
Abstract
α-Galactosidases are assigned to the class of hydrolases and the subclass of glycoside hydrolases (GHs). They belong to six GH families and include the only characterized α-galactosidases from yeasts (GH 27, Saccharomyces cerevisiae). The present study focuses on an investigation of the lactose-inducible α-galactosidase produced by Papiliotrema flavescens. The enzyme was present on the surface of cells and in the cytosol. Its temperature optimum was about 60 °C and the pH optimum was 4.8; the pH stability ranged from 3.2 to 6.6. This α-galactosidase also exhibited transglycosylation activity. The cytosol α-galactosidase with a molecular weight about 110 kDa, was purified using a combination of liquid chromatography techniques. Three intramolecular peptides were determined by the partial structural analysis of the sequences of the protein isolated, using MALDI-TOF/TOF mass spectrometry. The data obtained recognized the first yeast α-galactosidase, which belongs to the GH 36 family. The bioinformatics analysis and homology modeling of a 210 amino acids long C-terminal sequence (derived from cDNA) confirmed the correctness of these findings. The study was also supplemented by the screening of capsular cryptococcal yeasts, which produce the surface lactose-inducible α- and β-galactosidases. The production of the lactose-inducible α-galactosidases was not found to be a general feature within the yeast strains examined and, therefore, the existing hypothesis on the general function of this enzyme in cryptococcal capsule rearrangement cannot be confirmed.
Graphical Abstract

Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
α-Galactosidases (α-d-galactoside galactohydrolases, E.C. 3.2.1.22) are assigned to the class of hydrolases and the subclass of glycoside hydrolases (GHs) (Cao et al. 2009). All currently known α-galactosidases belong to the GH families 4, 27, 36, 57, 97 and 110, and include the only characterized α-galactosidases from yeasts (ascomycetous species Saccharomyces cerevisiae), which is assigned to the GH 27. However, two uncharacterized proteins of the basidiomycetous species Cryptococcus neoformans (UniprotKB accession numbers Q5K805 and Q55IG4), mentioned in the Carbohydrate Active Enzymes database (http://www.cazy.org/), may represent putative enzymes of the galactose metabolism and belong to the GH 36 family.
The mechanistic commonality of the GH 27 and the GH 36 families within clan GH-D was demonstrated by Comfort et al. (2007). The GH-D clan is characterized by the retaining mechanism of catalysis, the similar 3D structure status (β/α)8 and the catalytic nucleophile/base and proton donor represented by aspartic acids (http://www.cazy.org/; Cantarel et al. 2009). The GH 27 and GH 36 families comprehend enzymes with both hydrolytic and transglycosylation activities. α-Galactosidases ordinarily hydrolyse terminal, non-reducing α-d-galactose residues in α-d-galactosides (galactose oligosaccharides, galactomannans, galactolipids and α-d-fucosides).
Transglycosylations mediated by galactosidases have been found in some plants (Spangenberg et al. 2002), bacteria (Van Laere et al. 1999; Spangenberg et al. 2000), fungi (Saveľev et al. 1996; Eneyskaya et al. 1998; Brumer et al. 1999; Scigelová and Crout 2000; Puchart et al. 2000; Puchart and Biely 2005; Kurakake et al. 2011) and yeasts (Hashimoto et al. 1993, 1995). Among the yeasts, the transglycosylation exhibited by the surface α-galactosidase of Papiliotrema (Cryptococcus) laurentii has been described (Mastihuba et al. 2017).
The transglycosylating and hydrolytic activities of glycoside hydrolases, including α-galactosidases, may be involved in the galactose metabolism, which it has been suggested plays an important role in the virulent character of the pathogenic Cryptococcus species (Moyrand et al. 2007).
The pathogenic species Cryptococcus neoformans forms a capsule, composed of complex polysaccharides whose function in virulence is similar to those of classical encapsulated bacteria (Pirofski and Casadevall 1996). During the budding, release, and degradation of capsular polysaccharides, the capsule undergoes several changes. The general role of α-galactosidases in modifications of the cryptococcocal capsules has been suggested by Mészárosová et al. (2012).
