
Abstract
Members of the amino acid transporters (AATs) gene family transported amino acid across cellular membranes and participated in various aspects of normal plant growth and developmental processes as well as environmental responses. To date, no overall analysis or expression profiling of the AAT gene family in Populus has been reported. An investigation of the Populus genome revealed 100 putative AAT genes. These genes were classified into 11 subfamilies based on phylogenetic analysis. In each subfamily, the constituent parts of gene structure and motif were relatively conserved. A total of 100 genes were distributed on 19 chromosomes with 18-pair segmental duplication and 19-gene tandem duplication events, indicating that segmental and tandem duplications contribute almost equally to the expansion of the PtAAT gene family. Analysis of the Ka/Ks ratios showed that the duplicated genes of the AAT family basically underwent purifying selection. The expression levels of the 17 amino acid/auxin permease (AAAP) subfamily genes under abiotic stresses and in different tissues were investigated by quantitative real-time PCR (qRT-PCR) to explore their stress-related and tissue-specific expression patterns. The qRT-PCR results to explore the precise role of individual PtAAT gene. This study presents a thorough overview of the Populus AAT gene family and provides a new perspective on the evolution of this gene family. The results indicate that AAT family genes may be involved in many plant responses to stress conditions. Additionally, this study provides a solid foundation for uncovering the biological roles of AAT genes.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Amino acids have highly diverse and essential roles in plants; they provide important components to plants, such as compounds involved in plant metabolism and structure, and they contribute to the synthesis of a large variety of compounds critical to plant development, including nucleotides, chlorophyll, phytohormones and secondary metabolites (Tegeder 2012). Amino acid transporters (AATs) are the integral membrane proteins which play a key role in many essential biological processes. They can mediate long-distance amino acid transport and response to pathogen and abiotic stresses in higher plants (Wipf et al. 2002; Tegeder 2012; Tegeder et al. 1999), which are indispensable in various processes of plant growth and development. AAP1 was the first AAT identified in Arabidopsis thaliana, which can transport acidic, neutral, and basic amino acids, depending on the transporter (Frommer et al. 1993; Hsu et al. 1993). Based on heterologous complementation experiments and sequence homology, up to date, more than 60 putative amino acid transporters have been identified in Arabidopsis. Transporters have been further characterized by functional analysis in heterologous systems and by expression and localization studies. The AATs from plants are classified into two major families, the amino acid/auxin permease (AAAP) and amino acid–polyamine choline (APC) families, both of which belong to the APC superfamily (Saier 2009; Ortiz-Lopez et al. 2000). There are eight subfamilies in the AAAP family, including GABA transporters (GATs), proline transporters (ProTs), lysine histidine transporters (LHTs), amino acid permeases (AAPs), auxin transporters (AUXs), aromatic and neutral amino acid transporters (ANTs), and the amino acid transporter-like (ATLa and ATLb) subfamilies. The APC family consists of three subfamilies: cationic amino acid transporters (CATs), amino acid/choline transporters (ACTs), and polyamine H+-symporters (PHSs) (Fischer et al. 1998; Hunt et al. 2010; Okumoto and Pilot 2011). There are two domains in AAT family proteins, i.e., the Aa-trans and AA-permease domains.
Increasing evidence indicates that AATs are expressed in many tissues and play multiple, crucial roles in various aspects of plant growth and development. In Arabidopsis, the eight members (AtAAP1–AtAAP8) were found in the AtAAP subfamily, and six AtAAPs were demonstrated to transport neutral and charged amino acids with varying specificities and affinities (Couturier et al. 2010; Fischer et al. 1995; Okumoto et al. 2002). AtAAP1 was localized to the root tip and epidermis cells including root hairs and important in uptake of neutral and acidic amino acids (Lee et al. 2007; Svennerstam et al. 2011). AtAAP3 was mainly expressed in root vascular tissue and might be involved in amino acid uptake from the phloem or in retrieving amino acids from the soil (Okumoto et al. 2004). AtAAP5 has been shown to play a role in amino acid uptake by the root (Fischer et al. 1995; Hirner et al. 1998; Ortiz-Lopez et al. 2000). The expression of AtAAP6 has been detected in roots, sink leaves, cauline leaves, and xylem parenchyma, suggesting that AtAAP6 functioned in amino acid uptake from the xylem (Okumoto et al. 2002). AtAAP8 was expressed in young siliques and developing seeds and might play a crucial role in amino acid transport (Fischer et al. 1995; Schmidt et al. 2007). In addition, the AAP subfamily members from other species are also to be reported, such as StAAP1 (Koch et al. 2003), PvAAP1 (Tan et al. 2008), and OsAAP8 and OsAAP15 (Zhao et al. 2012). Recently, it was proposed that PtAAP11 may play a major role in xylogenesis by providing proline in Popular (Couturier et al. 2010).
AtLHT1 was a specific transporter for lysine and histidine, which mainly regulated root uptake of amino acids and supplied the leaf mesophyll with xylem-derived amino acids in Arabidopsis (Chen and Bush 1997; Hirner et al. 2006). Under the lower soil concentration, AtLHT1 and AtAAP5 played a role in uptake of neutral and acidic amino acids and basic amino acids, respectively (Svennerstam et al. 2008, 2011). AtProT2 was expressed in the root epidermis and cortex and involved in proline (compatible solute) acquisition by roots and was upregulated by salt stress (Grallath et al. 2005; Lehmann et al. 2011; Rentsch 1996). AtGAT1 was highly expressed in flowers and in response to wounding and senescence (Shelp 2006). AtANT1, which was specifically expressed in flowers and cauline leaves, functioned in the transport of aromatic and neutral amino acids as well as arginine and indole-3-acetic acid (Chen 2001). In the AtCAT subfamily, AtCAT5 functioned as a high-affinity, basic amino acid transporter in an amino acid transport in yeast (Frommer et al. 1995). AtCAT3, AtCAT6, and AtCAT8 preferentially transported neutral or acidic amino acids (Su et al. 2004). Meanwhile, AtBAT1, similar to a yeast GABA transporter (UGA4), was isolated as a bidirectional amino acid transporter (Dündar and Bush 2009). AtAUX1 functioned as an auxin influx carrier, which regulated root gravitropism and promoted lateral root formation by mediating the import of IAA (Bennett et al. 1996; Marchant 2002; Marchant et al. 1999).
Although numerous studies have revealed the roles of many AATs in plants, little information is available concerning the molecular characterization of AATs in some woody plants. Trees are very important among plants since they have extreme longevity and are able to generate woody biomass. Populus trichocarpa is a model woody species and for which genome sequencing was completed in 2006 (Tuskan et al. 2006). The availability of a genome sequence provided us with the perfect opportunity to conduct a comprehensive, genome-wide analysis of the AAT genes in P. trichocarpa. In this study, we performed bioinformatic analysis of 100 PtAAT genes. Among these, expression profiling of 17 AAP genes in different organs/tissues and under various abiotic stress treatments was performed by qRT-PCR. The results of this study provide a biological reference for further elucidating the roles of AATs in plants.
