudehri K, Renaud C, Capdeville G, Tauzin Y, Laigret F, Moing A (2007) Development of a second-generation genetic linkage map for peach [Prunus persica (L.) Batsch] and characterization of morphological traits affecting flower and fruit. Tree Genet Genomes 3:1–13. https://doi.org/10.1007/s11295-006-0053-1
Dirlewanger E, Cosson P, Howad W, Capdeville G, Bosselut N, Claverie M, Voisin R, Poizat C, Lafargue B, Baron O, Laigret F, Kleinhentz M, Arus P, Esmenjaud D (2004a) Microsatellite genetic linkage maps of myrobalan plum and an almond-peach hybrid - location of root-knot nematode resistance genes. Theor Appl Genet 109:827–838. https://doi.org/10.1007/s00122-004-1694-9
Dirlewanger E, Graziano E, Joobeur T, Garriga-Calder F, Cosson P, Howad W, Arus P (2004b) Comparative mapping and marker-assisted selection in Rosaceae fruit crops. PNAS 101:9891–9896. https://doi.org/10.1073/pnas.0307937101
Duval H, Hoerter M, Polidori J, Confolent C, Masse M, Moretti A, Van Ghelder C, Esmenjaud D (2014) High-resolution mapping of the RMia gene for resistance to root-knot nematodes in peach. Tree Genet Genomes 10:297–306. https://doi.org/10.1007/s11295-013-0683-z
Esmenjaud D, Minot JC, Voisin R, Bonnet A, Salesses G (1996) Inheritance of resistance to the root-knot nematode Meloidogyne arenaria in Myrobalan plum. Theor Appl Genet 92:873–879. https://doi.org/10.1007/bf00221900
Esmenjaud D, Minot JC, Voisin R, Pinochet J, Salesses G (1994) Inter- and intraspecific resistance variability in Myrobalan plum, peach, and peach-almond rootstock using 22 root-knot nematode populations. J Am Soc Hortic Sci 119:94–100
Fan S, Bielenberg DG, Zhebentyayeva TN, Reighard GL, Okie WR, Holland D, Abbott AG (2010) Mapping quantitative trait loci associated with chilling requirement, heat requirement and bloom date in peach (Prunus persica). New Phytol 185:917–930. https://doi.org/10.1111/j.1469-8137.2009.03119.x
Flint-Garcia SA, Thornsberry JM, Buckler ES (2003) Structure of linkage disequilibrium in plants. Annu Rev Plant Biol 54:357–374. https://doi.org/10.1146/annurev.arplant.54.031902.134907
Handoo ZA, Nyczepir AP, Esmenjaud D, van der Beek JG, Castagnone-Sereno P, Carta LK, Skantar AM, Higgins JA (2004) Morphological, molecular, and differential-host characterization of Meloidogyne floridensis n. sp. (Nematoda : Meloidogynidae), a root-knot nematode parasitizing peach in Florida. J Nematol 36:20–35
Hansen CJ, Lownsbery BF, Hesse CO (1956) Nematode resistance in peaches. Calif Agric 10:5–11
Hospital F (2005) Selection in backcross programmes. Philos Trans R Soc B 360:1503–1511. https://doi.org/10.1098/rstb.2005.1670
Jenkins JN, McCarty JC, Wubben MJ, Hayes R, Gutierrez OA, Callahan F, Deng D (2012) SSR markers for marker assisted selection of root-knot nematode (Meloidogyne incognita) resistant plants in cotton (Gossypium hirsutum L). Euphytica 183:49–54. https://doi.org/10.1007/s10681-011-0470-6
Joobeur T, Viruel MA, de Vicente MC, Jauregui B, Ballester J, Dettori MT, Verde I, Truco MJ, Messeguer R, Batlle I, Quarta R, Dirlewanger E, Arus P (1998) Construction of a saturated linkage map for Prunus using an almond x peach F2 progeny. Theor Appl Genet 97:1034–1041. https://doi.org/10.1007/s001220050988
Khallouk S, Voisin R, Portier U, Polidori J, Van Ghelder C, Esmenjaud D (2013) Multiyear evaluation of the durability of the resistance conferred by Ma and RMia genes to Meloidogyne incognita in Prunus under controlled conditions. Phytopathology 103:833–840. https://doi.org/10.1094/Phyto-09-12-0228-R
