
Abstract
Neutral proteases have broad application as additives in modern laundry detergents and therefore, thermostability is an integral parameter for effective production of protein crystals. To improve thermostability, the contribution of individual residues of Bacillus cereus neutral protease was examined by site-directed mutagenesis. The Lys11Arg and Lys211Arg mutants clearly possessed improved thermostabilities (Tm were 63 and 61 °C respectively) compared to the wild-type (Tm was 60 °C). MD simulations further revealed that the mutants had low RMSD and RMSF values compared to wild-type BCN indicating increased stability of the protein structure. Lys11Arg mutant particularly possessed the lowest RMSD values due to increased residue interactions, which resulted in enhanced thermostability. The mutants also displayed strong stability to most inhibitors, organic solvents and surfactants after incubation for 1 h. This study demonstrated Lys–Arg mutation enhanced thermostability of BCN and thus provides insight for engineering stabilizing mutations with improved thermostability for related proteins.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The complete genome sequences of Bacillus species possess many protease coding genes and the bacteria has widely been used as a source of many protein coding genes, as well as for expression of a variety of extracellular and intracellular proteases (Valbuzzi et al. 1999). Proteases are of prime commercial importance, accounting for approximately 60% market share of all industrial enzymes (Bouacem et al. 2015). Neutral proteases (NPs), also referred to as thermolysin-like proteinase (TLP), constitute the vast majority of industrial enzymes worldwide (Kasana et al. 2011). Neutral proteases have wide industrial applications as biocatalysts in various hydrolytic industrial processes, for example; they are broadly applied in the food (Ao et al. 2018; Yuzuki et al. 2015), pharmaceutical (Umeadi et al. 2008), leather (Jatavathu et al. 2011) and feed (Zhang et al. 2014) industries. This is mainly because of their comparatively high activity and stability under harsh operational conditions, such as high pH, temperature, and surfactants (Guleria et al. 2016; Rao et al. 1998). However, at high temperatures, neutral proteases are deactivated irreversibly because of autolysis, thus restricts their industrial application in processes that require high temperatures. Improving the biochemical properties (thermostability) of the proteases would therefore be advantageous during production of enzyme granules while surfactant stability of proteases is key for application in detergents. Two widely used approaches for engineering enzymes to improve thermal properties and activity or reduce enzyme inhibition include; rational design-, which exploits knowledge of the protein structure and mechanism of enzyme reaction to design effective site-directed mutations, and Random mutagenesis-, which mimics the natural evolution process in enzymes to generate variants (Baweja et al. 2016a).
Previous studies have indicated that the rate of autolysis in these proteases is influenced by local unfolding, making it vulnerable to auto proteolytic cleavage (Demidyuk et al. 2003). Furthermore, the role of the calcium ions found in the structure of NPs on protein stability have been well documented (Dahlquist et al. 1976) and reports have indicated that there is a release of one calcium ion (Ca3+ or Ca4+) during the initial steps of thermal inactivation (Roche and Voordouw 1978). Therefore, regions that partially unfold, for example; residues located at the protein surface are associated with protein stability since initiation of protein unfolding was speculated to occur at surface-located structures (Jackson and Fersht 1991). To satisfy the industrial demands, several mutations with significant stability effects on NPs have previously been engineered and notably majority of these mutations were located at the N-terminal (Veltman et al. 1996) confirming that thermal inactivation in neutral proteases begins at the N-terminal domain. Consequently, random mutagenesis prior to high through-put screening in directed evolution has been widely used (Gong 2014). By sequence and structural alignment of homologous neutral proteases, Argos et al. (1979) determined structural causes of thermostability and successfully designed stability by replacing Gly, Ser, Ser, Lys, and Asp in mesophiles with Ala, Ala, Thr, Arg, and Glu, respectively, as in thermophiles. Recently, a robust approach broadly adopted to improve enzyme properties is using computer-aided bioinformatics techniques to study the primary sequence, model the tertiary structure of proteins and predict protein interactions de novo (Wang et al. 2017). This rational structure engineering approach aids in determination of fundamental amino acid residues and their interactions then site-directed mutagenesis carried out, hence reducing library size. Recently, advancement in protein engineering techniques and computational tools has enabled design of efficient and stable biocatalysts through mutation of the amino acid residues (Vishal et al. 2017). De novo engineering of stabilizing mutations has been applied for design of suitable industrially valuable xylanase, proteases and inulinase enzymes (Shukla 2018). This study screened for potential protease producing strains, then explored heterologous expression and characterization of the 16SR sequenced Bacillus cereus neutral protease (BCN) in E. coli BL21, and finally analyzed the effects of Lys–Arg and Ser-Asp substitutions on thermostability of the protein.
Materials and methods
Bacterial strains, plasmid vectors and materials
The bacterial strain (E. coli BL21) used in this study was obtained from our laboratory stock collection. The isolate (B. cereus) was used for gene amplification while E. coli BL21 as the primary recombinant protein expression host. The restriction enzymes, PrimeSTAR HS DNA Polymerase and T4 DNA ligase were purchased from TaKaRa Bio Co. (Dalian, China). The mini-chromosome rapid isolation kit, mini plasmid rapid isolation kit, and Mini DNA rapid purification kit were obtained from Sangon Biotech Co., Ltd. (Shanghai, China). The AOE UV-1200 spectrophotometer was used for assaying activity. All other chemicals used are of high analytical grade.