Nevertheless, the presence of capsules has also been found within non-pathogenic species of the former polyphyletic genus Cryptococcus (Golubev 1991; David et al. 2007). Furthermore, capsule-independent induction of the fungal and yeast α-galactosidases by lactose has been found by various authors (Church and Mayers 1980; Flórez et al. 1981; Zeilinger et al. 1993).
However, up to now, the purification and biochemical characterization of the cryptococcal glycoside hydrolases have been reported only with endo-β-1,3-glucanase of Cr. neoformans. The role of this enzyme in cell wall rebuilding, as opposed to the rearrangement of the capsule, has been proposed (Maceková et al. 2006).
The purification and biochemical characterization of the cryptococcal glycoside hydrolases is a troublesome process due to the presence of polysaccharides (capsular, extracellular and intracellular) in high amounts, which can be removed with difficulty.
In our previous work, the production of α-galactosidase by the strain Cryptococcus laurentii CCY 17-3-29 (recently reclassified as Papiliotrema flavescens), was found to be dependent on the presence of the capsule and the type of C-source in the cultivation medium. The enzyme was present on the surface of cells and in the cytosol (CF) and was induced by the saccharides, which contained galactose bound in their structure. Moreover, the activities of this enzyme, as well as the optimization of its synthesis, were shown and discussed (Mészárosová et al. 2012).
This work represents the first report on the purification and characterization of α-galactosidase produced by the species of the former polyphyletic genus Cryptococcus. It focuses on the investigation of an inducible α-galactosidase of Papiliotrema (Cryptococcus) flavescens CCY 17-3-29.
The results we obtained were supplemented by the screening of capsular cryptococcal yeasts, which produce the surface lactose-inducible α- and β-galactosidases in general.
Materials and methods
In this study, the strains of the former polyphyletic genus Cryptococcus were used: Cryptococcus neoformans (CCY 17-1-2—serotype D, CCY 17-1-8—serotype A); Filobasidium magnum (Cryptococcus magnus) CCY 17-4-39; Naganishia globosa (Cryptococcus saitoi) CCY 17-3-18; Papiliotrema (Cryptococcus) flavescens (CCY 17-3-15, CCY 17-3-29, CCY 17-3-31, CCY 17-3-33, CCY 17-3-38); Papiliotrema (Cryptococcus) laurentii (CCY 17-3-2, CCY 17-3-9, CCY 17-3-17, CCY 17-3-24); Saitozyma flava (Cryptococcus flavus) CCY 17-3-5; Vishniacozyma (Cryptococcus) carnescens CCY 17-3-13; Vishniacozyma (Cryptococcus) victoriae CCY 17-3-26 as well as Bullera alba (CCY 17-3-35, CCY 17-3-37) and Filobasidium floriforme CCY 17-4-40. All strains were obtained from the Culture Collection of Yeasts (the Institute of Chemistry of the Slovak Academy of Sciences, Slovakia). Taxonomic classification of all strains was confirmed using D1/D2 LSUrDNA sequencing and/or MALDI TOF biotyping (Bruker), using the mass spectra of the strains sequenced as references. The species names follow the nomenclature of Liu et al. (2015).
The yeasts grew aerobically in a semisynthetic medium containing in 1 l: 20 g lactose, 1 g MgSO4·7H2O, 0.5 g KH2PO4, 5 g yeast autolysate, 3 g (NH4)2SO4 and 1 ml of solution with microelements (1000 mg of FeSO4·7H2O, 104 mg of MnSO4·H2O, 72 mg of (NH4)6Mo7O24·4H2O, 352 mg of Na2B4O7·10H2O, 60 mg of CuSO4·5H2O and 656 mg of ZnSO4 in 1 l of distilled water). Cultivation was carried out on an orbital shaker (100 rpm) at 28 °C.
The growth of cultures and the production of surface α/β-galactosidases were determined at regular intervals for a period of 7 days. All assays were carried out in triplicate and the results show their average value.
For the purification and characterization of the Pa. flavescens (CCY 17-3-29) α-galactosidase, the cultivation was performed for 48 h. The yeast cells were centrifuged at 3000×g, 4 °C for 15 min, washed using 50 mM Tris/HCl buffer, pH 7.5, twice and finally washed using the same buffer, containing 0.1 mM PMSF (phenylmethylsulfonyl fluoride, Sigma-Aldrich).