Materials and methods
Identification of AAT family genes in Populus
To identify AAT proteins in Populus, the P. trichocarpa genome database (release 3.0, http://www.phytozome.net/populus.php) was searched using Basic Local Alignment Search Tool algorithms (BLASTP), with the published Arabidopsis AAT protein sequences and their AAT 63 domains used as initial query sequences. Redundant sequences were then removed manually, and the Hidden Markov Model of the Pfam (http://pfam.sanger.ac.uk/search) and SMART (http://smart.embl-heidelberg.de/) databases were used to confirm each candidate sequence as a member of the AAT family (Finn et al. 2006, 2008; Letunic et al. 2004). A total of 63 Arabidopsis, 85 rice, and 96 maize AAT protein sequences were downloaded from Phytozome v10 (http://www.phytozome.net/). Populus AAT gene information, including the number of amino acids, ORF lengths, and chromosome locations, was obtained from the Phytozome database. Physicochemical parameters including the molecular weight (Mw) and isoelectric point (pI) of each gene product were calculated using compute pI/Mw tool from ExPASy (http://www.expasy.org/tools/), and parameter (resolution) was set to average (Gasteiger et al. 2003). Gene Structures Display Server (GSDS) (http://gsds.cbi.pku.edu.cn/) was used to illustrate the exon/intron structures of individual AAT genes by comparing their cDNAs and the corresponding genomic DNA sequences (Guo et al. 2007). To predict the putative TM regions in each PtAAT protein, the TMHMM Server v2.0 (http://www.cbs.dtu.dk/services/TMHMM/) was applied with default settings.
Phylogenetic and conserved motif analysis
The full-length amino acid sequences of all AAT proteins from Arabidopsis, rice, maize, and poplar were aligned using ClustalX 2.0 (Thompson et al. 1997). Bootstrap analysis was performed using 1000 replicates. The MEME motif search tool (http://meme.sdsc.edu/meme/intro.html) was used to identify motifs shared among related proteins within the PtAAT gene family with default settings (Bailey and Elkan 1995), except that the maximum number of motifs was defined as 20 and the maximum width was set to 300. The sequence conservation of PtAUX subfamily members in amino acid was analyzed by DNAMAN software and modified manually (Ma et al. 2011), and the conserved motifs and TM domains were annotated according to MEME analysis and TMHMM prediction, respectively.
Chromosomal location and gene duplication
An image of the chromosomal locations of PtAAT genes was generated using MapInspect (http://www.plantbreeding.wur.nl/uk/software_mapinspect.html) according to the chromosomal position information provided in the Phytozome database. To identify tandem and segmental duplications, two genes in the same species located in the same clade of the phylogenetic tree were defined as coparalogs. The Vista Synteny browser (pipeline.lbl.gov/cgibin/gateway2) was queried to detect the segmental duplication coordinates of the target genes. Coparalogs were deemed to result from segmental duplication if they were located on duplicated chromosomal blocks (Wei et al. 2007). Paralogs were deemed to be tandemly duplicated genes if two genes were separated by five or fewer genes in a 100-kb region (Wang et al. 2010). The local alignment of two protein sequences was calculated using the Smith–Waterman algorithm (http://www.ebi.ac.uk/Tools/psa/).
Identification of paralogs and orthologs
Paralogs and orthologs were identified following the method described by the report (Blanc 2004). For each species, all-against-all nucleotide sequence similarity searches were performed among the transcribed sequences using BLASTN software (Altschul et al. 1997). Sequences that aligned over 300 bp and showed at least 40 % identity were defined as pairs of paralogs. To identify putative orthologs between two species (A and B), each sequence from species A was searched against all sequences from species B using BLASTN. Additionally, each sequence from species B was searched against all sequences from species A. The two sequences were defined as orthologs if each of them was the best hit of the other and if more than 300 bp of the two sequences aligned.
Calculation of Ka/Ks values
Synonymous (Ks) and nonsynonymous substitution (Ka) rates were calculated according to a previous study (Hu et al. 2010). Pairs from the segmental duplication events were first aligned by ClustalX 2.0. Subsequently, the aligned sequences and the original cDNA sequences were analyzed with the PAL2NAL program (http://www.bork.embl.de/pal2nal/) using the CODEML program of PAML to estimate the Ks and Ka substitution rates (Suyama et al. 2006; Yang 1997). The divergence time (T) was calculated based on the Ks value and the number of substitutions per synonymous site per year as follows: T = Ks/2λ(λ = 9.1 × 10− 9) (Lynch 2000). A sliding window analysis of Ka/Ks ratios was performed with the following parameters: window size, 150 bp; step size, 9 bp.
Plant material and growth conditions
Asexually reproduced 8-week-old Populus euramericana cv. “Nanlin95” seedlings that were grown in a tissue culture laboratory under long day conditions (14 h light from 08:00 to 22:00) at 25–27 °C were used to assay gene expression levels in all experiments. Stem tips (internodes 1–3 from the top), young leaf (from internodes 1–3), mature leaf (from internodes 4–6), developing xylem (from the basal internodes), phloem (from the basal internodes), and root tissues were separately collected. For drought stress treatment, the plants were sprayed with 20 % PEG-6000 solution. For salt stress treatment, the plants were incubated in 200 mM NaCl solution at 25 °C. For cold stress treatment, the plants were incubated in a freezer at 4 °C. Parallel control samples were prepared at all five time points (1, 3, 6, 12, and 24 h). All samples were immediately frozen in liquid nitrogen and stored at −80 °C for RNA extraction after collection. Those without treatment were used as the control (0 h).
RNA isolation and qRT-PCR
Total RNA from stem tips, young leaves, and mature leaves were extracted using TRIzol reagent (Invitrogen, CA, USA) according to manufacturer’s instructions. Total RNA from differentiating xylem, phloem, and roots was isolated by the CTAB method with minor modifications (Chang et al. 1993). Total RNA was extracted from frozen samples using an RNAprep Pure Plant Kit (Tiangen) according to the manufacturer’s instructions. The first-strand cDNA was then synthesized using a PrimeScript™ RT Reagent Kit (TaKaRa). Gene-specific primers were designed using Primer Express 3.0, and their specificity was checked using information provided on the NCBI website. The poplar housekeeping ubiquitin gene (UBQ, gene id Potri.001G418500) was used as a reference for normalization due to its stable expression pattern. Real-time PCR was performed on an ABI 7300 Real-Time system (Applied Biosystems). Each reaction was performed in a final volume of 20 μl containing 12.5 μl of SYBR Green Master Mix reagent (Applied Biosystems), 1.5 μl of cDNA sample, and 1 μl of gene-specific primers. Specific primers for each PtAAP gene were designed to generate 90–150-bp products (Table S6). The qPCR reaction conditions were as follows: 50 °C for 2 min, 95 °C for 10 min, 40 cycles of 95 °C for 15 s, and annealing at 55–60 °C for 30 s. A melting curve was generated to analyze the specificity of the reactions, and three biological replicates were performed per treatment. Relative expression levels were calculated as 2− ΔΔCT[ΔC T = C T , Target − C T , CYP2. ΔΔCT = ΔC T , treatment − ΔC T , CK(0 h)]. The relative expression level (2−ΔΔCT, CK (0 h)) in control plants (without treatment) was normalized to 1 as described previously (Schmittgen and Livak 2008). Statistical analyses were performed using SDS software version 1.3.1 (Applied Biosystems).
