
Abstract
Ceratocystis wilt caused by the fungus Ceratocystis fimbriata, is currently one of the major diseases in commercial plantations of Eucalyptus trees in Brazil. Deployment of resistant genotypes has been the main strategy for effective disease management. The present study aimed at identifying genomic regions underlying the genetic control of resistance to Ceratocystis wilt in Eucalyptus by quantitative trait loci (QTL) mapping in an outbred hybrid progeny derived from a cross between (Eucalyptus dunnii × Eucalyptus grandis) × (Eucalyptus urophylla × Eucalyptus globulus). A segregating population of 127 individuals was phenotyped for resistance to Ceratocystis wilt using controlled inoculation under a completely randomized design with five clonal replicates per individual plant. The phenotypic resistance response followed a continuous variation, enabling us to analyze the trait in a quantitative manner. The population was genotyped with 114 microsatellite markers and 110 were mapped with an average interval of 12.3 cM. Using a sib-pair interval-mapping approach five QTLs were identified for disease resistance, located on linkage groups 1, 3, 5, 8, and 10, and their estimated individual heritability ranged from 0.096 to 0.342. The QTL on linkage group 3 overlaps with other fungal disease-resistance QTLs mapped earlier and is consistent with the annotation of several disease-resistance genes on this chromosome in the E. grandis genome. This is the first study to identify and attempt to quantify the effects of QTLs associated with resistance to Ceratocystis wilt in Eucalyptus.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Fast-growing species of Eucalyptus are currently planted across more than 100 countries worldwide due to their remarkable adaptability to different climatic conditions, rapid growth, high coppice capacity, and production of wood for diverse purposes (Iglesias and Wiltermann 2009). Along with the continued expansion of planted areas, many diseases have emerged causing economic losses, particularly those caused by pathogenic fungi (Alfenas et al. 2009). Among these, Ceratocystis wilt caused by Ceratocystis fimbriata Ellis and Halsted is currently one of the most important diseases that affect Eucalyptus plantations in Brazil (Fernandes et al. 2014).
Ceratocystis fimbriata is a soilborne ascomycete fungus and was originally described as the causal agent of black rot of sweet potato (Ipomoea batatas) in New Jersey, USA (Halsted 1890). Currently, C. fimbriata is recognized as a complex of many fungal species (Harrington et al. 2011). However, all strains in Brazil are considered to belong to a single species with a very broad host range that includes isolates varying greatly in aggressiveness to many cultivated hosts (Oliveira et al. 2015a, b). C. fimbriata mainly affects the xylem, where important symptoms are observed in stem cross sections exhibiting radial and longitudinal dark stripes. The pathogen infects the plant through fresh wounds in the trunk and roots causing wilting symptoms commonly followed by plant death (Ferreira et al. 2006).
The fungus was first reported in Brazil in 1997 in eucalypt clonal forests in southeastern Bahia, where plantations dominated by one particular hybrid combination (Eucalyptus urophylla × Eucalyptus grandis) displayed mortalities exceeding 40 % (Ferreira et al. 1999). Subsequently, the disease was reported in the Republic of Congo (Roux et al. 2000), Uganda (Roux et al. 2001), Uruguay (Barnes et al. 2003), South Africa (Roux et al. 2004), and China (Li et al. 2014). In addition to Eucalyptus, C. fimbriata infects other woody species of economic importance, such as mango (Mangifera indica), sycamore (Platanus spp.), gmelina (Gmelina arborea), coffee (Coffea arabica), rubber tree (Hevea brasiliensis), fig (Ficus carica), and acacia (Acacia spp.) (Baker et al. 2003; Harrington et al. 2011).
The management of Ceratocystis wilt is extremely difficult because of the vascular nature of the disease, the different forms of transmission, the long persistence of the fungus in the soil, and because the pathogen is native in many regions of Brazil (Ferreira et al. 2011). The existence of inter- and intraspecific genetic variability for resistance to Ceratocystis wilt in different species and clones of Eucalyptus has allowed the selection of resistant hybrid clones (Guimarães et al. 2010; Alfenas et al. 2004), and is currently the main method for managing the disease (Fonseca et al. 2010).
Genetic studies using hybrid progenies of E. grandis × E. urophylla showed that resistance to Ceratocystis wilt displays a high degree of additive genetic control with seemingly low dominance (Rosado et al. 2010a). Nevertheless, the identification of genomic regions involved in the resistance response to Ceratocystis wilt could help understand the genetic architecture of heritable resistance underlying the overall resistance phenotypes and possibly help accelerate the selection of resistant trees (Grattapaglia and Kirst 2008). Markers linked to resistance quantitative trait loci (QTL), once validated across additional pedigrees, could be used for the practice of early and indirect selection and for the identification of resistance genes in the now-available Eucalyptus genome sequence (Myburg et al. 2014). In this report, we describe the identification of genomic regions underlying the resistance response to Ceratocystis wilt in Eucalyptus using a QTL mapping approach by constructing a genetic map with transferable microsatellites and phenotyping trees with a controlled inoculation bioassay across biological replicates. Additionally, we show that the QTL detected for Ceratocystis wilt maps to the same genomic interval where major-effect QTL for Puccinia psidii rust resistance were recurrently mapped in earlier independent studies.