Isolation of α-galactosidase
The CF fraction was isolated from the cells of Pa. flavescens according to Ankel et al. (1970).
The washed cells were suspended in a homogenization solution composed of 100 mM Tris/HCl buffer, pH 7.5, 0.46 mM PMSF, 2.0 mM EDTA (ethylenediaminetetraacetic acid, Sigma) and homogenized in a rotatory disintegrator at − 10 °C (5 × 5 min with 1 min breaks). The disintegration of the cells was checked microscopically.
The disintegrated cells were centrifuged at 1300×g, 4 °C for 10 min. All subsequent steps were performed at 4 °C. The supernatant was centrifuged again at 12,000×g for 20 min, and afterwards, the supernatant from the previous step was spun at 100,000×g for 1 h (Optima™ L-90K, Beckman) to obtain the CF.
Purification of α-galactosidase
α-Galactosidase from the CF was purified using a combination of ion-exchange, affinity and gel-permeation chromatographies (Stratilová et al. 2005). In all fractions, the protein content and α-galactosidase activity were determined.
The pH of CF (10–40 ml) was adjusted to 3.8 and the solution was applied on a CM-Sephadex C-50 (Pharmacia, Sweden) column (size: 3 cm × 20 cm, flow rate: 16 ml/h, fractions: 4 ml) in starting eluent, 50 mM acetate buffer, pH 3.8. The bound proteins were released sequentially using: 100 mM acetate buffer, pH 4.4; 150 mM acetate buffer, pH 4.8; 200 mM acetate buffer, pH 5.4; and finally, the rest of proteins was released with 200 mM acetate buffer, pH 5.6 containing 1 M NaCl. The fractions with α-galactosidase activity were collected and subjected to next purification step.
The next step was performed on a Concanavalin A-SepharoseTM4B (GE Healthcare) column (size: 1 cm × 4 cm) in 50 mM acetate buffer, pH 5.0, with the addition of 1 M NaCl, 1 mM MnCl2, 1 mM MgCl2 and 1 mM CaCl2 (fractions 1 ml). Aliquots of the CF fractions with α-galactosidase activity, collected from CM-Sephadex C-50 (30–120 ml), were slowly applied to the column. After elution of all unbound proteins (using a starting buffer without microelements), 1 M methyl-α-d-mannopyranoside (α-MMP, Sigma) in 50 mM acetate buffer, pH 5.0, containing 1 M NaCl was used to release bound glycoproteins. The fractions with α-galactosidase activity were collected, dialyzed against distilled water, and freeze-dried.
Fast protein liquid chromatography and rechromatography (FPLC) of partially purified α-galactosidase was performed on Superose 12™ HR 10/30 (Pharmacia, Sweden) in 50 mM phosphate buffer, pH 7.0, 150 mM NaCl. The flow rate was 0.5 ml/min. The fractions which exhibited α-galactosidase activity were processed as described for the previous step. The protein composition was checked by SDS-PAGE (sodium dodecyl sulfate-polyacrylamide gel electrophoresis) and IEF-PAGE (isoelectric focusing-polyacrylamide gel electrophoresis), respectively.
Activity assay
The activity of α/β-galactosidase was generally assayed in a reaction mixture composed of 50 μl of the CF or the cultivation medium with cells, 175 μl of 100 mM citrate–phosphate buffer, pH 4.8 and 25 μl of 1% para-nitro-phenyl-α/β-galactopyranoside (PNP-α/β-galactopyranoside, Sigma) according to Mészárosová et al. (2012). The incubation was carried out at 37 °C for 30 min. The reaction was stopped by the addition of 1 ml 4% Na2CO3. An increase in color intensity, due to releasing of PNP from the substrate, was measured at 410 nm. The activity in fractions, obtained during the purification process, or on cells during the cultivation, was expressed as an increase in absorbance at 410 nm during 0.5 or 1 h. The specific activity of α-galactosidase was expressed in pmol of PNP, released from PNP-α-galactopyranoside, within one second (pcat) per milligram of the protein. A total activity of the enzyme was defined per the whole protein obtained or per the amount of cultivation medium used. They were determined by means of the standard graph for PNP. All assays were performed in triplicate and the results show their average value.