Results
Identification of AAT gene family in P. trichocarpa
In the present study, to gain insight into the AAT gene family in the model species P. trichocarpa, we first performed BLASTP analysis against the Populus genome database (release v3.0) using Arabidopsis AAT gene sequences as queries. Consequently, 136 candidates were selected. Next, these genes were analyzed against the Pfam database using Simple Modular Architecture Research Tool (SMART) to identify the presence of the conservative domains (Aa-trans and AA-permease domains) of AAT proteins in Populus. After removing redundant sequences, a total of 100 AAT genes were identified in the Populus genome, which is greater than that identified in other representative species, including Arabidopsis (63), rice (Oryza sativa; 85), and maize (Zea mays; 96) (Rentsch et al. 2007; Zhao et al. 2012). Of the 100 AAT genes, 71 belong to the AAAP subfamily and 29 belong to the APC subfamily. We designated these 71 and 29 AAT genes AAAP01 to AAAP71 and APC01 to APC29, respectively, according to their physical locations (from top to bottom) on chromosomes 1–19 (Table 1). The identified AAT genes in Populus encode proteins ranging from 108 to 643 amino acids (aa) in length, with an average size of 453 aa. Other characteristics of these AATs, including isoelectric point (pI), molecular weight (Mw), chromosome location, and exons, are shown in Table 1.
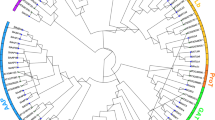
The numbers and positions of exons and introns were determined through comparing full-length cDNA sequences and the corresponding genomic DNA sequences of each PtAAT gene using GSDS (http://gsds.cbi.pku.edu.cn/). While introns are absent in 14 PtAAT genes, the remaining genes contain 1–14 introns (Fig. 1, right). Most members in the same subfamily share similar intron/exon structures and gene lengths. For example, the three members of the PtProT subfamily have seven exons and six introns and are nearly 400 bp in length. The putative transmembrane (TM) regions in PtAATs were predicted by TMHMM Server v2.0 (http://www.cbs.dtu.dk/services/TMHMM/). The number of TM regions in most PtAATs ranges from 8 to 13 (Figure S1), and PtAATs of the same subfamily have similar numbers of TM regions, such as 10 TMs in AUX and 11 TMs in ANT and ProTs. These observations demonstrate that the structures of members of the same subfamily are highly conserved.

Phylogenetic relationship and intron–exon structure of Populus AAT proteins. Left: Phylogenetic tree of PtAATs constructed by the neighbor-joining method. Bootstrap values from 1000 replicates are indicated at each node. The proteins on the tree are divided into 11 distinct subfamilies. The branches of different subfamilies are marked by different colors. Right: Exons and introns are indicated by yellow rectangles and gray lines, respectively. Untranslated regions (UTRs) are indicated by blue lines
Phylogenetic analysis and multiple-sequence alignment
To evaluate the evolutionary relationship among the 100 PtAATs, we performed phylogenetic analysis based on the alignment of full-length amino acid sequences of the 100 AAT proteins. The Populus AAT gene family was categorized into the AAAP and APC families (Fig. 1, left) based on phylogenetic analysis of AAT genes in Arabidopsis, rice, and maize. The AAAP family contains 71 genes comprising eight distinct subfamilies, namely the GABA transporters (GATs), proline transporters (ProTs), lysine histidine transporters (LHTs), amino acid permeases (AAPs), auxin transporters (AUXs), amino acid transporter-like (ATL), and aromatic and neutral amino acid transporters (ANTs) subfamilies. The ATL subfamily comprises two phylogenetic clades (ATLa and ATLb), while the APC family includes 29 genes subdivided into three distinct subfamilies, including the polyamine H+-symporters (PHSs), the cationic amino acid transporters (CATs), and the amino acid/choline transporters (ACTs).
Moreover, to further examine the diversification of Populus AAT genes, we used the MEME web server, revealing 20 distinct motifs (Fig. 2). Detailed information about the 20 putative motifs is shown in Table S1. Each of the putative motifs was annotated by searching Pfam and SMART. We found that only five motifs (9, 11, 14, 15, and 18) do not encode Aa-trans or AA-permease domains. Four motifs (8, 16, 17, and 19) encode AA-permease domain, while the remaining 11 motifs encode Aa-trans domain. Furthermore, several motifs are widespread among AAAP subfamily members (e.g., motif 3 and 11). By contrast, other motifs are specific to only one or two subfamilies. For example, motifs 1 and 14 are specific to AUXs, and motifs 4 and 18 are specific to AAPs and CATs, respectively. However, motif 17 is exclusively found in the APC subfamily, and motif 19 only exists in the PHS subfamilies. The domains in each subfamily are similar (Fig. 2). These results indicate that the structures of AAT family members are highly conserved, and they further validate our division of the AAT genes in Populus.

Schematic representation of the 20 conserved motifs in PtAAT proteins. Motifs of the PtAAT proteins were identified using the online MEME program. Different colored boxes represent different motifs, with their names in the center of the boxes. The colored boxes were ordered manually according to the results of MEME analysis. The length of each box in the figure does not represent the actual motif size
Alignment of the PtAAT amino acid sequences illustrated that most TM regions in the same subfamily are highly conserved. In addition, the differences in length and amino acid composition of several TM regions of different members are insignificant. An example of the alignment of PtAUX family members is shown in Fig. 3. There are five conserved motifs in PtAUXs, including motif 3, 1, 2, and 14. Motif 3 is located in the first TM region, while motif 1 comprises the first, second, third, and fourth TM regions and extends into the following sequences before the fifth TM region. In addition, motif 2 comprises the fifth, sixth, seventh, and eighth TM region. Motif 14 is located in the ninth and tenth TM regions.

Multiple sequence alignment and transmembrane region of PtAATs. Identical (100 %), conservative (75–99 %), and blocks (50–74 %) of similar amino acid residues are shaded in deep blue, cherry red, and light blue, respectively. The transmembrane regions are marked by black rectangles. The conserved motifs 3, 1, 2, and 14 are marked (in order) by red lines
Chromosomal location and gene duplication
Based on the starting position of each gene on the chromosomes, we mapped the 100 Populus AAT genes to the 19 Populus chromosomes. Most of the AAT genes are distributed on chromosomes 1–6 and 8–10. For example, chromosome 1 contains the highest number of genes (14 genes), while for chromosomes 2, 8, and 10, each contains 11 genes. Most of genes are located on the top of chromosomes 2 and 8 and the bottom of chromosomes 4, 9, and 10. By contrast, few AAT genes are present on the other chromosomes (Fig. 4). For example, there is only one AAT gene on chromosome 12.