Materials and methods
Plant material
Genetic map construction and QTL mapping was carried out using an outbred full-sib family of 127 plants originated from a cross between two interspecific F1 hybrid parents: [Eucalyptus dunnii (D) × E. grandis (G)] × [E. urophylla (U) × Eucalyptus globulus (GL)], generated during the Genolyptus Project (Grattapaglia 2003). Each plant of the DG × UGL mapping progeny was cloned via stem cuttings to enable inoculation on five replicates per individual. The cuttings at 90 days after setting were transplanted to pots of 2 L capacity containing a sterilized mixture of soil/sand/manure (3:1:1). The fertilizer Ouro Verde® (15-15-20 NPK) was applied once a week (100 mL plant−1 at 7.5 g L−1) up to 60 days after transplanting until the plants reached the stage of development suitable for inoculation.
Inoculation and disease evaluation
The isolate SBS-1 of C. fimbriata used in this study was obtained in 2007 from a hybrid E. grandis × E. urophylla clone exhibiting typical symptoms of the disease in the region of Teixeira de Freitas, Bahia, and used previously (Rosado et al. 2010a). The isolate was maintained in the mycology collection of the Forestry Pathology Lab at the Universidade Federal de Viçosa (UFV) in Brazil, on malt agar media at −80 °C. The fungus was grown in Petri dishes (9 cm diameter) containing MYEA (2 % malt extract, 0.2 % yeast extract and 2 % agar) for 8 days at 28 ± 1 °C under a photoperiod of 12 h (light intensity of 40 μmol s−1 m−2). Sterile distilled water was added to the plates to collect the spores (conidia + ascospores), and after scraping, the surface of the colony with Drigalski, the spore suspension was filtered through a double layer of cheesecloth. The inoculum concentration was determined using a Neubauer chamber in an optical microscope calibrated to 2.5 × 106 spores mL−1. The plants were wounded by making a 3 mm deep downward-slanting cut from the outer bark into the wood with a sterile scalpel at 2 cm above the ground line, and 0.5 mL of the spore suspension was then placed into the wound, which was subsequently covered with Parafilm®. A total of ten plants of the hybrid clone HGU1172 (E. grandis × E. urophylla) were used as susceptible controls. The inoculations were conducted in 2008 in a nursery at the Universidade Federal de Viçosa. Plants were evaluated for disease severity (%) 60 days after inoculation. Disease severity (%) was obtained by dividing the lesion length by plant height and multiplying by 100.
Microsatellite genotyping
DNA of the two parents and the progeny individuals was extracted according to the protocol described earlier (Grattapaglia and Sederoff 1994). The extracted DNA was resuspended in 50 μL of sterile distilled water and the concentration adjusted to 20 ng μL−1. Microsatellite genotyping was conducted as described by Faria et al. (2011), using multiplex systems of four to six co-amplified loci labeled with specific fluorochromes (FAM, HEX, or NED). PCR reactions in multiplexed systems were carried out using 1 μL of 10× QIAGEN Multiplex PCR Buffer (QIAGEN Inc., Valencia, CA, USA), equal final concentration of 0.1 μM for all primers of all microsatellite markers co-amplified, 1.0 μL of Q-Solution, 2.0 ng of genomic DNA, and RNase-free water to a final reaction volume of 10 μL. The recommended QIAGEN Multiplex PCR Handbook cycling protocol was used with an annealing temperature of 60 °C and 30 PCR cycles. Following PCR amplification, 1 μL of the reaction was added to 8 μL of Hi-Di formamide (Applied Biosystems) and 1 μL of size standard labeled with ROX (Brondani and Grattapaglia 2001). The DNA samples were denatured (95 °C for 5 min) and immediately placed on ice. Thereafter, the reaction was subjected to capillary electrophoresis on an ABI 3100XL automated sequencer (Applied Biosystems) according to the standard protocol used in the laboratory for the detection of fragments. The software programs GeneScan and Genotyper (Applied Biosystems) were used to analyze the raw data, and AlleloBin (Indury and Cardon 1997) was used to calibrate the allele size values in the base pairs to integers.
Linkage analyses and construction of the genetic map
Testing for departures from Mendelian assumptions (Mendelian segregation with no segregation distortion) for segregating markers was performed using a chi-square test (χ 2) with a 20 % probability of type I error for the entire genome (experiment wise). This error rate is acceptable from a genome-wide standpoint. In addition, Bonferroni multiple testing correction was applied at the individual-marker level. The integrated map was constructed using the software GQMOL (Cruz 2008). Linkage group numbering followed the Eucalyptus reference map (Brondani et al. 2006), which in turn corresponds to chromosome numbering in the Eucalyptus reference genome (Myburg et al. 2014). Maximum recombination frequency of 30 % and minimum LLR score (logarithm of the likelihood ratio) of 3 were used to determine linkage between markers. The distances between markers (in cM) were calculated using the Haldane mapping function.