Medium and culture conditions
For isolation and screening of strains with protease activity, the modified medium used consisted of; 0.55% peptone, 0.5% glucose, 0.2% magnesium sulphate, 2% agar, 0.2%, potassium dihydrogen phosphate, 0.2% sodium carbonate, 1% casein and 1% skim milk at pH 7.4 (Baweja et al. 2016b). For construction of the recombinant strains, E. coli BL21 harboring the pET28a+ plasmid vector was grown overnight at 37 °C (180 rpm) in Luria–Bertani (LB), pH to 7.4, supplemented with 50 µg/mL kanamycin whenever necessary (Esakkiraj et al. 2016).
Screening of bacterial strains for protease activity
Isolates from soil samples were used in quantitative screening for protease activity as previously described (Kamran et al. 2015). Colonies on plates with clear zones due to hydrolysis of casein and skim milk were selected as potential protease activity strains and the individual colonies used for further study (Omrane Benmrad et al. 2016; Qoura et al. 2015).
The 16S rDNA sequence, phylogenetic analysis
The 16S rRNA of the isolate was amplified by PCR using the universal primers 1.27F and 1.149R (Table S1). The resultant sequences were compiled and BLASTx program in NCBI GenBank database applied to obtain homologous sequences (Johnson et al. 2008). The PSI-BLAST tool was used to generate homologous sequences at 1e−03 E-value. Total 250 homologous sequences selected were aligned using MUSCLE, then the phylogenetic tree generated in ClustalW2 (Edgar and Batzoglou 2006). The phylogenetic tree was modified using FigTree v1.4.4 and finally edited by online iTOL server (https://itol.embl.de/) (Letunic and Bork 2011).
Cloning of the BCN gene
The B. cereus ATCC 4342 (GenBank accession No. CP009628.1) was used for designing primers 28a-bcn-F and 28a-bcn-R, flanked with BamHI and XhoI restriction sites respectively. Then the full-length cDNA of the isolate was amplified by PCR. The resulting amplicon (purified) and the expression vector pET28a+ were digested with BamHI and XhoI restriction enzymes, prior to ligation and transformation into E. coli BL21 competent cells as described previously (Jia et al. 2013). Successful ligation was confirmed by double digestion and sequencing. The resulting recombinant BL21/pET28-BCN was cultured in LB medium for protein expression.
Heterologous expression and purification of BCN
The recombinant BL21pET28-BCN was grown overnight on 10 mL LB medium supplemented with 5 µg/mL kanamycin at 37 °C, 180 rpm, then the overnight cells were expanded in 50 mL media with 25 µg/mL kanamycin, cultured in a shaker for 2 h under the same conditions above, prior to induction for protein expression with IPTG. Cells were harvested by centrifugation at 8000 rpm, 4 °C for 10 min, then washed with 0.01 M phosphate buffer and sonicated for 15 min, then further centrifugation for 20 min. Protein purification was accomplished by Ni2+-affinity chromatography on an AKTA purifier system (GE Healthcare, Sweden) fitted with a HisTrap™ HP column purchased from GE Life Sciences, USA. The crude enzyme (5 mL) was loaded onto the column at a loading rate of 0.5 mL/min with Binding Buffer consisting of 0.02 M Tris–HCl buffer and 0.5 M NaCl, at pH 7.4, then the protease enzyme was eluted under a linear gradient of imidazole concentrations from 0 to 0.5 M at 1 mL/min. Finally, excess imidazole was removed from the purified protein by dialysis using 0.05 M Tris–HCl buffer at pH 7.0 (Li et al. 2018). The purified protease enzyme was used for further activity assay, sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) analysis (12% acrylamide), and stored in 10% glycerol at − 40 °C for further analysis (Esakkiraj et al. 2016).
Enzyme activity assay
Protease activity was determined through a modified method described by (Baweja et al. 2016b). The purified enzyme solution (0.5 mL) was mixed with 1% casein solution prepared in 2.5 mls Tris–HCl buffer (pH 7.4). The reaction mixture was incubated at 37 °C for 15 min. For the blank solution, TCA was added to the substrate before the enzyme. After incubation, 2.5 mL of 110 mM TCA was added and kept at room temperature for 30 min. The mixture was then centrifuged at 10,000 rpm for 10 min, then 2.5 mL of 0.5 M sodium carbonate added. Afterward, 0.5 mL of 0.5 M Folin ciocalteu reagent was added to the mixture and kept at room temperature for 30 min, before absorbance measurements were done at 660 nm. One unit of protease activity was expressed as the amount of enzyme required to form 1 µL/mL tyrosine from 1% casein at optimum conditions. Bradford assay was applied for protein concentration determination with bovine serum albumin (BSA) used as the standard (Ernst and Zor 2010).
Characterization of purified recombinant BCN enzyme
The optimum pH was determined using several buffers (pH 4.0–6.0, 0.05 M acetate buffer; pH 6.0–7.0, 0.05 M phosphate buffer; pH 7.0–9.0, 0.05 M Tris–HCl buffer; and pH 9.0–10.0, 0.05 M glycine–NaOH buffer) and the optimum temperature examined in 0.05 M Tris–HCl buffer (pH 9.0) at temperatures between 30 and 90 °C using the standard reaction mixture and conditions. The thermostability of the protease was measured by pre-incubating the enzyme without substrate at various temperatures (50–70 °C) for 1 h then monitoring the residual enzyme activities under optimum conditions. Effects of various compounds on enzyme activity were further examined in the standard reaction mixture supplemented with various cations at concentrations up to 1 mM. The enzyme activity of the recombinant protease without the addition of metal ions was used as the control. Kinetic parameters were performed in 0.05 M Tris–HCl buffer (pH 9) at 60 °C by changing the concentration of the casein from (1–5 mM). Lineweaver–Burk plots used to calculate kinetic parameters Km and Vmax according to the enzyme reactions were generated by Hyper32 software.