Chromosomal localization and gene duplication events of PtAAT genes. Respective chromosome numbers are indicated above each bar. The scale on the left is in megabases (Mb). The duplicated paralogous pairs of AAT genes in the segmental duplicated blocks and tandem duplication blocks are indicated by small boxes of the same color and connected by gray lines and red rectangles, respectively
We identified duplicated genes using the Vista Synteny browser, revealing that 55 % of the genes arose from duplication events, including 18 gene pairs derived from segmental duplication and 19 genes derived from tandem duplication. Most of these duplicated genes are located on chromosomes 8 and 10. A total of 14 genes derived from segmental duplication are present on chromosomes 8 and 10. On the other hand, 19 PtAAT genes arose from tandem duplication (when genes separated by five or fewer gene loci in a 100-kb region are considered to be tandemly duplicated). Most of these genes are present on chromosome 1. Among these 19 genes, there are six pairs of genes, each containing two genes that come from tandem duplication (PtAPC01–PtAPC02, PtAAAP04–PtAAAP03, PtAPC06–PtAPC07, PtAAAP15–PtAAAP16, PtAAAP28–PtAAAP29, PtAPC28–PtAPC29). In addition, there are two blocks that contain three and four tandem duplication genes, respectively, such as, PtAAAP05–PtAAAP06–PtAAAP07 and PtAAAP58–PtAAAP59–PtAAAP60–PtAAAP60. These results indicate that segmental and tandem duplications have contributed almost equally to the expansion of the PtAAT gene family.
Over the course of evolution, duplicated genes can undergo non-functionalization (loss of original functions), neo-functionalization (acquisition of novel functions), or sub-functionalization (partition of original functions) during subsequent evolution (He and Zhang 2005; Lynch 2000; Vandepoele et al. 2003). To assess the selection mode of the duplicated AAT genes, we estimated the average rate of nonsynonymous substitution (Ka) versus synonymous substitution of nucleotides (Ks) by calculating the Ka/Ks ratio for each pair of duplicated AAT genes. In general, Ka/Ks <1 suggests that the fixation rate of amino acid change was reduced by purifying selection or functional constraint, while Ka/Ks = 1 indicates neutral selection and Ka/Ks >1 suggests that these genes may have been subject to positive selection, which favors the accumulation of adaptive genetic variations. In the current study, the Ka/Ks ratios were less than 0.3 (Table 2), indicating that the 18 gene pairs experienced negative or purifying selection during the process of evolution. Based on the divergence rate of 9.1 × 10−9 synonymous mutations per synonymous site per year as previously proposed for Populus (Tuskan et al. 2006), the duplications that gave rise to the 18 paralogous gene pairs were estimated to have occurred between 3.93 and 18.98 million years ago (Mya) (Table 2).
To determine the Ka/Ks ratios of different loci in the coding sequences, we preformed sliding-window analysis of the 18 gene pairs (Figure S2). The results show that the AAT domains have undergone strongly positive selection (Ka/Ks <<1), while most loci and regions have undergone moderately or strongly negative selection, just as the overall Ka/Ks ratio has predicted.
Comparative analysis of the AAT genes in Populus, Arabidopsis, rice, and maize
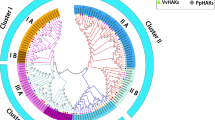
Most previous studies have focused on analyzing the AAT gene family in plants such as Arabidopsis, rice, and maize. To further analyze the evolutionary relationships of AAT genes among these four plants, we first constructed a NJ phylogenetic tree with ClustalX 2.0 using 344 full-length AAT protein sequences, including 63 sequences from Arabidopsis, 85 from rice, 96 from maize, and 100 from Populus. The characteristics of the 344 AAT genes, including isoelectric point (pI), molecular weight (Mw), chromosome location, ORF length, and amino acids (aa), are shown in Table S2. In Arabidopsis, the AAT gene family is divided into 12 subfamilies, with AtTTP1 and AtTTP2 serving as the outgroup. Therefore, the phylogenetic tree clearly shows that the 344 AAT proteins could be divided into 12 subfamilies. While the Tryptophan/tyrosine permease (TTP) subfamily contains only the Arabidopsis AAT proteins, other subfamilies contain Arabidopsis, rice, maize, and Populus AAT proteins. We counted the number of AAT proteins in every subfamily among the four species (Table S3), revealing that AAP is the largest subfamily in every species.
Both subfamilies (except TTP subfamily) contain Arabidopsis, rice, maize, and Populus AAT genes, suggesting that the main characteristics of this family in plants were generated before the dicot–monocot split. To clarify the paralogous and orthologous relationships among this family, the paralogous and orthologous extensins within and between species are listed in Table S4 and Table S5. In Arabidopsis and rice, there were 12 paralogous groups containing 24 extensins. In Populus, there were 34 paralogous groups containing 68 extensins identified. In maize, there were 11 paralogous groups containing 22 extensins identified (Fig. 5, Table S4). The combined phylogenetic tree reveals that most genes in the AAT family, especially the duplicated genes, are contained in paralogous pairs in each species, which supports the occurrence of lineage-specific AAT gene duplication events. And then, these two species shared the 28 orthologous pairs, followed by rice and maize. There were also nine orthologous pairs (AtVAAT3/PtAAAP3, AtVAAT2/PtAAAP69, AtCAT4/PtAPC28, AtCAT9/PtAPC09, AtLHT10/PtAAP04, AtLHT4/PtAAAP62, AtLAT5/PtAPC02, AtANT1/PtAAAP66, and AtAAP6/PtAAAP12) detected between Populus and Arabidopsis. Only two pairs of orthologous (ZmAPC17–PtAPC17 and ZmAAAP40–PtAAAP65) were detected between Populus and maize (Fig. 5, Table S5).

Phylogenetic tree of full-length AAT protein sequences from Populus, Arabidopsis, rice, and maize. The tree was generated with ClustalX 2.0 using the NJ method. Populus, Arabidopsis, rice, and maize proteins are indicated with different-colored dots. Yellow rectangles represent the orthologous pairs from rice and maize, crimson rectangles represent the orthologous pairs from Populus and Arabidopsis, black rectangles represent the orthologous pairs from Populus and maize
Differential expression profiling of PtAAP genes
As mentioned above, the Arabidopsis genes clustered in the AAP subfamily were participated in various processes of plant growth and development, and they can regulate the transport of amino acid and play an indispensable role in responses to abiotic stress (Tegeder 2012). The phylogenetic analysis indicated that AAP subfamily contains 17 Populus AAT genes and that these genes are closely related to the Arabidopsis stress-responsive genes (Fig. 5). This observation prompted us to investigate possible stress-responsive genes of the 17 PtAAP genes by qRT-PCR. We investigated the expression levels of 17 PtAAP genes in roots, young leaves, mature leaves, stem tips, xylem, and phloem of young poplar trees (“Nanlin95”) using qRT-PCR. PtAAAP05, PtAAAP06, PtAAAP07, PtAAAP29, PtAAAP34, and PtAAAP45 were significantly expressed in stem tips and phloem (Fig. 6a). PtAAAP58, PtAAAP59, and PtAAAP60 were expressed at high levels in phloem and at low levels in other tissues (Fig. 6b). Meanwhile PtAAAP09, PtAAAP10, and PtAAAP25 were mainly expressed in stem tips (Fig. 6c). PtAAAP12, PtAAAP33, PtAAAP43, and PtAAAP61 were highly expressed in some tissues, while PtAAAP28 was only expressed in differentiating xylem (Fig. 6d, e).