Biochemical characterization of α-galactosidase
The temperature optimum and the thermal stability of the α-galactosidase from the CF were measured at temperatures ranging from 20 to 80 °C. The enzyme-buffer mixtures were incubated at temperatures within this range in 10 °C steps. PNP-α-galactopyranoside was added for 10 min at 1-h intervals from 0 to 6 h. The released PNP was then measured as described. The values obtained were compared with those for a temperature optimum (the 0 h assay; 100%) and expressed in % (relative activity).
Both the pH optimum and the stability of the α-galactosidase were assayed in a range of 3.2–7.0, at intervals of 0.2, using McIlvaine buffers (100 mM). The enzyme was exposed to these buffers over 4 days at 4 °C. The activity assay was performed every day, after incubation at 37 °C for 10 min. The values obtained were compared with those for a pH optimum (the 0-day assay; 100%) and expressed in %.
Various concentrations of PNP-α-galactopyranoside (0.3–6.7 mM) were used in the activity assay for kinetic analysis. The activity assay was carried out at 37 °C after 10 min incubation. The data from the kinetic assays were processed and the respective kinetic constants Km and Vmax were calculated by non-linear regression (Hill function) of v0 [mol/l s] = f(S) [mol/l] using Origin 6.0 (OriginLab Corporation, MA 01060, USA) software.
The reaction progress of the transglycosylation with α-galactosidase was monitored by thin-layer chromatography (TLC on Silicagel 60 plates F 254, Merck, developed twice in 1-butanol:ethanol:water, 10:8:7 by vol., and then sprayed with 1% orcinol in 10%, by vol., sulphuric acid in ethanol, followed by heating at 100 °C for about 10 min). The reaction mixture contained 60 mM PNP-α-galactopyranoside, 80 mM galactose and CF (2:1:1, by vol.). The incubation was carried out at 37 °C for 24 h. PNP, PNP-α-galactopyranoside and galactose were used as standards.
The approximate relative molecular masses (Mr) of the enzymes were determined by gel filtration on a Superose 12 HR 10/30 (Pharmacia, Sweden) connected to a FPLC device (Pharmacia, Sweden) in a 50 mM phosphate buffer, 150 mM NaCl, pH 7.0. Proteins from the calibration kit MW-GF-1000 (Sigma) were used as standards.
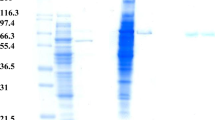
SDS-PAGE, for the visualization of the purification progress of the α-galactosidase from the CF, was performed on a Mini-Protean 3 Electrophoresis System (Bio-Rad Laboratories, Hercules, California) in a 7–10% gel (Laemmli 1970), under reducing conditions (with β-mercaptoethanol). Coomassie Blue staining was used for the band visualization of crude CF proteins and silver-staining for partially purified α-galactosidase (PageSilverTMSilver Staining Kit, Fermentas). Calibration proteins, ranging from 15 to 170 kDa (Spectra™ Multicolor High Range Protein Ladder, Fermentas), were used as standards.
Ultrathin-layer isoelectric focusing in polyacrylamide gels on polyester films (IEF-PAGE) was performed in the pH region 3–10, as described by Radola (1980). The standard proteins (Protein Test Mixture for pI-Determination, pH 3–10, Serva, Germany) were visualized with Coomassie Blue. The α-galactosidase activity in the gel was visualized by spraying the gel with 1% PNP-α-galactopyranoside, followed by spraying with 4% Na2CO3 after 5 min.