Sequence and structure analysis
BLASTx program (https://www.ncbi.nlm.nih.gov/BLAST/) was used to identify the cDNA sequence of BCN and Clustal Omega (https://www.ebi.ac.uk/Tools/msa/clustalo) used to align the nucleotide and amino acid sequences. Proparam tool (https://web.expasy.org/protparam) was used to predict the molecular mass and theoretical isoelectric point (pI) of the enzyme and SWISS-MODEL webserver (https://swissmodel.expasy.org) applied for building the homology model of protease. Finally, the catalytic amino acid residues, amino acids frequency and β-factor sites were predicted by Hotspot Wizard (https://loschmidt.chemi.muni.cz/hotspotwizard/) (Sumbalova et al. 2018).
Site-directed mutagenesis and BCN mutant characterization
The recombinant protein was subjected to site-directed mutagenesis by overlap-extension PCR with long chain primers (Table S1) (Ke and Madison 1997; Ling and Robinson 1997; Sambrook and Russell 2006). The wild-type recombinant plasmid pET28a-BCN was used as a template for PCR reaction with high-fidelity enzyme. A mutation point was introduced via the upstream primers, and the downstream primer used as a universal primer (pET28a-2254-R). The amplified product from the first step of PCR served as a large fragment primer containing the mutated sequence. The denatured extension in the second round of PCR yielded a full-length plasmid containing the entire mutated gene. The reaction conditions were as follows: pre-denaturation (95 °C for 3 min), denaturation (95 °C for 30 s), annealing (55 °C for 1 min), extension (72 °C for 3 min, 5 cycles), denaturation (95 °C for 30 s), extension (68 °C for 6 min, 20 cycles), and final extension at 68 °C for 12 min. The complete PCR product was digested with DpnI at 37 °C for 2 h and then transformed into E. coli BL21. The positive transformants were verified by double digestion prior to sequencing by Talen Bio, China.
Molecular dynamics (MD) simulation
The protein structural model obtained by homology modelling (Fig. 2b) using SWISS-MODEL was analyzed and validated using the WHATIF web interface (Hekkelman et al. 2010) (https://swift.cmbi.umcn.nl/servers/html/index.html). Molecular dynamic (MD) simulations and energy minimization were performed using Gromacs (Pronk et al. 2013). The protein model was immersed in a cubic box, with the distance between protein atoms and the edge of the box set at more than 1.0 nm. The energy of the system was minimized using the steepest descent method, prior to MD simulations at 300 K for 30 ns. The RMSD and RMSF values of residues were plotted using GraphPad prism8. Finally, the software PYMOL was used to analyze variations in the molecular tertiary structure (Alexander et al. 2011).
Results
Screening of protease-producing strains
Isolates with clear zones formed after culturing on casein-skimmed milk agar media at pH 7.4 for 24 h were identified as potential protease producers. The strain that produced clear zones of comparably larger diameter (Fig. 1a) than the colony was selected as the high protease activity strain and thus used for further experiments. The 16S rRNA sequence of the amplified gene was MRSRPERVIGHTGTETRPRLLREAAVGNLPQWTKV and standard nucleotide BLAST analysis in NCBI revealed 97.39% sequence identity to Bacillus cereus H17 16SR gene (KC441786.1).

Strain isolation, identification and Phylogenetic tree analysis. a Plate containing Isolation media (casein-skimmed milk agar); the clear zones represent enzyme hydrolysis of casein (indicating protease activity). b Phylogenetic tree of Bacillus Cereus thermolysin-like protease (BCN) generated in BLASTx program and Clustal Omega
Sequence alignment and structure analysis of BCN
The complete genome of Bacillus cereus ATCC 4342 (GenBank accession No. CP009628.1) was used for designing PCR primers. The resultant 1701 bp PCR amplicon (Fig. 2a) encoding neutral protease was used for cloning, protein expression and to model the protein structure. The modelled protein structure had 96.53% homology to a neutral protease (1npc) with a predicted molecular mass of 61 kDa and a theoretical isoelectric point (pI) of 5.82. Quality analysis of the modelled structure showed Ramachandran favored value of 96.19%, hence of good quality and assessment of the model on the Hotspot Wizard 3.0 web server revealed Ala78, Thr81, Tyr82, Ser93, Thr146, Val149, Arg91, Tyr85, Ile94, Leu101 and Leu145 as residues surrounding the catalytic pocket. The phylogenetic tree and the sequence alignment with known NPs (neutral proteases) are shown in Figs. 1b and 2c respectively. Results indicated that BCN had 48.9% sequence identity to Geobacillus stearothermophilus neutral protease, 73.19% Bacillus thermoproteolyticus neutral protease and 84.59% to Bacillus cereus DSM139 neutral protease (Fig. 2c).