QTL mapping
QTL detection was carried out with the previously constructed maps by single-marker analysis (ANOVA and contrasts between means or multiple comparisons, using trait means of the different locus-specific genotypic categories for each parent) and interval-mapping using the sib-pair approach described by Fulker and Cardon (1994). Analysis of variance (ANOVA) was performed to check for significant differences compared to the mapping population average, grouped according to the marker genotypic classes, allowing for multiple comparisons. QTL detection using ANOVA was carried out using a simple linear model with only one factor to decompose the variation among and within genotypic classes of markers:
where
- y ij :
-
Characteristic value in the j-th individual (j = 1, 2 ... n i ) carrier of the ith marker genotype (i = 1, 2, 3, 4 - to fully informative families);
- μ :
-
Mean of the population;
- M i :
-
Effect of the ith allele of the markers;
- ε ij :
-
Random error∼N(0, σ 2).
With fully informative markers (i.e., all four genotypic classes discernible, with three or four alleles segregating) the statistical test associated with the ANOVA is an F test with 3 degrees of freedom.
To decompose the influence of the alleles from each parent over the trait, contrasts were done by testing alleles individually. The contrast between means (δ) based on the t statistic was used to assess the existence of significant differences between means for individuals with specific alleles.
To test H o : δ = 0, we used the statistic t defined by the expression:
\( t=\frac{\widehat{\delta}}{\sqrt{\widehat{V}\left(\widehat{\delta}\right)}}=\frac{{\widehat{\mu}}_1-{\widehat{\mu}}_2}{\sqrt{\widehat{V}\left({\widehat{\mu}}_1\right)+\widehat{V}\left({\widehat{\mu}}_2\right)}} \), associated with n 1 + n 2 − 2 degrees of freedom,
Where:
- V(δ):
-
Variance of contrast between means;
- n 1 and n 2 :
-
Number of individuals in the full-sib family having the A1 and A2 alleles, respectively;
- V(μ 1) and V(μ 2):
-
Variance of individuals in the full-sib family having the A1 and A2 alleles, respectively.
Analogously, the contrast was evaluated considering the marker information coming from the second parent.
For the sib-pair interval method, the proportion of identical by descent (IBD) alleles in adjacent markers on a linkage group was used to estimate the proportion of alleles IBD at a putative QTL over the interval between the markers. The position that provides a better fit of the regression was taken as the estimated position of the QTL on the linkage group. Thus, it was possible to separately obtain estimates of the effect of the QTL on the additive genetic variance and its position (Cruz et al. 2009). This methodology is based on estimates of the IBD alleles of two linked markers, M 1 and M 2 at r 12 centimorgans, denoted \( {\widehat{\pi}}_{1j} \) and \( {\widehat{\pi}}_{2j} \), to estimate the values of the rate of IBD alleles among sibling pairs for the site Q, defined by π qj .
In positions with a better fit of the regression model, the estimated \( {\widehat{\beta}}_q \), which was associated with the optimal choice criterion of the regression equation, was used to infer the most likely position of the QTL and to estimate the value of the contribution of the QTL to the additive variance of the trait, given by \( {\widehat{\sigma}}_{\mathrm{AQTL}}^2=-\frac{{\widehat{\beta}}_q}{2} \). The heritability of the QTL within families of sibs was estimated by the expression: \( {h}_{QTL}^2=\frac{\sigma_{\mathrm{AQTL}}^2}{2{\sigma}_F^2} \), with \( {\sigma}_F^2 \) as the phenotypic variance within family and \( {\sigma}_{\mathrm{AQTL}}^2 \) as the additive variance of the QTL. QTL detection using this sib-pair regression method of Fulker and Cardon (1994) was performed using the software GQMOL (http://www.ufv.br/dbg/gqmol/gqmol.htm).
Results
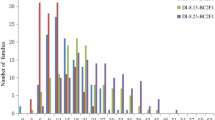
The variation of resistance response to Ceratocystis wilt in the DG × UGL family was continuous, with no clear separation between resistant and susceptible plants (Fig. 1). The progeny showed a disease average phenotypic severity of 12 %, with minimum and maximum values equal to 4 and 44 %, respectively. The susceptible comparator (clone HGU1172) had a disease severity of 47 %, demonstrating that the conditions were favorable to infection. The minimum severity value found (4 %) in some plants was equivalent in length to the injury caused as a simple response to the wound made during inoculation. The plants with a disease severity greater than the minimum value exhibited typical symptoms caused by C. fimbriata, such as xylem discoloration, wilting, die-back, and in some cases, eventual death of the plant (Fig. 2).