Real-time PCR analysis of tissue-specific expression of AAP subfamily genes. Relative mRNA levels of individual genes normalized to UBQ10 are shown. Y-axis indicates relative mRNA expression levels. The X-axis indicates different organs. Error bars, 6 ± SE. R roots, YL young leaves, ML mature leaves, ST stem tips, X differentiating xylem, P phloem. a Genes with preferential expression in JT and P. b, c, e Genes with preferential expression in P, ST, and X, respectively. d Genes with preferential expression in some tissues
Since expression patterns can provide important clues for possible gene function, we further investigated the expression levels of these genes in response to various abiotic stresses. Therefore, we performed qRT-PCR to study the expression levels of 17 selected AAP subfamily members (PtAAAP05, PtAAAP06, PtAAAP07, PtAAAP09, PtAAAP10, PtAAAP12, PtAAAP25, PtAAAP28, PtAAAP29, PtAAAP33, PtAAAP34, PtAAAP43, PtAAAP45, PtAAAP58, PtAAAP59, PtAAAP60, and PtAAAP61) in response to PEG, salt, and cold treatment. For the PEG (drought) treatment, seven of the 17 genes (PtAAAP05, PtAAAP12, PtAAAP33, PtAAAP34, PtAAAP43, PtAAAP45, and PtAAAP61) were upregulated in response to PEG (drought) treatment. PtAAAP05, PtAAAP12, PtAAAP33, PtAAAP43, PtAAAP45, and PtAAAP61 exhibited the highest expression levels during early (1 h) treatment; the expression of PtAAAP12 and PtAAAP45 at 1 h was 8-fold that at 0 h. The expression of only one gene (PtAAAP34) peaked at 24 h. PtAAAP06, PtAAAP07, PtAAAP10, PtAAAP28, PtAAAP58, PtAAAP59, and PtAAAP60 were downregulated during early PEG (drought) stress treatment and upregulated at later time points. For example, PtAAAP10, PtAAAP28, and PtAAAP59 were strongly upregulated (>4-fold) at 3 h, as PtAAAP28 and PtAAAP59 were at 6 h. PtAAAP09, PtAAAP25, and PtAAAP29 were downregulated at later time points (Fig. 7). The qRT-PCR analysis also revealed that PtAAP genes are also regulated by cold stress. The highest expression levels of PtAAAP05 and PtAAAP06 were found at 1 h after treatment, while PtAAAP10 and PtAAAP45 were most strongly expressed at 6 h after treatment. PtAAAP07, PtAAAP12, and PtAAAP33 exhibited major changes in expression, with almost no expression detected at the three earliest time points and dramatically upregulated expression detected thereafter. PtAAAP58 exhibited only slight differences in expression over the 24-hour time course. The expression of PtAAAP28, PtAAAP43, and PtAAAP59 began to decline quickly when subjected to cold stress, followed by a return to near pretreatment levels. The expression of PtAAAP09 and PtAAAP34 significantly decreased gradually at all later time points, while the expression of PtAAAP29, PtAAAP60, and PtAAAP61 increased gradually at these time points (Fig. 8). Under NaCl treatment, the expression of 12 genes (PtAAAP05, PtAAAP06, PtAAAP07, PtAAAP09, PtAAAP10, PtAAAP25, PtAAAP28, PtAAAP29, PtAAAP43, PtAAAP45, PtAAAP59, and PtAAAP60) peaked at 24 h after treatment, while the expression of PtAAAP33, PtAAAP34, and PtAAAP58 peaked at 3 and 6 h, respectively. PtAAAP05, PtAAAP07, PtAAAP09, PtAAAP29, and PtAAAP60 had the highest expression levels (more than 4-fold that of control levels) at 24 h, while PtAAAP45 had the highest expression level more than 14-fold that of control levels at 24 h (Fig. 9).

Expression patterns of 17 selected AAP subfamily genes under drought stress revealed by qRT-PCR. The Y-axis indicates relative expression levels. The X-axis indicates the time courses of stress treatments. Error bars, 6 ± SE

Expression patterns of 17 selected AAP subfamily genes under cold stress revealed by qRT-PCR. The Y-axis indicates relative expression levels. The X-axis indicates the time courses of stress treatments. Error bars, 6 ± SE

Expression patterns of 17 selected AAP subfamily genes under salinity stress revealed by qRT-PCR. The Y-axis indicates relative expression levels. The X-axis indicates the time courses of stress treatments. Error bars, 6 ± SE
Together, the qRT-PCR results indicate that only PtAAAP05 was upregulated under all three stress treatments, and PtAAAP10, PtAAAP25, PtAAAP28, and PtAAAP59 were downregulated under these treatment, implying that some PtAAT genes play important roles in regulating the responses to drought, cold, and salt stress. Moreover, while some duplicated genes within a sister pair exhibited similar expression patterns, differential expression patterns between two duplicated genes was also observed. For example, under PEG (drought) stress, the highest expression level of PtAAAP60 was observed at 12 h, while that of PtAAAP61 was observed at 1 h. Under cold stress treatment, PtAAAP06 exhibited major changes in expression, while PtAAAP58 exhibited only minor changes in expression. Finally, PtAAAP12 was downregulated under salt stress, while PtAAAP43 was upregulated.
Discussion
The plant AAT gene family has been comprehensively analyzed in Arabidopsis, rice, and maize. However, this family has not previously been studied in Populus. In the current study, we performed an overall analysis of the 100 PtAAT genes, including analysis of their phylogeny, chromosomal location, gene structure, conserved motifs, and expression profiles. A total of 100 full-length AAT genes were identified in the Populus genome, which exceeds the number identified in Arabidopsis, rice, and maize, implying that there was genome expansion of the Populus AAT counterparts. The 100 PtAAT genes were divided into 11 subfamilies based on their phylogenetic relationships with AAT proteins from Arabidopsis, rice, and maize. The number of members in each subfamily differs (Table S3). The largest subfamily, AAP, contains 17 genes, while the smallest subfamily, ProT, contains only three genes. However, a previous report estimated that there are 14 genes in the AAP subfamily (Couturier et al. 2010). There are two possible reasons for this discrepancy. First, an increasing number of sequences have been assembled and introduced into the Populus genome database. Second, in the current study, we performed genome-wide analysis of the AAT family, while the previous study focused on a subfamily of AAT genes.
Chromosomal mapping of PtAAT family genes revealed their variable distribution on 19 Populus chromosomes, but most members are localized on chromosomes 1, 2, 3, 4, 5, 6, 8, 9, and 10. Meanwhile, tandemly duplicated genes are mainly present on chromosomes 1, 5, and 11, and duplicated segments are mainly localized on chromosomes 10 and 16 (Fig. 4). In each subfamily, the characteristics of exon/intron structures and motif compositions were relatively conserved in recent paralogs (Figs. 1, right, and 2), which indicates their close evolutionary relationship and helps confirm the classification of the subfamilies.
Gene duplication is one of the major evolutionary mechanisms for generating novel genes, which helps organisms adapt to different environments (Bowers et al. 2003; Gu et al. 2003). The Populus genome has undergone at least three rounds of genome-wide duplication, followed by multiple segmental duplications, tandem duplications, and transposition events (He and Zhang 2005; Prince and Pickett 2002). Transposition events include retroposition and replicative transposition (Kong et al. 2007). As many as 14,000 of the 45,000 predicted genes in Populus are retained in duplicated pairs resulting from the salicoid duplication event (Hurst 2002). In the present study, our analysis of gene duplication revealed that 55 of the 100 Populus AAT genes (55 %) arose from duplication events, including 18 pairs from segmental duplication and 19 from tandem gene duplication, indicating that segmental and tandem duplications have contributed almost equally to the expansion of the PtAAT gene family. We further calculated the duplication dates of the 18 pairs of genes derived from segmental duplication. The Ka/Ks ratios were less than 0.3 (Table 2), indicating that these 18 gene pairs experienced negative or purifying selection during the process of evolution, which occurred between 3.93 and 18.98 Mya.