Agarose gel, SDS-page gel, amino acid sequence alignment and protein structure of BCN. a Agarose gel electrophoresis analysis of PCR amplicon showing the target gene Lane M; DNA Marker DL2000; Lane 1–3; show the 1701 bp amplicon and the 12% SDS-PAGE of the protease. Lane M is the Protein Marker-Broad (Takara, China); Lane 1–3 purified recombinant protease. b Protein structure model generated from SWISS-MODEL. c Sequence alignment of BCN with three Neutral proteases from Bacillus thermoproteolyticus, Bacillus Cereus DSM139, and Geobacillus stearothermophilus. The dots (red) indicate sites of mutation according to sequence alignment with thermolysin
Expression and purification of the BCN
The Bacillus cereus ATCC 4342 DNA fragment coding for the thermolysin-like protease was successfully expressed in E. coli BL21 as an intracellular protein which was then further purified as a His tagged protein on an AKTA Prime (Japan). Analysis of the molecular weight of the protein by 12% SDS-PAGE revealed an approximately 36.6 kDa protein band (Figs. 2a and S1A), deviating from the predicted theoretical mass of 61 kDa. This result suggests that post- translational changes likely took place during activation or maturation of the protein. Discrepancies between experimental and theoretical results have previously been reported for several proteins, arousing interest and number of studies on protein folding, N-glycosylation and maturation (Guan 2015; Stanley 2011).
Characterization of purified BCN
The optimum temperature of the mature protein peptide (purified) was 60 °C (Fig. 3a). Incubating the enzyme at 60 °C for 1 h showed that it possessed great stability over a broad pH range from 5–11, with more than 75% activity retained (Fig. S1B). Several alkaline proteases isolated from Bacillus species reported in literature (Esakkiraj et al. 2016; Qoura et al. 2015) possess high optimal temperatures for their respective activities making these enzymes characteristically valuable for industrial application, example; as additives in detergents and in the leather industry for tanning. Other valuable alkaline proteases having high optimum temperature of 60 °C with broad application in the detergent industry include; subtilisin Carlberg, successfully expressed in Bacillus licheniformis and Bacterial Protease Nagase (BPN), in Bacillus amyloliquefaciens (Qoura et al. 2015).

Characterization of the expressed recombinant BCN and its mutants. a Shows the effects of temperature on enzyme activity of BCN and its mutants; wild-type BCN (black), Lys11Arg (red), and Lys211Arg (blue). The enzyme was incubated in the presence of CaCl2 at temperatures 30–90 °C. Residual protease activity was determined after 60 min. b The effect of pH on the protease. c Indicates effects of metal ions on BCN and its mutants. d Represents the effects of three commercial detergents on the enzyme activity of BCN. Each value represents the mean of three independent experiments
The protein exhibited highest activity at pH 9 thus is alkaline (Fig. 3b). Previous studies have also reported that neutral proteases (thermolysin-like proteases) from fungal sources were either acidic or alkaline rather than neutral proteases; widely reported examples include the acid protease from Aspergillus oryzae HG76 (Li et al. 2014).
The effect of different concentrations (1–10 mM) of metal ions, organic solvents, surfactants and inhibitors on BCN activity was assessed and the results (Fig. 3c) showed that the presence of calcium (Ca2+), magnesium (Mg2+), copper (Cu2+), manganese (Mn2+), barium (Ba2+), zinc (Zn+) ions in the essay enhanced enzyme activity when compared to the control. The optimum pH and temperature of the wild type enzyme was 9.0 and 60 °C respectively. Similar to our results, Esakkiraj et al., cloned and expressed a 27 kDa thermostable, detergent stable, thiol dependent alkaline protease from Bacillus cereus PMW8 with an optimum temperature of 60 °C and pH of 9.0.
The enzyme also displayed exceptional stability and compatibility toward different metal ions, detergents, oxidizing, and reducing agents. These results indicated that these bivalent ions evidently elevated enzyme activity (with residual activities ˃ 100%). On the contrary, enzyme activity was greatly affected by the presence of nickel (Ni2+) retaining ˂ 65% activity. The inhibitory effect of heavy metal ions on enzyme activity is widely recorded in literature. Bouacem et al. 2015 reported that Ni2+ and mercury (Hg2+) inhibited the enzyme activity of a detergent-stable serine alkaline protease from Caldicoprobacter guelmensis [3]. Jaouadi et al. (2008) also reported that the presence of Hg2+ entirely inhibited the activity of the purified SAPB protease from Bacillus pumilus CBS.
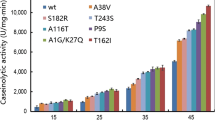
Additionally, the expressed recombinant BCN protein displayed strong stability to most inhibitors, organic solvents and surfactants tested and enzyme activity was enhanced by β-mercaptoethanol, Tween-80, Toulene and SDS as shown in Table 1. It further retained more than 80% activity in the presence of DMF, EDTA, iodoacetamide, DTT, PMSF, hexane and DMSO. However, only 61% enzyme activity was retained when 10% benzene was added into the reaction mixture.