Structural characterization of α-galactosidase
After SDS-PAGE, the protein bands from the gels were processed according to the protocol of Shevchenko et al. (2006). Briefly, the excised gel pieces (about 5 × 2 mm), containing separated proteins, were dehydrated with 450 μl acetonitrile (5 min) and a supernatant was removed. The proteins were reduced in-gel using 50 μl 10 mM DTT (dl-Dithiothreitol, Sigma-Aldrich) in 100 mM NH4HCO3 (at 56 °C for 30 min). The mixture was homogenized after the addition of 450 μl acetonitrile and the supernatant was removed. The protein alkylation was performed with 50 μl 55 mM iodoacetamide (Sigma-Aldrich) in 100 mM NH4HCO3 (in the dark at room temperature for 20 min). The pieces of gel were washed with 450 μl 100 mM NH4HCO3 (5 min) and dehydrated with 450 μl acetonitrile (5 min), the supernatant was removed. The gel pieces were rehydrated at 4 °C (30 min) with the solution containing 10 mM NH4HCO3, and 13 μg/ml porcine trypsin (Promega, modified sequencing grade). Subsequently, the digestion buffer (solution containing 10 mM NH4HCO3) was added to fully cover the gel pieces (about 30–50 μl) and digestion was carried out overnight at 37 °C. The supernatant from in-gel digestion was acidified with 10 μl 1% TFA and purified using self-made reversed-phase micro columns which contained frit from C18 Empore Disks (3M, Minneapolis, MN) and POROS R3 50 μm material (Rappsilber et al. 2003). The in-gel tryptic digest of protein, with a molecular weight about 110 kDa, obtained after purification on CM-Sephadex C-50 column, was processed further using manual microgradient chromatographic separation (Franc et al. 2012), employing a capillary column (length: 30 mm, i.d. 250 μm), packed with 5 μm particles from a Poroshell 300 Extended-C18 column (Agilent Technologies) or a capillary column (length: 27 mm, i.d. 250 μm) packed with 2.7 μm particles from Ascentis Express Peptide ES-C18 column (Sigma-Aldrich). After MALDI-TOF/TOF MS analysis, selected peptide fractions from MALDI target spots were redissolved in 60% acetonitrile and transferred to a 0.5 ml tube, dried in a vacuum centrifuge and esterified using 50 μl of the mixture of acetylchloride/ethanol (8:50, v/v) for 2 h at room temperature. The mixture was dried in the vacuum centrifuge, redissolved in the 2 μl 60% acetonitrile and reanalyzed using MALDI-TOF/TOF MS, to confirm the peptide sequences derived from MALDI-TOF/TOF MS/MS analysis of underivatized peptides.
MALDI-TOF/TOF MS measurements, in the positive reflectron mode, were performed with a 4800 Proteomics Analyzer (Applied Biosystems). MS spectra were acquired using a dual-stage reflectron mirror. The accelerating voltage applied for the MS measurements was 20 kV. The raw spectral data were further processed using Data Explorer 4.8 software (Applied Biosystems). For the mass determination of the protein digest, a solution of α-cyano-4-hydroxycinnamic acid (5 mg/ml) in acetonitrile/0.1% TFA (3:2, v/v) was used. A purified peptide mixture (0.5 μl) was mixed with a matrix solution (0.5 μl) on a target and dried at room temperature. The peak m/z values from the MS and MS/MS analyses were submitted to the Mascot protein identification program package (local installation, version 2.1) and the data was searched for. The parameters used for the data searching were as follows: database—NCBInr (ver. 5.1.2011); taxonomy—all entries; enzyme—trypsin; allowed missed cleavages—1; fixed modifications—carbamidomethyl (C); variable modifications—Oxidation (M), Pyro-cmC (N-term camC), Pyro-glu (N-term E), Pyro-glu (N-term Q); peptide tolerance − 50 ppm; MS/MS tolerance − 250 mmu; peptide charge—(+ 1); monoisotopic masses; instrument—MALDI-TOF-PSD. Similarity based searches were done using BLAST programs (http://www.uniprot.org/?tab=blast; http://blast.ncbi.nlm.nih.gov).
RNA isolation
Yeasts from 48 h-old culture in a lactose medium were sampled by repeated centrifugation into 2 ml micro tubes (1.28 × 108 cells − 11.8 mg per tube). Each sample of yeast cells was homogenized with 340 µl glass beads (treated with HNO3) in 1000 µl of TRI reagent (Ambion) using the homogenizer Vortex Genie 2 (Scientific Industries), with 8 mixing cycles each for 1 min, followed by 2 min cooling on ice. The RNA was isolated according to Ambion’s protocol, recommended for samples rich in polysaccharides. The RNA was precipitated using 250 µl isopropanol and 250 µl of a solution containing 0.8 M sodium citrate and 1.2 M NaCl in DEPC—treated water. RNA was dissolved in 150 µl DEPC—treated water and its quality was verified spectrophotometrically and by agarose gel electrophoresis.