To examine the phylogenetic relationship among the AAT domain proteins in Populus, Arabidopsis, maize, and rice, we performed a genome-wide comparison of plant AAT members from these four plants to explore how the AAT gene family has evolved. There were many paralogous pairs in each subfamily, especially in Populus, wherein more than half of the extensins were associated with intraspecific duplication, suggesting that the AAT proteins have many from the four species appearing to be more closely related to each other than to those from the same species in different subfamilies (Fig. 5, Table S4). The presence of 11 distinct subfamilies (except TTP subfamily) of AAT genes and the presence of both monocots and dicots containing members in all 11 subfamilies indicate AAT genes have diversified before the monocot–dicot split. There are 39 orthologous pairs, including nine pairs from Populus and Arabidopsis, while 28 pairs from rice and maize, which further verifies the validity of the phylogenetic tree, in which PtAATs are more closely grouped with AtAATs than with OsAATs and ZmAATs. Because the genetic relationship between rice and maize is closer than Populus and Arabidopsis, as we all know, rice and maize are both monocots and belong to Gramineae, while Populus and Arabidopsis are both dicots but belong to different families. Arabidopsis comes from Cruciferae while Populus comes from Salicaceae. Most orthologous showed a close relationship than paralogs between Populus and Arabidopsis AAT genes, indicating that these genes which formed the orthologous pairs might have originated from their common ancestor, in which ancient duplication events occurred predating Populus–Arabidopsis divergence. Populus and Arabidopsis lineages diverged about 100–120 million years ago (Mya) (Tuskan et al. 2006). In the combined tree of AAT genes in all four species, we identified only two monocot and dicot orthologous pairs (ZmAPC17–PtAPC17 and ZmAAAP40–PtAAAP65) from maize and Populus (Fig. 5, Table S5), suggesting that the orthologous pair originated from common ancestral genes that existed before the divergence of monocots and dicots.
Most of the amino acid transporters from plants that have been functionally characterized belong to the AAAP superfamily, with the amino acid permease (AAP) family being the subfamily that has been studied most. In Arabidopsis, there were eight genes in the AAP subfamily, and only AtAAP7 has not been studied so far. The functions of the remaining seven (AtAAP1 to AtAAP6 and AtAAP8) genes have been reported. Based on the genetic relationship between Arabidopsis and Populus, we speculated the function of the PtAAPs. The expression profile of a gene can provide a valuable clue for its functional study (Zhao et al. 2010). Our real-time PCR analysis of tissue-specific expression of AAP subfamily reveal that the expression patterns of the 17 PtAAP genes could be divided into five major groups. Some preferential or tissue-specific expression of PtAAP genes were also identified. The tissue expression results showed that six genes (PtAAAP05, PtAAAP06, PtAAAP07, PtAAAP29, PtAAAP34, and PtAAAP45) were significantly expressed in stem tips and phloem. PtAAAP58, PtAAAP59, and PtAAAP60 were expressed at high levels in phloem (Fig. 6). The PtAAAP43 and PtAAAP61 were highly expressed in root. In Arabidopsis, AtAAP3 with mainly expression in root vascular tissue may be involved in amino acid uptake from phloem (Okumoto et al. 2004). Similar expression patterns suggest that these root preferentially or specifically expressed genes might play important roles in root formation and development. Our data showed that PtAAAP12, an ortholog of AtAAP6, which had high expression levels in some tissues especially in young leaves, differentiating xylem and phloem, and PtAAAP28 was almost preferentially expressed in differentiating xylem. It is known that AtAAP6 was found to be expressed in the xylem parenchyma and might be responsible for amino acid uptake from xylem (Fischer et al. 2002). So we speculated that PtAAAP12 and PtAAAP28 were responsible for amino acid uptake from xylem. AtAAP1 regulates the import of amino acids into developing Arabidopsis embryos (Sanders et al. 2009). AtAAP2 may play critical role in the long-distance transport of amino acid (Hirner et al. 1998; Okumoto and Pilot 2011). Therefore, combining their phylogenetic relationship and role of the AtAAPs, we infer that some PtAAP genes might participate in the uptake and long-distance transport of amino acid.
It is known that amino acid transport is highly regulated by environmental signals, such as light, low temperature, high salt, and/or drought (Grallath et al. 2005). In spite of a few, the amino acid permease (AAP) subfamily genes have been reported to play positive roles in response to abiotic stress in many plants (Ueda et al. 2001). However, no AAP gene response to abiotic stresses was reported in Populus. For this purpose, we investigated the expression patterns of Populus AAP genes under PEG (drought), cold, and salt stresses. The subfamily of PtAAP genes shows significant and differential expression pattern under three abiotic stresses (Figs. 7–9). The results demonstrated that most of them were downregulated by salt stress. Moreover, we also found that the PtAAP genes were either increase or repressed under the PEG and cold treatment. Our investigation suggests that the PtAAT genes may play a critical role in abiotic stress signaling in Populus. Taken all of the results by abiotic stresses, we can detect that most genes within the same subclass of the phylogenetic tree showed the similar expression patterns. For example, three pairs of duplicated genes (PtAAAP07–PtAAAP59, PtAAAP09–PtAAAP29, and PtAAAP12–PtAAAP43) showed the similar expression patterns under PEG stress. PtAAAP25–PtAAAP34, PtAAAP60–PtAAAP61, and PtAAAP07–PtAAAP59 showed the similar expression patterns under cold stress, as well as two pairs of duplicated genes (PtAAAP09–PtAAAP29 and PtAAAP07–PtAAAP59) showed the similar expression patterns under salt stress (Fig. 7). These duplicated genes in the same subclass share high sequence similarity and had the similar expression patterns, indicating that the regulatory sequences that respond to the stress conditions did not diverge much along with the evolution of each gene after duplication. In contrast, there were some pairs of duplicated genes expressed differently, indicating that the regulatory sequences that respond to the stress conditions had diverged much along with the evolution of each gene after duplication.
Conclusions
In conclusion, the results of this study display the genomic framework, classification, duplication manner, Ka/Ks ratios, and conserved motifs of the 100 PtAAT members. We selected 17 amino acid/auxin permease (AAAP) subfamily genes to explore their stress-related and tissue-specific expression patterns under abiotic stresses and in different tissues were investigated by quantitative real-time PCR (qRT-PCR). The qRT-PCR results to explore the precise role of individual PtAAT gene. This study presents a thorough overview of the Populus AAT gene family and provides a new perspective on the evolution of this gene family. These data will provide an insight into further understanding of functions of AAT members and their roles in Populus growth and development.