Lys–Arg mutation improved the thermostability of BCN
Thermostability of thermolysin compared to B. cereus neutral protease is not largely determined by differences in the metal binding sites, but rather attributed to increased hydrophobicity of β-pleated sheet and β-turn, higher bulkiness of α-helical regions and increased polarity of the salt bridges as a result of numerous amino-acid exchanges distributed over the whole molecule (Chou and Fasman 1974; Sidler et al. 1986). Therefore, identification of individual regions or residues that confer protein stability was paramount to successful design of stable industrial biocatalysts. Considering the C-terminal of proteins has been reported to often influence protein thermostability (Hardy et al. 1994; Veno et al. 2017; Zheng et al. 2016) (Sidler et al. 1986), Ser312 residue of BCN (corresponding to Asp242 of thermolysin) was through site-directed mutagenesis changed to Asp. Results showed there was no significant difference in thermostability of the Ser312Asp mutant after incubation at 50 to 70 °C for 60 min. Although the Ser312Asp mutation resulted in the formation of a stronger ionic bond (2.7 Å) between Ser312 located on the α-helix (302–314) and Arg286 on the adjacent α-helix (282–297) (Fig. 4a, b), the additional salt bridge formed (16 in total) compared to the wild-type (15 salt bridges) had a marginal stabilizing effect on the protein structure. However, Argos et al. (1979) determined by statistical approach the preferred cold–hot substitutions on the entire neutral proteinase system and concluded that Gly-Ala, Ser-Thr, Ser-Asp, and Lys–Arg mutations highly improved the thermostability of the protein. Therefore, mutating Lys211 residue (located in the C-terminal) to Arg resulted in considerable increase in thermostability of BCN (61 °C) compared to 60 °C of the wild-type, albeit with a slightly decreased specific activity of 1508.70 U/mg compared to 1517.87 U/mg of the wild-type (Table 2).

Structure analysis of BCN and its mutants Lys11Arg, Lys 211 and Ser312Asp. a Polar contacts between Ser312 and Asp286 prior to mutation. b Interactions between mutated Asp312 and Arg286 after mutation showing an additional polar contact between residues. c Local interactions of residues within 4 Å of Lys11. d Additional polar contacts within the Arg11 residue after mutation showed interactions with Asn64, which further interacted with Asp68
Moreover, previous reports cited the crucial role of hydrophobic interactions between residue 63 and the side chains of residues that form 8–12, 15–20, 60–63 β-sheets (Va19, Arg11, Gln17 and Gln61) in the stability of BCN (Van den Burg et al. 1994). Based on sequence similarities between B. cereus neutral protease (BCN), Geobacillus stearothermophilus neutral protease, and Bacillus thermoproteolyticus neutral protease (thermolysin), site directed mutation on Lys11 to Arg was performed. Results indicated that the Lys11Arg mutant had a thermostability of 63 °C, after incubation in a water bath at 50 to 70 °C for 60 min compared to 60 °C of the wild-type BCN with a specific activity of 1543.5 U/mg, 1.02 times that of the wild-type BCN. Notably, the mutations had no significant effect on the protease activity when incubated in a reaction mixture supplemented with commercial detergents (Ariel, Omo and Tide, although the mutants (Lys11Arg and Lys211Arg) retained slightly higher specific activities compared with the wild-type BCN, as shown in Fig. 3d. Therefore, there is still need to design mutations that will significantly confer stability to BCN for improved potential industrial application in detergents.
The BCN mutants also showed significant stability after incubation at 40–90 °C for 1 h, with the Lys11 mutant retaining approximately 70% residual activity, the Lys211Arg mutant 62%, compared to the residual activity of the wild-type BCN of 58% at 70 °C (Fig. S1B). Finally, MD simulation was performed on Gromacs (Pronk et al. 2013) to explore the mechanism of improved thermostability of mutants. The Root mean square deviation (RMSD) for backbone atoms and Root Mean Square Fluctuation (RMSF) values of mutants were calculated and structure analysis of the selected mutants done to assess structural modifications to the wild-type BCN (Zhao et al. 2015). As shown in Fig. 5a, b, results showed that the RMSD and RMSF values of the mutants (Lys11Arg and Lys211Arg) were lower compared to the wild-type BCN, indicating improved rigidity and stability of the protein structure.

MD simulation on the wild-type BCN and its variants. a RMSF value of the protein for the wild-type BCN (black), Lys11Arg (red), and Lys211Arg (blue) during the MD simulations. b Backbone RMSD for the wild-type BCN (black) and its mutants, Lys11Arg (red), and Lys211Arg (blue). MD simulations were carried out in AMBER99SB-ILDN protein force fields for 30 ns
Discussion
This study reported expression of a B. cereus neutral protease (BCN). The recombinant enzyme (BCN) and its mutants had enhanced relative activity in the presence of various metal ions and this was associated to probable modification of the enzyme structure making it resistant to thermal denaturation, hence preserving its active form at higher temperatures. Certainly, the modelled protein structure showed the predicted active site of the protein flanked by three Ca2+ ions and one Zn2+ ion. Previous studies reported that the removal of Ca2+ from the metal-binding site resulted in a significant decline in thermostability, confirming that the Ca2+ probably plays a crucial role in the structural integrity and stability of the enzyme. The inhibitory effect of heavy metal ions on enzyme activity was attributed to denaturation of proteins due to the binding of these toxic metal ions to various organic ligands and amino acid residues within the active site. For example, it is well documented that Mercury ions tend to react with amino acid residues (histidine, tryptophan) and interact with protein thiol groups forming mercaptides, causing permanent denaturation of the enzyme (Nascimento and Martins 2004).