Severity of Ceratocystis wilt in 127 individuals of the mapping family DG × UGL (E. dunnii × E. grandis) × (E. urophylla × E. globulus) inoculated with isolate SBS1 of Ceratocystis fimbriata. Each gray bar represents the mean severity of five replicates of each genotype. The black bar corresponds to the susceptible comparator HGU1172

Symptoms of Ceratocystis wilt caused by Ceratocystis fimbriata on Eucalyptus spp. a Initial development of wilt symptoms (white arrow). b Plant with wilt symptoms 45 days after inoculation. c Outer discoloration of the stem. d Xylem discoloration
Of the 114 microsatellite markers genotyped, 110 were mapped, 89 (78 %) were completely informative (segregating 1:1:1:1), 6 were informative only in the male parent UGL (1:1 segregation), and 19 were informative only in the female parent DG (1:1 segregation). Using the χ 2 statistic, we found that 24 of the 110 mapped markers (21 %) showed segregation distortion (SD) from the expected genotypic proportions. Of these, 9 had gametic distortion for both parents (DG and UGL) and/or the parent in which it was informative. The other 15 had gametic distortion only in one parent: two markers had distortion in the gametes from female parents, and 13 had distortion in the male parents (Table 1). The test of gametic segregation in markers with SD showed that this occurs in a higher percentage in the male parent compared to the female (86 and 13 %, respectively) when considering only the fully informative markers (Table 1). The 110 markers were mapped on 15 linkage groups (LGs), whose lengths varied from 12.3 to 191.5 cM. The greatest distance between two markers (gap) was 43.1 cM. The total map length was 1352 cM with an average interval of 12.29 cM between markers.
QTL analysis via single-marker analysis of variance based on the data of individual phenotype severity identified four markers (Eg98, Embra22, Embra388, and Embra674) with significant effects (t test, P ≤ 0.05). Mean comparisons based on the contrast method, showed two of these markers (Eg98 and Embra388) associated with effects inherited from the female parents (DG) and two others (Embra22 and Embra674) associated to effects inherited from the male parent (UGL) (Table 2).
The sib-pair method of Fulker and Cardon (1994), confirmed the existence of the four QTLs identified by single-marker analysis, in addition to the identification of another QTL on LG1 associated with marker EN6 (Table 3 and Fig. 3). This QTL is located between markers En06 and Embra11, with estimated heritability (\( {\widehat{h}}_{QTL}^2 \)) of 0.11. The estimated effect of allelic replacement in the female and male parents was 1.84 and 0.09, respectively. In other words, this allele contributed an average of 1.84 and 0.09 % increase in lesion length depending on the parent. On LG3, the QTL was mapped between markers Embra1656 and Eg98, with the highest value of \( {\widehat{h}}_{QTL}^2 \) (0.34). The estimated effect of allelic replacement in the female parent was −4.53, thus contributing to the reduction of lesion length by 4.5 % on average. On LG5, the QTL was mapped between markers Embra388 and Embra41, with \( {\widehat{h}}_{QTL}^2 \) of 0.16. The estimated effect of allelic replacement in the female and male parents was 3.89 and 0.12, respectively. The QTL detected on LG8 was mapped between markers Embra674 and Embra203, with \( {\widehat{h}}_{QTL}^2 \) of 0.9. The estimated effect of allelic replacement in the male parent was −3.19. The QTL on LG10 was mapped between markers Embra22 and Embra127, with \( {\widehat{h}}_{QTL}^2 \) of 0.11. The estimated effects of allelic substitution in female and male parents were −1.42 and −4.37, respectively.

Linkage groups with the respective QTLs for resistance to Ceratocystis wilt in the mapping family DG × UGL (Eucalyptus grandis × E. dunni) × (E. urophylla × E. globulus). Black bars indicate the most likely QTL locations, and h 2 Q is the heritability of the trait. Distances (in cM) were calculated using the Haldane mapping function
The total genetic variance of the trait from phenotypic analysis was 114.9 and the sum of the variance of significant QTL effects was 106.4, which matches with the estimate for the trait genetic variance. The respective total heritabilities were 0.88 and 0.82 for the trait and sum of QTL effects, respectively, which are coherent.
Discussion
Five QTLs associated with resistance to Ceratocystis wilt were identified and their effects tentatively quantified in a hybrid full-sib family of Eucalyptus spp. Three QTLs identified on LG3, LG8, and LG10 involved alleles positively contributing to the resistance response to Ceratocystis wilt. The two QTLs identified on LG1 and LG5, on the other hand, contributed negatively to resistance in the population studied. These alleles targeted by specific microsatellites could be further investigated toward its use in breeding. The QTL effects on LG3 were inherited from the female parent, while the QTL on LG8 originated from the male parent. Allelic effects from both parents were detected for the QTL on LG10. These results demonstrate that both parents (DG and UGL) and their respective species compositions were able to contribute with usable variation in resistance response to Ceratocystis wilt.