References
Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25:3389–3402
Bailey TL, Elkan C (1995) The value of prior knowledge in discovering motifs with MEME. Proceedings/International Conference on Intelligent Systems for Molecular Biology; ISMB International Conference on Intelligent Systems for Molecular Biology 3:21–29
Bennett MJ, Marchant A, Green HG, May ST, Ward SP, Millner PA, Walker AR, Schulz B, Feldmann KA (1996) Arabidopsis AUX1 gene: a permease-like regulator of root gravitropism. Science 273:948–950
Blanc KH (2004) Widespread paleopolyploidy in model plant species inferred from age distributions of duplicate genes. Plant Cell 16:1667–1678
Bowers JE, Chapman BA, Rong J, Paterson AH (2003) Unravelling angiosperm genome evolution by phylogenetic analysis of chromosomal duplication events. Nature 422:433–438
Chang S, Puryear J, Cairney J (1993) A simple and efficient method for isolating RNA from pine trees. Plant Mol Biol Report 11:113–116
Chen L (2001) ANT1, an aromatic and neutral amino acid transporter in Arabidopsis. Plant Physiol 125:1813–1820
Chen L, Bush DR (1997) LHT1, a lysine- and histidine-specific amino acid transporter in Arabidopsis. Plant Physiol 115:1127–1134
Couturier J, De FE, Fitz M, Wipf D, Blaudez D, Chalot M (2010) PtAAP11, a high affinity amino acid transporter specifically expressed in differentiating xylem cells of poplar. J Exp Bot 61:1671–1682(12)
Dündar E, Bush DR (2009) BAT1, a bidirectional amino acid transporter in Arabidopsis. Planta 229:1047–1056
Finn RD, Mistry J, Schuster-Böckler B, Griffiths-Jones S, Hollich V, Lassmann T, Moxon S, Marshall M, Khanna A, Durbin R (2006) Pfam: clans, web tools and services. Nucleic Acids Res 34:D247–D251
Finn RD, Tate J, Mistry J, Coggill PC, Sammut SJ, Hotz HR, Ceric G, Forslund K, Eddy SR, Sonnhammer ELL (2008) The Pfam protein families database. Nucleic Acids Res 36:263–266(4)
Fischer WN, Kwart M, Hummel S, Frommer WB (1995) Substrate specificity and expression profile of amino acid transporters (AAPs) in Arabidopsis. J Biol Chem 270:16315–16320
Fischer WN, André B, Rentsch D, Krolkiewicz S, Tegeder M, Breitkreuz K, Frommer WB (1998) Amino acid transport in plants. Trends Plant Sci 3:188–195
Fischer WN, Loo DDF, Koch W, Ludewig U, Boorer KJ, Tegeder M, Rentsch D, Wright EM, Frommer WB (2002) Low and high affinity amino acid H+-cotransporters for cellular import of neutral and charged amino acids. Plant J 29:717–731
Frommer WB, Hummel S, Riesmeier JW (1993) Expression cloning in yeast of a cDNA encoding a broad specificity amino acid permease from Arabidopsis thaliana. Proc Natl Acad Sci 90:5944–5948
Frommer WB, Hummel S, Unseld M, Ninnemann O (1995) Seed and vascular expression of a high-affinity transporter for cationic amino acids in Arabidopsis. Proc Natl Acad Sci 92:12036–12040
Gasteiger E, Gattiker A, Hoogland C, Ivanyi I, Appel RD, Bairoch A (2003) ExPASy: the proteomics server for in-depth protein knowledge and analysis. Nucleic Acids Res 31:3784–3788
Grallath S, Weimar T, Meyer A, Gumy C, Suter-Grotemeyer M, Neuhaus JM, Rentsch D (2005) The AtProT family. Compatible solute transporters with similar substrate specificity but differential expression patterns. Plant Physiol 137:117–126
Gu Z, Steinmetz LM, Gu X, Scharfe C, Davis RW, Li WH (2003) Role of duplicate genes in genetic robustness against null mutations. Nature 421:63–66
Guo AY, Zhu QH, Chen X, Luo JC (2007) GSDS: a gene structure display server. Hereditas 29(8):1023–1026
He X, Zhang J (2005) Rapid subfunctionalization accompanied by prolonged and substantial neofunctionalization in duplicate gene evolution. Genetics 169:1157–1164
Hirner B, Fischer WN, Rentsch D, Kwart M, Frommer WB (1998) Developmental control of H+/amino acid permease gene expression during seed development of Arabidopsis. Plant J 14:535–544(10)
Hirner A, Ladwig F, Stransky H, Okumoto S, Keinath M, Harms A, Frommer WB, Koch W (2006) Arabidopsis LHT1 is a high-affinity transporter for cellular amino acid uptake in both root epidermis and leaf mesophyll. Plant Cell 18:1931–1946
Hsu LC, Chiou TJ, Chen L, Bush DR (1993) Cloning a plant amino acid transporter by functional complementation of a yeast amino acid transport mutant. Proc Natl Acad Sci 90:7441–7445
Hu R, Qi G, Kong Y, Kong D, Qian G, Zhou G (2010) Comprehensive analysis of NAC domain transcription factor gene family in Populus trichocarpa. BMC Plant Biol 10:145
Hunt E, Gattolin S, Newbury HJ, Bale JS, Tseng HM, Barrett DA, Pritchard J (2010) A mutation in amino acid permease AAP6 reduces the amino acid content of the Arabidopsis sieve elements but leaves aphid herbivores unaffected. J Exp Bot 61:55–64
Hurst LD (2002) The Ka/Ks ratio: diagnosing the form of sequence evolution. Trends Genet 18:486–487
Koch W, Kwart M, Laubner M, Heineke D, Stransky H, Frommer WB, Tegeder M (2003) Reduced amino acid content in transgenic potato tubers due to antisense inhibition of the leaf H+ /amino acid symporter StAAP1. Plant J 33:211–220
Kong H, Landherr LL, Frohlich MW, Leebens-Mack J, Ma H, dePamphilis CW (2007) Patterns of gene duplication in the plant SKP1 gene family in angiosperms: evidence for multiple mechanisms of rapid gene birth. Plant J 50(5):873–885
Lee YH, Foster J, Chen J, Voll LM, Weber APM, Tegeder M (2007) AAP1 transports uncharged amino acids into roots of Arabidopsis. Plant J 50:305–319
Lehmann S, Gumy C, Blatter E, Boeffel S, Fricke W, Rentsch D (2011) In planta function of compatible solute transporters of the AtProT family. J Exp Bot 62:787–796(10)
Letunic I, Copley RR, Schmidt S, Ciccarelli FD, Doerks T, Schultz J, Ponting CP, Bork P (2004) SMART 4.0: towards genomic data integration. Nucleic Acids Res 32:D142–D144
Lynch M (2000) The evolutionary fate and consequences of duplicate genes. Science 290:1151–1155
Ma H, Zhao H, Liu Z, Zhao J (2011) The phytocyanin gene family in rice (Oryza sativa L.): genome-wide identification, classification and transcriptional analysis. PloS One 6(10):e25184
Marchant A (2002) AUX1 promotes lateral root formation by facilitating indole-3-acetic acid distribution between sink and source tissues in the Arabidopsis seedling. Plant Cell 14:589–597
Marchant A, Kargul J, May ST, Muller P, Delbarre A, Perrot-Rechenmann C, Bennett MJ (1999) Aux1 regulates root gravitropism in Arabidopsis by facilitating auxin uptake within root apical tissues. EMBO J 18:2066–2073
Okumoto S, Pilot G (2011) Amino acid export in plants: a missing link in nitrogen cycling. Mol Plant 4:453–463