Kochba J, Spiegel-Roy P (1975) Inheritance of resistance to the root-knot nematode (Meloidogyne javanica Chitwood) in bitter almond progenies. Euphytica 24:453–457. https://doi.org/10.1007/bf00028213
Lalli DA, Decroocq V, Blenda AV, Schurdi-Levraud V, Garay L, Le Gall O, Damsteegt V, Reighard GL, Abbott AG (2005) Identification and mapping of resistance gene analogs (RGAs) in Prunus: a resistance map for Prunus. Theor Appl Genet 111:1504–1513. https://doi.org/10.1007/s00122-005-0079-z
Lu ZX, Reighard GL, Nyczepir AP, Beckman TG, Ramming DW (2000) Inheritance of resistance to root-knot nematodes (Meloidogyne sp.) in Prunus rootstocks. HortScience 35:1344–1346
Lu ZX, Sossey-Alaoui K, Reighard GL, Baird WV, Abbott AG (1999) Development and characterization of a codominant marker linked to root-knot nematode resistance, and its application to peach rootstock breeding. Theor Appl Genet 99:115–122. https://doi.org/10.1007/s001220051215
Mackay TFC, Stone EA, Ayroles JF (2009) The genetics of quantitative traits: challenges and prospects. Nat Rev Genet 10:565–577. https://doi.org/10.1038/nrg2612
Maquilan MAD (2017) Rootstock breeding for resistancce to the peach root-knot nematode (Meloidogyne floridensis). Dissertation, University of Florida
Miah G, Rafii MY, Ismail MR, Puteh AB, Rahim HA, Islam KN, Latif MA (2013) A review of microsatellite markers and their applications in rice breeding programs to improve blast disease resistance. Int J Mol Sci 14:22499–22528. https://doi.org/10.3390/ijms141122499
Mnejja M, Garcia-Mas J, Audergon JM, Arus P (2010) Prunus microsatellite marker transferability across rosaceous crops. Tree Genet Genomes 6:689–700. https://doi.org/10.1007/s11295-010-0284-z
Okie WR, Beckman TG, Nyczepir AP, Reighard GL, Newall WC, Zehr EI (1994) BY520-9, a peach rootstock for the southeastern United States that increases scion longevity. HortScience 29:705–706
Olmstead M, Chaparro J, Ferguson J (2015) Rootstocks for Florida stone fruit. HS1110. Gainesville: University of Florida Institute of Food and Agricultural Sciences. http://edis.ifas.ufl.edu/hs366 Accessed 15 January 2016
Ramming DW, Tanner O (1983) ‘Nemared’ peach rootstock. HortScience 18:376
Reighard G, Loreti F (2008) Rootstock development. p. 193–220. In: Layne, D. and Bassi, D. (eds) The peach: botany, production and uses. CABI publishing, Wallingford
Rubio-Cabetas MJ, Lecouls AC, Salesses G, Bonnet A, Minot JC, Voisin R, Esmenjaud D (1998) Evidence of a new gene for high resistance to Meloidogyne spp. in Myrobalan plum, Prunus cerasifera. Plant Breed 117:567–571. https://doi.org/10.1111/j.1439-0523.1998.tb02209.x
Sharpe R (1967) Root-knot nematode populations on peaches in Florida. Proc Fla State Hort Soc 80:342–344
Sharpe RH (1957) Okinawa peach shows promising resistance to root-knot nematodes. Fla Agric Exp 657:320–322
Sherman WB, Lyrene PM, Sharpe RH (1991) Flordaguard peach rootstock. HortScience 26:427–428
Tautz D (1989) Hypervariability of simple sequences as a general source for polymorphic DNA markers. Nucleic Acids Res 17:6463–6471. https://doi.org/10.1093/nar/17.16.6463
Taylor AL, Sasser JN (1978) Biology, identification and control of root-knot nematodes (Meloidogyne species). N.C. State Univ. Dep. Plant Path. and USAID, Raleigh, p 111p