Thus far, QTLs associated with disease-resistance in Eucalyptus have been described for Mycosphaerella leaf spot (Mycosphaerella cryptica), rust (P. psidii), and Calonectria leaf blight (Calonectria pteridis) (reviewed by Grattapaglia et al. 2012). With the exception of rust, in which only one QTL located on LG3 was identified (Mamani et al. 2010; Rosado et al. 2010b), QTLs for Mycosphaerella leaf spot and Calonectria leaf blight were found distributed across different LGs (Freeman et al. 2008; Zarpelon et al. 2015). This same pattern of distribution of QTLs across different LGs was found for Ceratocystis wilt, suggesting that resistance to these three diseases is more likely polygenic.
Interestingly, the QTL found in our study was mapped on LG3 in essentially the same genomic segment as the major-effect QTL Ppr1 for P. psidii resistance initially detected by Junghans et al. (2003) and later positioned on LG 3 (Mamani et al. 2010). Microsatellite markers Eg98 and Embra1656 flanked the Ppr1 QTL in that study as well as the QTL for Ceratocystis wilt mapped in this study. One of the five QTLs detected for Mycosphaerella resistance was also mapped to the same genomic region in E. globulus, although the interval for its most likely position was wider (Freeman et al. 2008). These results taken together apparently point to a syntenic fungal disease-resistance locus on linkage group 3 across Eucalyptus species. This finding is in line with the recent description of the unique features of Eucalyptus chromosome 3, which corresponds to LG3. It is the only chromosome not showing inter-chromosomal segmental duplications, while containing nearly exclusively the ancestral eudicot chromosome and thus a much conserved gene content and order even when compared to Populus. More importantly, however, is the fact that this chromosome was found to contain several clusters of shared syntenic genes related to perennial habit, including several syntenic sets of disease-resistance genes (Myburg et al. 2014).
The hypothesis of the existence of genes with pleiotropic action conferring resistance to multiple pathogens in Eucalyptus as P. psidii, Teratoshaeria nubilosa, and C. fimbriata is not unanticipated. There are several examples of genes conferring multi-pathogen resistance. For example, loci Lr34/Yr18/Pm38 and Lr46/Yr29/Pm39 simultaneously confer resistance to stripe rust and powdery mildew in wheat (Lillemo et al. 2008). The Mi1 gene in tomato confers resistance to root-knot nematodes, aphids, whitefly, viruses, bacteria, and fungi (Milligan et al. 1998). The Dm3 gene in lettuce (Meyers et al. 1998) confers resistance to downy mildew and aphid; and the Pto gene in tomato, when overexpressed, confers resistance to Pseudomonas syringae pv. Tomato, Xanthamonas campestris pv. vesicatoria, and Cladosporium fulvum (Chen and Roland 2011; Tang et al. 1999). However, the most likely polygenic nature of disease-resistance and the limited resolution of QTL mapping data collectively indicate that there is a reasonable probability of only apparent co-location of resistance loci to different diseases.
QTL detection was carried out using two alternative methods: single-marker analysis (ANOVA) and the sib-pair regression method of Fulker and Cardon (1994). Although ANOVA allows the detection of markers associated with QTLs, it does not identify which of the parents contributes favorable alleles to the resistance phenotype. Thus, an additional analysis based on contrasting the average effects is necessary to identify the effect of the segregating alleles from each parent on resistance.
A high number (21 %) of the mapped markers showed segregation distortion (SD) from the expected genotypic proportions. This was not unexpected, considering that the two parents were interspecific hybrids themselves, respectively of E. dunnii with E. grandis and E. urophylla with E. globulus. Potential problems in the pairing of homologous chromosomes might happen during meiosis in such interspecific hybrids resulting in segregation distortion; although there is evidence that the genome of eucalypt species are largely syntenic and collinear (Hudson et al. 2012). However, small differences between species are possible and indeed, two putative small translocations or duplications were reported between E. grandis and E. globulus linkage maps (Hudson et al. 2012). Our rate of segregation distortion (21 %) is consistent with a segregation distortion amounting to 27 % of the markers reported in backcross families involving an E. grandis × E. globulus F1 hybrid parent (Myburg et al. 2004). The gametic segregation test carried out also revealed that SD occurred in a higher percentage in the E. urophylla × E. globulus male parent. This could be explained by a variety of genetic factors such as the presence of lethal genes and sterility hybrid mismatch, a combination commonly called as the partial lethal factor (Cheng et al. 1998). On the other hand, the extent of distortion (21 %) was equivalent to the type 1 error (20 %) rate adopted, therefore warranting further investigation.
The genetic map built in this study was based on a joint analysis that considered both the fully informative markers segregating 1:1:1:1 together with the partially informative pseudo-testcross 1:1 markers. A single integrated map for both parents was built, which subsequently allowed a more informative QTL mapping analysis with the sib-pair interval-mapping approach using exclusively the fully informative markers. It is important to note that when the partially informative pseudo-testcross markers are used and QTL mapping is carried out in separate parental maps, the QTL analysis is less powerful (Cruz et al. 2009). The observation of four additional linkage groups relative to the expected haploid number of chromosomes of Eucalyptus spp. was likely due to the absence of sufficient segregating markers in certain genomic regions precluding the linkage of these additional smaller groups to the expected 11. Nevertheless, this did not impact the quality of the maps and the subsequent QTL analyses because the order of the mapped markers was maintained when compared to the reference map for Eucalyptus (Brondani et al. 2006).