Sequence characterization of 3′ region of α-galactosidase cDNA
The 3′ part of cDNA was prepared using the partially modified 3′ RACE procedure (Frohman et al. 1988). Two primers were used: a universal anchor-oligo(dT)-adaptor primer (AOdTA; 5′-GACTCGAGTCGACATCGATTTTTTTTTTTTTTTTTV-Wobbles-3′) and a consensus-degenerate hybrid α-GAL primer (AGAL3f; 5′-TACCCTGATGTCCTCTGGGARGGNTGYGC-3′). The AGAL3f primer was designed according to the CODEHOP method (COnsensus-DEgenerate Hybrid Oligonucleotide Primer method—Rose et al. 2003) on the basis of “YPDVLWEGCASGGGR” peptide sequence derived from a MALDI-TOF/TOF sequencing of purified α-galactosidase.
First strand cDNA (fs-cDNA) was synthetised by RevertAid H Minus First Strand cDNA Synthesis Kit (Thermo Scientific, USA) using 0.8 µg of total RNA and 100 pmol AOdTA primer in 20 µl reaction mixture. PCR amplifications of cDNA were performed with Phusion Hot Start II High-Fidelity DNA Polymerase (Thermo Scientific, USA), using 3 µl of fs-cDNA, 20 pmol of AOdTA and 26 pmol of AGAL3f primers in 50 µl reaction mixture. The PCR reaction was performed according to the following conditions: an initial denaturation at 98 °C for 30 s, followed by 35 cycles at 98 °C for 10 s, 66 °C for 30 s and 72 °C for 30 s, and a final extension at 72 °C for 10 min.
The α-Gal 3′ cDNA fragment of expected size was purified by preparative agarose gel electrophoresis using a GeneJET™ Gel Extraction Kit (Thermo Fisher Scientific) and cloned into a pJET 1.2/blunt vector (CloneJET PCR Cloning Kit, Thermo Scientific) in Escherichia coli XL10 Gold (Agilent Technologies, USA). Plasmid DNAs were isolated from several transformants using a GeneJET™ Plasmid Miniprep Kit (Thermo Scientific, USA) and sequenced by commercial service.
Bioinformatics analyses and homology modeling
The sequence of the α-Gal 3′ cDNA fragment was assembled from the data obtained employing the BioEdit program (http://www.mbio.ncsu.edu/bioedit/bioedit.html). The associated entire dataset from this study can be accessed via the European Nucleotide Archive (ENA accession number PRJEB20951). The cDNA translation was carried out by the Translator program (http://www.justbio.com/).
The protein sequences were aligned using http://www.uniprot.org (blast) and Clustal Omega tools. Homology modeling of the C-terminal fragment was performed with the Modeller 9v16 program, using the crystal structure of the GH36 alpha-galactosidase, AgaB of Geobacillus stearothermophilus (4fnq.pdb), as a template (Merceron et al. 2012). The quality of the model was expressed by means of its RMSD value (root-mean-square deviation of backbone atomic positions).
Results
The growth of yeasts and the production of a surface α-galactosidase by Pa. flavescens (CCY 17-3-29) and two strains of Cr. neoformans (CCY 17-1-8 and CCY 17-1-2), on a lactose containing medium, were compared, to verify a general role of inducible α-galactosidase in capsule modifications within the former polyphyletic genus Cryptococcus (Fig. 1). The standard deviations (SD) of the data points from the growth curves were within ± 8.2%, and the SD of the data points from the enzyme assays were within ± 4.5%. The induction of α-galactosidase by lactose was observed only with the strain Pa. flavescens. To explain this result, 16 other strains were tested: B. alba (2 strains), Fil. floriforme (1 strain), Fil. magnum (1 strain), Na. globosa (1 strain), Pa. flavescens (4 strains), Pa. laurentii (4 strains), Vis. carnescens (1 strain), Vis. victorie (1 strain) and Sait. flava (1 strain). Surface α-galactosidase, the growth curves, and β-galactosidase activity, required to break down of lactose, were determined (Table 1).