Van Ghelder C, Esmenjaud D (2016) TNL genes in peach: insights into the post-LRR domain. BMC Genomics 17:317. https://doi.org/10.1186/s12864-016-2635-0
Van Ghelder C, Lafargue B, Dirlewanger E, Ouassa A, Voisin R, Polidori J, Kleinhentz M, Esmenjaud D (2010) Characterization of the RMja gene for resistance to root-knot nematodes in almond: spectrum, location, and interest for Prunus breeding. Tree Genet Genomes 6:503–511. https://doi.org/10.1007/s11295-010-0268-z
Van Ooijen J (2006) JoinMap 4.1, software for the calculation of genetic linkage maps in experimental populations of diploid species. Kyazma B. V, Wageningen
Van Ooijen J (2009) MapQTL 6, software for the mapping of quantitative trait loci in experimental populations of diploid species. Kyazma B. V, Wageningen
Verde I, Abbott AG, Scalabrin S, Jung S, Shu S, Marroni F, Zhebentyayeva T, Dettori MT, Grimwood J, Cattonaro F (2013) The high-quality draft genome of peach (Prunus persica) identifies unique patterns of genetic diversity, domestication and genome evolution. Nat Genet 45:487–496. https://doi.org/10.1038/ng.2586
Verde I, Bassil N, Scalabrin S, Gilmore B, Lawley CT, Gasic K, Micheletti D, Rosyara UR, Cattonaro F, Vendramin E, Main D, Aramini V, Blas AL, Mockler TC, Bryant DW, Wilhelm L, Troggio M, Sosinski B, Aranzana MJ, Arús P, Iezzoni A, Morgante M, Peace C (2012) Development and evaluation of a 9K SNP array for peach by internationally coordinated SNP detection and validation in breeding germplasm. PLoS One 7:e35668. https://doi.org/10.1371/journal.pone.0035668
Voorrips RE (2002) MapChart: software for the graphical presentation of linkage maps and QTLs. J Hered 93:77–78. https://doi.org/10.1093/jhered/93.1.77
Yamamoto T, Shimada T, Imai T, Yaegaki H, Haji T, Matsuta N, Yamaguchi M, Hayashi T (2001) Characterization of morphological traits based on a genetic linkage map in peach. Breed Sci 51:271–278. https://doi.org/10.1270/jsbbs.51.271
Yamamoto T, Yamaguchi M, Hayashi T (2005) An integrated genetic linkage map of peach by SSR, STS, AFLP and RAPD. J Japan Soc Hort Sci 74:204–213. https://doi.org/10.2503/jjshs.74.204
Data archiving statement
(Peach × P. kansuensis linkage maps to be submitted to the Genome Database for Rosaceae. Accession numbers will be provided once available.)
Funding
This study was funded in part by the Florida Department of Agriculture and Consumer Services under the Specialty Crop Block Grant Nos. 18004 and 20727.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by C. Chen
Electronic supplementary material
ESM 1.
Figure S2 Microsatellite SSR markers screened for polymorphism and selected for mapping the Mf resistance locus on each of the eight linkage groups (LG1-LG8) of the ‘Texas’ almond x ‘Earlygold’ (TxE) peach reference map. Marker names and positions (Kosambi map distances, cM) are indicated on the right and left side of bars, respectively. Markers selected for linkage analyses are denoted by bold, uppercase letters. Three markers (colored red and green) in LG 4 and LG5 were mapped to other interspecific Prunus linkage maps at distances beyond the length of the TxE map. Non-selected markers denoted by lowercase letters are characterized by one of the following: 1) non-polymorphic (enclosed in parentheses), 2) polymorphic but are spaced at very close intervals (bold letters), and 3) polymorphic but are difficult to score (italicized letters) (DOCX 29 kb)
ESM 2.