In conclusion, this is the first study to map QTLs for Ceratocystis wilt resistance in Eucalyptus. It represents the initial step toward understanding the genetic architecture of the resistance response to C. fimbriata in species of the genus. The microsatellite markers linked to the QTLs identified in this study shall contribute to direct future QTL validation efforts in unrelated pedigrees. Clearly, however, as pointed out before (Zarpelon et al. 2015), QTLs mapped in single bi-parental pedigrees, while useful to demonstrate that discrete genomic regions can make relatively important contributions to trait variation, they may not be directly applicable in marker-assisted selection at the population level due to the uncertainties of the QTL effects across genetic background and environments. The within-family analysis uses only co-segregation information and so, it is expected to be useful only for within-family selection. However, if denser marker panels and multiple families are used, results could include population level linkage disequilibrium information (Azevedo et al. 2015), and potentially become applicable across the breeding population. Furthermore, if the QTLs mapped in this study ultimately get validated across families, they could provide grounds for assigning additional weight to markers located in those regions in genomic prediction models (Zhang et al. 2014), possibly increasing accuracy of genomic selection for disease-resistance.
References
Alfenas AC, Zauza EAV, Mafia RG, Assis TF (2009) Clonagem e doenças do eucalipto, 2nd edn. Ed. UFV, Viçosa
Azevedo CF, Resende MDV, Silva FF, Viana JMS (2015) Ridge, lasso and Bayesian additive-dominance genomic models. BMC Genet 16:105. doi:10.1186/s12863-015-0264-2
Baker C, Harrington T, Krauss U, Alfenas A (2003) Genetic variability and host specialization in the Latin American clade of Ceratocystis fimbriata. Phytopathology 93:1274–1284
Barnes I, Roux J, Wingfield BD, Neill MO, Wingfield MJ (2003) Ceratocystis fimbriata infecting Eucalyptus grandis in Uruguay. Australas Plant Pathol 32:361–366
Brondani RPV, Grattapaglia D (2001) Cost-effective method to synthetise a fluorescent internal DNA standard for automated fragment sizing. Biotechniques 31:793–795
Brondani RPV, Williams ER, Brondani C, Grattapaglia D (2006) A microsatellite-based consensus linkage map for species of Eucalyptus and a novel set of 230 microsatellite markers for the genus. BMC Plant Biol 6:20
Chen X, Roland PC (2011) Innate immunity in rice. Trends Plant Sci 16:451–459
Cheng R, Kleinhofs A, Ukai Y (1998) Method for mapping a partial lethal-factor locus on a molecular-marker linkage map of a backcross and doubled-haploid population. Theor Appl Genet 97:293–298
Cruz CD (2008) GQMOL: Programa para análise de genética quantitativa molecular. Versão 2008.6.1. Disponível em: <http://www.ufv.br/dbg/gqmol/gqmol.htm>, Desenvolvido pelo setor de Genética da Universidade Federal de Viçosa
Cruz CD, God PIVG, Bhering LL (2009) Mapeamento de QTLs em populações exogâmicas. In: Borém A (ed) Marcadores moleculares. Ed. UFV, Viçosa, pp. 443–481
Faria DA, Mamani EMC, Pappas GJ, Grattapaglia D (2011) Genotyping systems for Eucalyptus based on tetra-, penta-, and hexanucleotide repeat EST microsatellites and their use for individual fingerprinting and assignment tests. Tree Genet Genomes 7:63–77
Fernandes BV, Zanuncio AJV, Furtado EL, Andrade HB (2014) Damage and loss due to Ceratocystis fimbriata in Eucalyptus wood for charcoal production. Bioresources 9:5473–5479
Ferreira FA, Demuner AMM, Demuner NL, Pigato S (1999) Murcha de Ceratocystis em eucalipto no Brasil. (Abstr.). Fitopatol Bras 24:284
Ferreira FA, Maffia LA, Barreto RW, Demuner NL, Pigatto S (2006) Sintomatologia da murcha de Ceratocystis fimbriata em eucalipto. Rev Árvore 30:155–162
Ferreira MA, Harrington TC, Alfenas AC, Mizubuti ESG (2011) Movement of genotypes of Ceratocystis fimbriata within and among Eucalyptus plantations in Brazil. Phytopathology 101:1005–1012