Okumoto S, Schmidt R, Tegeder M, Fischer WN, Rentsch D, Frommer WB, Koch W (2002) High affinity amino acid transporters specifically expressed in xylem parenchyma and developing seeds of Arabidopsis. J Biol Chem 277:45338–45346
Okumoto S, Koch W, Tegeder M, Fischer WN, Biehl A, Leister D, Stierhof YD, Frommer WB (2004) Root phloem-specific expression of the plasma membrane amino acid proton co-transporter AAP3. J Exp Bot 55:2155–2168
Ortiz-Lopez A, Chang H, Dr B (2000) Amino acid transporters in plants. Biochimica Et Biophysica Acta 1465:275–280
Prince VE, Pickett FB (2002) Splitting pairs: the diverging fates of duplicated genes. Nat Rev Genet 3:827–837
Rentsch D (1996) Salt stress-induced proline transporters and salt stress-repressed broad specificity amino acid permeases identified by suppression of a yeast amino acid permease-targeting mutant. Plant Cell 8:1437–1446
Rentsch D, Schmidt S, Tegeder M (2007) Transporters for uptake and allocation of organic nitrogen compounds in plants. Febs Lett 581:2281–2289
Saier SM Jr (2009) The transporter classification database: recent advances. Nucleic Acids Res 37:D274–D278
Sanders A, Collier R, Trethewy A, Gould G, Sieker R, Tegeder M (2009) AAP1 regulates import of amino acids into developing Arabidopsis embryos. Plant J 59:540–552
Schmidt R, Stransky H, Koch W (2007) The amino acid permease AAP8 is important for early seed development in Arabidopsis thaliana. Planta 226:805–813
Schmittgen TD, Livak KJ (2008) Analyzing real-time PCR data by the comparative CT method. Nat Protoc 3:1101–1108
Shelp BJ (2006) Extracellular gamma-aminobutyrate mediates communication between plants and other organisms. Plant Physiol 142:1350–1352
Su YH, Frommer WB, Ludewig U (2004) Molecular and functional characterization of a family of amino acid transporters from Arabidopsis. Plant Physiol 136:3104–3113
Suyama M, Torrents D, Bork P (2006) PAL2NAL: robust conversion of protein sequence alignments into the corresponding codon alignments. Nucleic Acids Res 34:W609–W612
Svennerstam H, Ganeteg U, Näsholm T (2008) Root uptake of cationic amino acids by Arabidopsis depends on functional expression of amino acid permease 5. New Phytol 180:620–630
Svennerstam H, Jämtgård S, Ahmad I, Huss‐Danell K, Näsholm T, Ganeteg U (2011) Transporters in Arabidopsis roots mediating uptake of amino acids at naturally occurring concentrations. New Phytol 191:459–467
Tan Q, Grennan AK, Pélissier HC, Rentsch D, Tegeder M (2008) Characterization and expression of French bean amino acid transporter PvAAP1. Plant Sci 174:348–356
Tegeder M (2012) Transporters for amino acids in plant cells: some functions and many unknowns. Curr Opin Plant Biol 15:315–321
Tegeder M, Wang XD, Frommer WB, Offler CE, Patrick JW (1999) Sucrose transport into developing seeds of Pisum sativum L. Plant J 18:151–161(11)
Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG (1997) The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res 25:4876–4882
Tuskan GA, Difazio S, Jansson S, Bohlmann J, Grigoriev I, Hellsten U, Putnam N, Ralph S, Rombauts S, Salamov A (2006) The genome of black cottonwood, Populus trichocarpa (Torr. & Gray). Science 313:1596–1604
Ueda A, Shi W, Sanmiya K, Shono M (2001) Functional analysis of salt-inducible proline transporter of barley roots. Plant Cell Physiol 42:1282–1289(8)
Vandepoele K, Simillion C, Van de Peer Y (2003) Evidence that rice and other cereals are ancient aneuploids. Plant Cell 15:2192–2202
Wang L, Kai G, Yu L, Tu Y, Hu H, Wang B, Cui X, Peng L (2010) Expression profiling and integrative analysis of the CESA/CSL superfamily in rice. BMC Plant Biol 10:443–462
Wei F, Jr EHC, Nelson W et al (2007) Physical and genetic structure of the maize genome reflects its complex evolutionary history. PLoS Genet 3:e123
Wipf D, Ludewig U, Tegeder M, Rentsch D, Koch W, Frommer WB (2002) Conservation of amino acid transporters in fungi, plants and animals. Trends Biochem Sci 27:139–147
Yang Z (1997) PAML: a program package for phylogenetic analysis by maximum likelihood. Comput Appl Biosci 13:555–556
Zhao X, Huang J, Yu H, Lei W, Xie W (2010) Genomic survey, characterization and expression profile analysis of the peptide transporter family in rice (Oryza sativa L.). BMC Plant Biol 10:92
Zhao H, Ma H, Li Y, Xin W, Jie Z (2012) Genome-wide survey and expression analysis of amino acid transporter gene family in rice (Oryza sativa L.). PloS One 7:e49210
Acknowledgments
This work was supported by fundings from National Natural Science Foundation (31370561), Specialized Research Fund for the Doctoral Program of Higher Education (20133418110005) Anhui Provincial Natural Science Foundation (1308085MC36), and Anhui Agricultural University Disciplinary Construction Foundation (XKTS2013001).
Conflict of interest
The authors declare that they have no competing interests.
Data archiving statement
All identified amino acid transporter gene sequences were deposited into the Phytozome database (http://www.phytozome.net/). The accession numbers are listed in Table 1.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by A. Brunner
This article is part of the Topical Collection on Gene Expression
Electronic supplementary material
Below is the link to the electronic supplementary material.
Table S1
Major MEME motif sequences in Populus AAT proteins. (DOC 31 kb)
Table S2
Number of AAT genes in the Populus, Arabidopsis, rice and maize genomes. (DOC 373 kb)
Table S3
The general information and sequence characterization of AAT genes in Arabidopsis, rice and maize. (DOCX 13 kb)
Table S4
Paralogous extensins identified in four modern plants. (DOCX 15 kb)
Table S5
Orthologous extensins identified in four modern plants. (DOCX 13 kb)
Table S6
Primers used in qRT-PCR analysis of PtAAP genes. (DOC 55 kb)
Figure S1
Prediction of the transmembrane regions of 100 PtAATs. The transmembrane regions of 100 PtAATs were predicted by using the TMHMM Server v2.0 (http://www.cbs.dtu.dk/services/TMHMM/). (TIFF 11622 kb)
Figure S2
Sliding window plots of representative duplicated AAT genes in Populus. As shown in the key, dark gray blocks and light gray blocks indicate the positions of the Aa-trans and AA-permease domain, respectively. The window size is 150 bp, and the step size is 9 bp. (TIFF 1465 kb)
Rights and permissions
About this article

Cite this article
Wu, M., Wu, S., Chen, Z. et al. Genome-wide survey and expression analysis of the amino acid transporter gene family in poplar. Tree Genetics & Genomes 11, 83 (2015). https://doi.org/10.1007/s11295-015-0908-4
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11295-015-0908-4