File S4 Microsatellite allele configurations and genotype cross combinations for F2 and BC1F1 peach x P. kansuensis interspecific progenies inferred from segregation patterns of marker band and electropherogram data (XLSX 35 kb)
ESM 3.
Figure S5 Combined SSR linkage map based on two F2 mapping populations derived from ‘Flordaguard’ (FG) x Prunus kansuensis (PK). Microsatellite markers were assigned into eight linkage groups (LG1-LG8) and ordered in accordance with the ‘Texas’ almond x ‘Earlygold’ peach linkage map. Numbers to the left side of the bars represent the estimated genetic distances (centiMorgan) of the microsatellite markers (DOCX 20 kb)
ESM 4.
Combined SSR linkage map based on five F2 mapping populations derived from ‘Okinawa’ (OK) x Prunus kansuensis (PK). Microsatellite markers were assigned into eight linkage groups (LG1-LG8) and ordered in accordance with the ‘Texas’ almond x ‘Earlygold’ peach linkage map. Numbers to the left side of the bars represent the estimated genetic distances (centiMorgan) of the microsatellite markers (DOCX 20 kb)
ESM 5.
Figure S7 Combined SSR linkage map based on three BC1 mapping populations derived from peach (‘Flordaguard’ [FG] and ‘UFSharp’ [SH]) x F1 (‘Okinawa’ [OK] x Prunus kansuensis [PK]). Microsatellite markers were assigned into eight linkage groups (LG1-LG8) and ordered in accordance with the ‘Texas’ almond x ‘Earlygold’ peach linkage map. Numbers to the left side of the bars represent the estimated genetic distances (centiMorgan) of the microsatellite markers (DOCX 20 kb)
ESM 6.
Figure S8 Combined SSR linkage map based on two BC1 mapping populations derived from peach (‘UFSharp’ [SH]) x F1 (‘Flordaguard’ [FG] x Prunus kansuensis [PK]). Microsatellite markers were assigned into eight linkage groups (LG1-LG8) and ordered in accordance with the ‘Texas’ almond x ‘Earlygold’ peach linkage map. Numbers to the left side of the bars represent the estimated genetic distances (centiMorgan) of the microsatellite markers (DOCX 20 kb)
ESM 7.
Table S1 Origins of the microsatellite markers tested for polymorphism and number of markers included in the linkage maps (DOCX 21 kb)
ESM 8.
Table S3 Characteristics of the polymorphic microsatellite markers used to map the Mf resistance locus in peach x Prunus kansuensis interspecific progenies (DOCX 22 kb)
ESM 9.
Table S9 Combined linkage map based on five F2 mapping populations derived from crosses between ‘Okinawa’ x Prunus kansuensis (OK x PK) compared with Prunus ‘Texas’ x ‘Earlygold’ (TxE) reference map (DOCX 14 kb)
Table S10
Combined linkage map based on two F2 mapping populations derived from crosses between ‘Flordaguard’ x Prunus kansuensis (FG x PK) compared with Prunus ‘Texas’ x ‘Earlygold’ (TxE) reference map (DOCX 14 kb)
Table S11
Combined linkage map based on three BC1 mapping populations of peach (‘Flordaguard’ or ‘UFSharp’) x F1 (‘Okinawa’ x Prunus kansuensis) (FG/SH x [OK x PK]) compared with Prunus ‘Texas’ x ‘Earlygold’ (TxE) reference map (DOCX 14 kb)
Table S12
Combined linkage map based on two BC1 mapping populations of peach (‘UFSharp’) x F1 (‘Flordaguard’ x Prunus kansuensis) (SH x [FG x PK]) compared with Prunus ‘Texas’ x ‘Earlygold’ (TxE). (DOCX 14 kb)
Rights and permissions
About this article

Cite this article
Maquilan, M.A.D., Olmstead, M.A., Olmstead, J.W. et al. Genetic analyses of resistance to the peach root-knot nematode (Meloidogyne floridensis) using microsatellite markers. Tree Genetics & Genomes 14, 47 (2018). https://doi.org/10.1007/s11295-018-1260-2
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11295-018-1260-2