Site directed mutagenesis at position 11 (Lys11Arg) and 211 (Lys211Arg) enhanced thermostability of the protein to differing degrees (63 and 61 °C respectively), while Ser312Asp mutation did not yield any significant improvement in thermostability. Conforming to previous reports (Fersht and Serrano 1993), the observed marginal effect of the formed salt bridge in the Ser312Asp mutant suggests that thermal inactivation of BCN is not influenced by unfolding of the C-terminal domain (Veltman et al. 1996). On the contrary, we speculated that the small beneficial stabilizing effect of Lys211Arg mutation at the 209–212 α-helix on the C-terminal could be due to enhanced packing of the protein molecule as a result of increased bulkiness of α-helical and random coil structures. Analysis of the Lys211Arg mutant structure in VMD showed two formed salt bridges at (Asp208–Arg211 and Glu188–Arg211), compared to only one salt bridge (Glu188–Lys211) observed in the wild-type (data not shown). Therefore, in line with previous reports, our results suggest that determining factors for stability do not adhere to statistical rules but instead each single mutation ought to be examined in its specific context (Veltman et al. 1996). Moreover, the Lys11–Arg mutation located at the end of the 4–12 strand that forms part of the N-terminal β-hairpin with the 16–25 strand had improved thermostability (63 °C), because of increased interactions with residue Phe63 (Fig. 4d), whose role in protein stability of neutral proteases has been previously demonstrated (Hang et al. 2017). The conformation of the Arg11 side chain was reported to be mainly restricted by interactions with other residues (Veltman et al. 1997), therefore, mutating Lys11 to Arg enhanced hydrophobic interactions between the Phe63 phenyl ring and the aliphatic part of the Arg11 side chain resulting in increased rigidity and altered flexibility of the main-chain, hence improved thermostability of BCN. Indeed, a tight network of hydrogen bonds and residue interactions play a key role in maintaining the putative structural environment of the protein (Jonsdottir et al. 2014). Furthermore, structural analysis for residue interactions within 4 Å of mutation Lys11Arg clearly indicated increased hydrophobic interactions between nearby residues, for example; Arg11 formed two polar contacts with Phe63 and Asn64, which further interacted with Asp68 (Fig. 4b, c).
In conclusion, the lower RMSF and RMSD of the Lys11Arg and Lys211Arg mutants was attributed to increased bulkiness and polar interactions between residues in this region hence the improved thermostability. Finally, in agreement with previous reports, this study revealed that the N-terminal residues evidently contributed to global protein stability confirming their role in protein unfolding of NPs (Veltman et al. 1996; Wijma et al. 2013).
References
Alexander N, Woetzel N, Meiler J (2011) bcl::Cluster: A method for clustering biological molecules coupled with visualization in the Pymol Molecular Graphics System. In: 2011 IEEE 1st international conference on computational advances in bio and medical sciences (ICCABS), pp 13–18
Ao XL et al (2018) Purification and characterization of neutral protease from Aspergillus oryzae Y1 isolated from naturally fermented broad beans. AMB Express 8:96. https://doi.org/10.1186/s13568-018-0611-6
Argos P, Rossman MG, Grau UM, Zuber H, Frank G, Tratschin JD (1979) Thermal stability and protein structure. Biochemistry 18:5698–5703
Baweja M, Nain L, Kawarabayasi Y, Shukla P (2016a) Current technological improvements in enzymes toward their biotechnological applications. Front Microbiol. https://doi.org/10.3389/Fmicb.2016.00965
Baweja M, Tiwari R, Singh PK, Nain L, Shukla P (2016b) An alkaline protease from Bacillus pumilus MP 27: functional analysis of its binding model toward its applications as detergent additive. Front Microbiol 7:1195. https://doi.org/10.3389/fmicb.2016.01195
Bouacem K et al (2015) Biochemical characterization of a detergent-stable serine alkaline protease from Caldicoprobacter guelmensis. Int J Biol Macromol 81:299–307. https://doi.org/10.1016/j.ijbiomac.2015.08.011
Chou PY, Fasman GD (1974) Prediction of protein conformation. Biochemistry 13:222–245
Dahlquist FW, Long JW, Bigbee WL (1976) Role of calcium in the thermal stability of thermolysin. Biochemistry 15:1103–1111
Demidyuk IV, Zabolotskaya MV, Safina DR, Kostrov SV (2003) Molecular mechanisms of stabilization of proteolytic enzymes: a model of thermolysin-like microbial metalloproteases. Russ J Bioorg Chem 29:418–425. https://doi.org/10.1023/A:1026093207604
Edgar RC, Batzoglou S (2006) Multiple sequence alignment. Curr Opin Struct Biol 16:368–373. https://doi.org/10.1016/j.sbi.2006.04.004
Ernst O, Zor T (2010) Linearization of the Bradford protein assay. J Vis Exp: JoVE. https://doi.org/10.3791/1918
Esakkiraj P, Meleppat B, Lakra AK, Ayyanna R, Arul V (2016) Cloning, expression, characterization and application of protease produced by Bacillus cereus PMW8. RSC Adv 6:38611–38616. https://doi.org/10.1039/c5ra27671c