The growth of Pa. flavescens CCY 17-3-29 (triangle), Cr. neoformans: CCY 17-1-2 (square) and CCY 17-1-8 (circle) in the medium containing glucose (full symbols) and lactose (empty symbols) (a); production of the surface α-galactosidase by these strains (b)
A culture medium which contained lactose was not suitable for all the strains tested: Pa. flavescens (CCY 17-3-29), Sait. flava (CCY 17-3-5), Vis. victoriae (CCY 17-3-26), B. alba (CCY 17-3-35) grew very slowly, whereas Pa. laurentii (CCY 17-3-2 and CCY 17-3-6) exhibited a long adaptation phase. A comparison of the growth and the production of the surface β-galactosidase showed that the link between the lactose medium and the production of this enzyme does not exist in general. The results confirmed neither the expected impact of this enzyme on the growth of the strains nor the anticipated induction of β-galactosidase by lactose. Although of the strains tested, the strain Vis. carnescens (CCY 17-3-13) grew the most rapidly, its surface β-galactosidase exhibited only very low activity. An increased production of this enzyme was observed on the cell surface of Pa. laurentii (CCY 17-3-2, a type strain), Pa. flavescens (CCY 17-3-31) and Sait. flava (CCY 17-3-5). The production of the lactose-inducible α-galactosidase by capsular yeasts was strain-dependent, with the exception of Pa. flavescens. Therefore, the α-galactosidase produced by Pa. flavescens was subjected to further studies. The cytosol was used as an enzyme source instead of the cell walls, because they are coated with capsular polysaccharides.
The average total activity of the cytosolic inducible α-galactosidase, obtained from individual cultivations of Pa. flavescens CCY 17-3-29 on a lactose-containing medium (10 l), reached 2669 ± 276 pcat. Before purification, α-galactosidase was characterized by its temperature and pH optima, and thermal and pH stabilities (Fig. 2). The enzyme exhibited maximum activity at 60 °C (Fig. 2a). Its activity declined after 6 h incubation: to 88.9% at 50 °C; to 71.4% at 60 °C; and to 4.2% at 70 °C (half-time of inactivation ranged from 1 to 2 h). The enzyme was completely inactivated at 80 °C within the first hour. The optimum pH of the enzyme was 4.8 (Fig. 2b). A slight decrease in activity during the 4-day storage was only observed in the pH region near neutral. The SD of the all assays was within ± 4.5%.

Characterization of the Pa. flavescens α-galactosidase obtained from the cytosol: pH optimum versus pH stability during 4-day incubation of enzyme in buffers with corresponding pH (a); temperature optimum versus thermal stability of enzyme during 6 h incubation at corresponding temperature (b); thin-layer chromatography of transglycosylating reactions (St standards—PNP, PNP-α-galactopyranoside and galactose; line 1, the reaction mixture composed of PNP-α-galactopyranoside, galactose and CF at time zero; line 2, the reaction mixture after 24 h) (c)
Transglycosylating activity in CF was carried out using both PNP-α-galactopyranoside and galactose as substrates. The production of galactobiose, PNP-galactobiose, galactotriose and PNP-galactotriose was observed on a TLC plate (Fig. 2c). The precipitate of a crude protein was separated by SDS-PAGE and the main proteome (composed of isocitrate dehydrogenase, phosphoglycerate kinase, HSP60, phosphoglycerate mutase and phosphoglucomutase) was recognized, using sequence similarity analysis of fragments obtained after tryptic cleavage of proteins in the gel (results not shown). α-Galactosidase, which was evidently a minor protein, was not detected.
To obtain homogeneous α-galactosidase from Pa. flavescens, four purification steps, including rechromatography on a Superose 12 column (Figs. 3, 4a), were required. The efficiency of the individual purification steps is given in the purification table (Table 2). The molecular weight of this enzyme, based on the activity assay, was about 120 kDa (estimated after a calibration of the last column, Superose 12) (Fig. 3c). IEF-PAGE indicated that the isoelectric point for this protein was about 4.2 (Fig. 4b). Kinetic analysis of purified α-galactosidase, on PNP-α-galactopyranoside as a substrate, showed Km, 1.28 mM and Vmax 300.98 pcat.