Fonseca SM, Resende MDV, Alfenas AC, Guimarães LMS, Assis TF, Grattapaglia D (2010) Manual Prático de Melhoramento Genético do Eucalipto. Editora UFV, Viçosa, 200p
Freeman JS, Potts BM, Vaillancourt RE (2008) Few Mendelian genes underlie the quantitative response of a forest tree, Eucalyptus globulus, to a natural fungal epidemic. Genetics 178:563–571
Fulker DW, Cardon LR (1994) A sib-pair approach to interval mapping of quantitative trait loci. Am J Hum Genet 5:1092–1103
Grattapaglia D (2003) Genolyptus. In: Borém A, Giudice M, Sediyama T (eds) Melhoramento Genômico. Editora UFV, Viçosa, pp. 51–71
Grattapaglia D, Kirst M (2008) Eucalyptus applied genomics: from gene sequences to breeding tools. New Phytol 179:911–929
Grattapaglia D, Sederoff RR (1994) Genetic linkage maps of Eucalyptus grandis and E. urophylla using a pseudo-testcross mapping strategy and RAPD markers. Genetics 137:1121–1137
Grattapaglia D, Vaillancourt RE, Shepherd M, Thumma BR, Foley W, Külheim C, Potts BM, Myburg A (2012) Progress in Myrtaceae genetics and genomics: Eucalyptus as the pivotal genus. Tree Genet Genomes 8:463–508
Guimarães LMS, Resende MDV, Lau D, Rosse LN, Alves AA, Alfenas AC (2010) Genetic control of Eucalyptus urophylla and E. grandis resistance to canker caused by Chrysoporthe cubensis. Genet Mol Biol 33:525–531
Halsted BD (1890) Some fungus diseases of the sweet potato. The black rot. N J Agric Exp Station Bull 76:7–14
Harrington TC, Thorpe DJ, Alfenas AC (2011) Genetic variation and variation in aggressiveness to native and exotic hosts among Brazilian populations of Ceratocystis fimbriata. Phytopathology 101:555–566
Hudson CJ, Kullan ARK, Freeman JS, Faria D, Grattapaglia D, Kilian A, Myburg AA, Potts BM, Vaillancourt RE (2012) High synteny and collinearity among Eucalyptus genomes revealed by high-density comparative genetic mapping. Tree Genet Genomes 8:339–352
Iglesias I, Wiltermann D (2009) In eucalyptologics information resources on eucalypt cultivation worldwide http://www.git-forestry.com (GIT Forestry Consulting, retrieved, 29 March 2009)
Indury RM, Cardon LR (1997) A simple method for automated allele binning in microsatellite markers. Genome Res 7:1104–1109
Junghans DT, Alfenas AC, Brommonschenkel SH, Oda S, Mello EJ Grattapaglia D (2003) Resistance to rust (Puccinia psidii Winter) in Eucalyptus: mode of inheritance and mapping of a major gene with RAPD markers. Theoretical and Applied Genetics 108:175–180
Li J, Zhang Y, Xu KC, Yang JY, Han YH, Sun YX, Huang Q (2014) First report of wilt of Eucalyptus caused by Ceratocystis fimbriata in China. Plant Dis 98:1744
Lillemo M, Asalf B, Singh RP, Huerta-Espino J, Chen XM, He ZH, Bjornstad A (2008) The adult plant rust resistance loci Lr34/Yr18 and Lr46/Yr29 are important determinants of partial resistance to powdery mildew in bread wheat line Saar. Theor Appl Genet 116:1155–1166
Mamani EMC, Bueno NW, Faria DA, Guimarães LMS, Lau D, Alfenas AC, Grattapaglia D (2010) Positioning of the major locus for Puccinia psidii rust resistance (Ppr1) on the reference linkage map for Eucalyptus and validation across unrelated pedigrees. Tree Genet Genomes 6:953–962
Meyers BC, Chin DB, Shen KA, Sivaramakrishnan S, Lavelle DO, Zhang Z, Michelmore RW (1998) The major resistance gene cluster in lettuce is highly duplicated and spans several megabases. Plant Cell 10:1817–1832
Milligan SB, Bodeau J, Yaghoobia J, Kaloshian I, Zabel P, Williamson VM (1998) The root-knot nematode resistance gene Mi from tomato is a member of leucine zipper, nucleotide binding, leucinerich repeat family of plant genes. Plant Cell 10:1307–1319
Myburg AA, Grattapaglia D, Tuskan GA, Hellsten U, Hayes RD, Grimwood J, Jenkins J, Lindquist E, Tice H, Bauer D, Goodstein DM, Dubchak I, Poliakov A, Mizrachi E, Kullan AR, Hussey SG, Pinard D, van der Merwe K, Singh P, van Jaarsveld I, Silva-Junior OB, Togawa RC, Pappas MR, Faria DA, Sansaloni CP, Petroli CD, Yang X, Ranjan P, Tschaplinski TJ, Ye CY, Li T, Sterck L, Vanneste K, Murat F, Soler M, Clemente HS, Saidi N, Cassan-Wang H, Dunand C, Hefer CA, Bornberg-Bauer E, Kersting AR, Vining K, Amarasinghe V, Ranik M, Naithani S, Elser J, Boyd AE, Liston A, Spatafora JW, Dharmwardhana P, Raja R, Sullivan C, Romanel E, Alves-Ferreira M, Kulheim C, Foley W, Carocha V, Paiva J, Kudrna D, Brommonschenkel SH, Pasquali G, Byrne M, Rigault P, Tibbits J, Spokevicius A, Jones RC, Steane DA, Vaillancourt RE, Potts BM, Joubert F, Barry K, Pappas GJ, Strauss SH, Jaiswal P, Grima-Pettenati J, Salse J, Van de Peer Y, Rokhsar DS, Schmutz J (2014) The genome of Eucalyptus grandis. Nature 510:356–362