Fersht AR, Serrano L (1993) Principles of protein stability derived from protein engineering experiments. Curr Opin Struct Biol 3:75–83. https://doi.org/10.1016/0959-440x(93)90205-Y
Gong Y, Xu GC, Zheng GW, Li CX, Xu JH (2014) A thermostable variant of Bacillus subtilis esterase: characterization and application for resolving dl-menthyl acetate. J Mol Catal B. https://doi.org/10.1016/j.molcatb.2014.07.014
Guan YH, Zhu QF, Huang DL, Zhao SY, Li JL, Peng JR (2015) An equation to estimate the difference between theoretically predicted and SDS PAGE-displayed molecular weights for an acidic peptide. Sci Rep. https://doi.org/10.1038/srep13370
Guleria S, Walia A, Chauhan A, Shirkot CK (2016) Purification and characterization of detergent stable alkaline protease from Bacillus amyloliquefaciens SP1 isolated from apple rhizosphere. J Basic Microbiol 56:138–152. https://doi.org/10.1002/jobm.201500341
Hang F, Wang Q, Hong Q, Gao C, Zhang H, Chen W (2017) Structural insight into a novel neutral metalloproteinase from Paenibacillus spp. BD3526: implications for mechanisms of rapid inactivation and calcium-dependent stability. Int J Biol Macromol 95:1082–1090. https://doi.org/10.1016/j.ijbiomac.2016.10.098
Hardy F, Vriend G, van der Vinne B, Frigerio F, Grandi G, Venema G, Eijsink VG (1994) The effect of engineering surface loops on the thermal stability of Bacillus subtilis neutral protease. Protein Eng 7:425–430
Hekkelman ML, Beek TAHT, Pettifer SR, Thorne D, Attwood TK, Vriend G (2010) WIWS: a protein structure bioinformatics Web service collection. Nucleic Acids Res 38:W719–W723. https://doi.org/10.1093/nar/gkq453
Jackson SE, Fersht AR (1991) Folding of chymotrypsin inhibitor 2. 1. Evidence for a two-state transition. Biochemistry 30:10428–10435
Jaouadi B, Ellouz-Chaabouni S, Rhimi M, Bejar S (2008) Biochemical and molecular characterization of a detergent-stable serine alkaline protease from Bacillus pumilus CBS with high catalytic efficiency. Biochimie 90:1291–1305. https://doi.org/10.1016/j.biochi.2008.03.004
Jatavathu M, Jatavathu S, Raghavendra RMV, Sambasiva RKRS (2011) Efficient leather dehairing by bacterial thermostable protease. Int J Bio-Sci Bio-Technol 3:11–26
Jia M, Xu M, He B, Rao Z (2013) Cloning, expression, and characterization of L-asparaginase from a newly isolated Bacillus subtilis B11–06. J Agric Food Chem 61:9428–9434. https://doi.org/10.1021/jf402636w
Johnson M, Zaretskaya I, Raytselis Y, Merezhuk Y, McGinnis S, Madden TL (2008) NCBI BLAST: a better web interface. Nucleic Acids Res 36:W5–W9. https://doi.org/10.1093/nar/gkn201
Jonsdottir LB, Ellertsson BO, Invernizzi G, Magnusdottir M, Thorbjarnardottir SH, Papaleo E, Kristjansson MM (2014) The role of salt bridges on the temperature adaptation of aqualysin I, a thermostable subtilisin-like proteinase. Biochim Biophys Acta 1844:2174–2181. https://doi.org/10.1016/j.bbapap.2014.08.011
Kamran A, Ur Rehman H, Ul Qader SA, Baloch AH, Kamal M (2015) Purification and characterization of thiol dependent, oxidation-stable serine alkaline protease from thermophilic Bacillus sp. J Genet Eng Biotechnol 13:59–64. https://doi.org/10.1016/j.jgeb.2015.01.002
Kasana RC, Salwan R, Yadav SK (2011) Microbial proteases: detection, production, and genetic improvement. Crit Rev Microbiol 37:262–276. https://doi.org/10.3109/1040841X.2011.577029
Ke SH, Madison EL (1997) Rapid and efficient site-directed mutagenesis by single-tube 'megaprimer' PCR method. Nucleic Acids Res 25:3371–3372
Letunic I, Bork P (2011) Interactive tree of life v2: online annotation and display of phylogenetic trees made easy. Nucleic Acids Res 39:W475–W478. https://doi.org/10.1093/nar/gkr201
Li CH, Xu DF, Zhao MM, Sun LJ, Wang YL (2014) Production optimization, purification, and characterization of a novel acid protease from a fusant by Aspergillus oryzae and Aspergillus niger. Eur Food Res Technol 238:905–917. https://doi.org/10.1007/s00217-014-2172-5
Li X et al (2018) Simultaneous cell disruption and semi-quantitative activity assays for high-throughput screening of thermostable L-asparaginases. Sci Rep 8:7915. https://doi.org/10.1038/s41598-018-26241-7
Ling MM, Robinson BH (1997) Approaches to DNA mutagenesis: an overview. Anal Biochem 254:157–178. https://doi.org/10.1006/abio.1997.2428
Nascimento WCAD, Martins MLL (2004) Production and properties of an extracellular protease from Thermophilic Bacillus sp. Braz J Microbiol 35:91–96
Omrane Benmrad M et al (2016) A novel organic solvent- and detergent-stable serine alkaline protease from Trametes cingulata strain CTM10101. Int J Biol Macromol 91:961–972. https://doi.org/10.1016/j.ijbiomac.2016.06.025
Pronk S et al (2013) GROMACS 4.5: a high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics 29:845–854. https://doi.org/10.1093/bioinformatics/btt055
Qoura F, Kassab E, Reisse S, Antranikian G, Brueck T (2015) Characterization of a new, recombinant thermo-active subtilisin-like serine protease derived from Shewanella arctica. J Mol Catal B 116:16–23. https://doi.org/10.1016/j.molcatb.2015.02.015
Rao MB, Tanksale AM, Ghatge MS, Deshpande VV (1998) Molecular and biotechnological aspects of microbial proteases. Microbiol Mol Biol Rev: MMBR 62:597–635
Roche RS, Voordouw G (1978) The structural and functional roles of metal ions in thermolysin. CRC Crit Rev Biochem 5:1–23
Sambrook J, Russell DW (2006) Rapid and efficient site-directed mutagenesis by the single-tube megaprimer PCR method. CSH Protoc. https://doi.org/10.1101/pdb.prot3467
Shukla P (2018) Editorial: futuristic protein engineering: developments and avenues. Curr Protein Pept Sci 19:3–4. https://doi.org/10.2174/138920371901171121142734
Sidler W, Niederer E, Suter F, Zuber H (1986) The primary structure of Bacillus cereus neutral proteinase and comparison with thermolysin and Bacillus subtilis neutral proteinase. Biol Chem Hoppe-Seyler 367:643–657
Stanley P (2011) Golgi glycosylation. Cold Spring Harbor Perspect Biol. https://doi.org/10.1101/cshperspect.a005199
Sumbalova L, Stourac J, Martinek T, Bednar D, Damborsky J (2018) HotSpot Wizard 3.0: web server for automated design of mutations and smart libraries based on sequence input information. Nucleic Acids Res 46:W356–W362. https://doi.org/10.1093/nar/gky417
Umeadi C, Kandeel F, Al-Abdullah IH (2008) Ulinastatin is a novel protease inhibitor and neutral protease activator. Transplant Proc 40:387–389. https://doi.org/10.1016/j.transproceed.2008.01.034
Valbuzzi A, Ferrari E, Albertini AM (1999) A novel member of the subtilisin-like protease family from Bacillus subtilis. Microbiology 145(11):3121–3127. https://doi.org/10.1099/00221287-145-11-3121
Van den Burg B, Dijkstra BW, Vriend G, Van der Vinne B, Venema G, Eijsink VG (1994) Protein stabilization by hydrophobic interactions at the surface. Eur J Biochem 220:981–985
Veltman OR, Vriend G, Middelhoven PJ, van den Burg B, Venema G, Eijsink VG (1996) Analysis of structural determinants of the stability of thermolysin-like proteases by molecular modelling and site-directed mutagenesis. Protein Eng 9:1181–1189
Veltman OR, Vriend G, Hardy F, Mansfeld J, VandenBurg B, Venema G, Eijsink VGH (1997) Mutational analysis of a surface area that is critical for the thermal stability of thermolysin-like proteases. Eur J Biochem 248:433–440. https://doi.org/10.1111/j.1432-1033.1997.00433.x
Veno J, Ahmad Kamarudin NH, Mohamad Ali MS, Masomian M, Raja Abd Rahman RNZ (2017) Directed evolution of recombinant C-terminal truncated Staphylococcus epidermidis Lipase AT2 for the enhancement of thermostability. Int J Mol Sci. https://doi.org/10.3390/ijms18112202
Vishal K, Mehak B, Hao L, Pratyoosh S (2017) Microbial enzyme engineering: applications and perspectives. Recent Adv Appl Microbiol. https://doi.org/10.1007/978-981-10-5275-0_13
Wang X et al (2017) Thermostability improvement of a Talaromyces leycettanus xylanase by rational protein engineering. Sci Rep 7:15287. https://doi.org/10.1038/s41598-017-12659-y
Wijma HJ, Floor RJ, Janssen DB (2013) Structure- and sequence-analysis inspired engineering of proteins for enhanced thermostability. Curr Opin Struct Biol 23:588–594. https://doi.org/10.1016/j.sbi.2013.04.008
Yuzuki M, Matsushima K, Koyama Y (2015) Expression of key hydrolases for soy sauce fermentation in Zygosaccharomyces rouxii. J Biosci Bioeng 119:92–94. https://doi.org/10.1016/j.jbiosc.2014.06.015
Zhang H, Zhang BR, Zheng YB, Shan AS, Cheng BJ (2014) Neutral protease expression and optimized conditions for the degradation of blood cells using recombinant Pichia pastoris. Int Biodeterior Biodegrad 93:235–240. https://doi.org/10.1016/j.ibiod.2014.05.024
Zhao Y, Zeng C, Massiah MA (2015) Molecular dynamics simulation reveals insights into the mechanism of unfolding by the A130T/V mutations within the MID1 zinc-binding Bbox1 domain. PLoS ONE 10:e0124377. https://doi.org/10.1371/journal.pone.0124377
Zheng F, Huang J, Liu X, Hu H, Long L, Chen K, Ding S (2016) N- and C-terminal truncations of a GH10 xylanase significantly increase its activity and thermostability but decrease its SDS resistance. Appl Microbiol Biotechnol 100:3555–3565. https://doi.org/10.1007/s00253-015-7176-y
Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant Nos. 21778024, 31870066, 31570085), the National Key Research and Development Program of China (Grant No. 2018YFA090039), National First-Class Discipline Program of Light Industry Technology and Engineering (Grant No. LITE 2018-06), Fundamental Research Funds for the Central Universities (Grant No. JUSRP51708A), the 111 Project (Grant No. 111-2-06), and the Priority Academic Program Development of Jiangsu Higher Education Institution. We further acknowledge the Collaborative guidance from lab mates and friends.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that the research was conducted without any conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Osire, T., Yang, T., Xu, M. et al. Lys–Arg mutation improved the thermostability of Bacillus cereus neutral protease through increased residue interactions. World J Microbiol Biotechnol 35, 173 (2019). https://doi.org/10.1007/s11274-019-2751-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11274-019-2751-5