Myburg AA, Vogl C, Griffin AR, Sederoff RR, Whetten RW (2004) Genetics of postzygotic isolation in Eucalyptus: whole-genome analysis of barriers to introgression in a wide interspecific cross of Eucalyptus grandis and E. globulus. Genetics 166:1405–1418
Oliveira LSS, Guimarães LMS, Ferreira MA, Nunes AS, Pimenta LVA, Alfenas AC (2015a) Aggressiveness, cultural characteristics and genetic variation of Ceratocystis fimbriata on Eucalyptus spp. For Pathol. doi:10.1111/efp.12200
Oliveira LSS, Harrington TC, Ferreira MA, Damacena MB, Al-Sadi AM, Al-Mahmooli HIS, Alfenas AC (2015b) Species or genotypes? Reassessment of four recently described species of the Ceratocystis wilt pathogen, C. fimbriata, on Mangifera indica. Phytopathology. doi:10.1094/PHYTO-03-15-0065-R
Rosado CCG, Guimarães LMS, Titon M, Lau D, Rosse L, Resende MDV, Alfenas AC (2010a) Resistance to ceratocystis wilt (Ceratocystis fimbriata) in parents and progenies of Eucalyptus grandis x E. urophylla. Silvae Genet 59:99–106
Rosado TB, Tomaz RS, Junior MFR, Rosado AM, Guimarães LMS, Araújo EF, Alfenas AC, Cruz CD (2010b) Detection of QTL associated with rust resistance using IBD-based methodologies in exogamic Eucalyptus spp. populations. Crop Breed Appl Biotechnol 10:321–328
Roux J, Coutinho TA, Mujuni Byabashaija D, Wingfield MJ (2001) Diseases of plantation Eucalyptus in Uganda. S Afr J Sci 97:16–18
Roux J, Van Wyk M, Hatting H, Wingfield MJ (2004) Ceratocystis species infecting stem wounds on Eucalyptus grandis in South Africa. Plant Pathol 53:414–421
Roux J, Wingfield MJ, Bouillet JP, Wingfield BD, Alfenas AC (2000) A serious new wilt disease of Eucalyptus caused by Ceratocystis fimbriata in Central Africa. For Pathol 30:175–184
Tang X, Xie M, Kim YJ, Zhou J, Klessig DF, Matin GB (1999) Overexpression of Pto activates defence responses and confers broad resistance. Plant Cell 11:15–29
Zarpelon TG, Guimarães LMS, Faria DA, Coutinho MM, Neto BC, Teixeira RU, Grattapaglia D, Alfenas AC (2015) Genetic mapping and validation of QTLs associated with resistance to Calonectria leaf blight caused by Calonectria pteridis in Eucalyptus. Tree Genet Genomes 11:803
Zauza EAV, Alfenas AC, Harrington TC, Mizubuti ES, Silva JF (2004) Resistance of Eucalyptus clones to Ceratocystis fimbriata. Plant Dis 88:758–760
Zhang Z, Ober U, Erbe M, Zhang H, Gao N, He J, Li J, Simianer H (2014) Improving the accuracy of whole genome prediction for complex traits using the results of genome wide association studies. PLoS One 9:e93017
Acknowledgments
This work was supported by financial grants from FAPEMIG (Fundação de Amparo à Pesquisa de Minas Gerais), CNPq (Conselho Nacional de Desenvolvimento Científico e Tecnológico) and FAP-DF (Nextree Project). The authors are grateful to the Genolyptus project and Veracel S.A. for providing the genetic material of the mapping population DG × UGL and Leonardo S. S. Oliveira for review.
Data archiving statement
All individual genotype and phenotype data used for analysis are available at: https://dendrome.ucdavis.edu/tgdr/index.php. Markers names, linkage group, GenBank ID, and Linkage positions in cM are available at: http://dendrome.ucdavis.edu/cgi-bin/cmap/map_set_info?map_set_acc=TG161
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by R. Burdon
Rights and permissions
About this article

Cite this article
Rosado, C.C.G., da Silva Guimarães, L.M., Faria, D.A. et al. QTL mapping for resistance to Ceratocystis wilt in Eucalyptus . Tree Genetics & Genomes 12, 72 (2016). https://doi.org/10.1007/s11295-016-1029-4
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11295-016-1029-4