Purification stages of the cytosolic Pa. flavescens α-galactosidase. Proteins are expressed as A280 (—) and the activity of the enzyme expressed as A410/30 min (O—O): CM-Sephadex C-50 column, starting eluent: 0.05 M acetate buffer, pH 3.8, ↓a: 0.10 M acetate buffer, pH 4.4, ↓b: 0.15 M acetate buffer, pH 4.8 (a); Concanavalin A-SepharoseTM4B column, starting eluent: 0.05 M acetate buffer, pH 5.0 with the addition of 1 M NaCl, 0.001 M MnCl2, 0.001 M MgCl2 and 0.001 M CaCl2, ↓a: starting buffer without microelements, ↓b: 1 M methyl-α-d-mannopyranoside in 0.05 M acetate solution, pH 5.0 with 1 M NaCl (b); Superose 12 column, eluent: 0.05 M phosphate buffer, pH 7.0 with 0.15 M NaCl (c)

SDS-PAGE of proteins after individual purification steps for the α-galactosidase from the CF (Mr molecular mass of markers, St standard, CM purified CF after CM-Sephadex C-50; Sup1, after Superose 12; Sup2, after rechromatography on Superose 12) (a); IEF-PAGE with the activity detection (St standard, pI isoelectric points of standards, CF activity staining of partially purified CF. Activity was detected by spraying the gel with PNP-α-galactopyranoside, followed by spraying with 4% Na2CO3 after 5 min) (b)
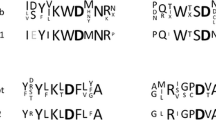
After separation on CM-Sephadex C-50, three sequences corresponding to α-galactosidase were identified using mass spectrometry de-novo sequencing of a tryptic digest of the weaker protein band on SDS-PAGE gel (Figs. 3a, 4a—line CM). An additional separation of tryptic digests, using manual chromatographic separation coupled to MALDI-TOF/TOF MS (Franc et al. 2012), was required. The “YPDVLWEGCASGGGR” sequence was used to design a primer for the preparation of a 3′ α-Gal cDNA fragments. The sequencing of four identical 3′ α-Gal cDNA fragments clearly resulted in the indentification of 210 amino acids long C-terminal sequence of the α-galactosidase. Two other protein sequences (“TTPLEFR” and “DAIPGLVALGER”) were used to verify the correctness of the amino acid sequence derived from the cDNA. A comparison of the sequence with those found in the UniProt database showed the highest homology with fungal α-galactosidases from Hirsutella minnesotensis (72.2%—A0A0F8A6K7_9HYPO), Pseudogymnoascus sp. (71.5%—A0A093YDB1_9PEZI), Trichoderma reesei (71.0%—A0A024RZH1_HYPJE), Trichoderma virens (71.0%—G9N147_HYPVG), Ophiocordyceps sinensis (70.5%—T5AP68_OPHSC), Byssochlamys spectabilis (70.0%—V5FPY1_BYSSN) and Trichoderma harzianum (70.0%—A0A0F9XT47_TRIHA) (Fig. 5a). All these seven proteins are members of the GH 36 family.

Alignment of the Pa. flavescens α-galactosidase C-terminal sequence with sequences of other proteins found in the UniProt database which showed mutually the highest homology: A0A0F8A6K7_9HYPO (Hirsutella minnesotensis—72.2%), A0A093YDB1_9PEZI (Pseudogymnoascus sp.—71.5%), A0A024RZH1_HYPJE (Trichoderma reesei—71.0%), G9N147_HYPVG (Trichoderma virens—71.0%), T5AP68_OPHSC (Ophiocordyceps sinensis—70.5%), V5FPY1_BYSSN (Byssochlamys spectabilis—70.0%), A0A0F9XT47_TRIHA (Trichoderma harzianum—70.0%) (a); alignment of the Pa. flavescens α-galactosidase C-terminal sequence with the Cr. neoformans GH 36 proteins Q5K805_CRYNJ and Q55IG4_CRYNB (b); De-novo determined sequences of the Pa. flavescens α-galactosidase are given in black boxes
The sequence determined was aligned with the sequence of proteins Q5K805 and Q55IG4 (http://www.uniprot.org), which was found to play a part in the galactose metabolism of Cr. neoformans (Loftus et al. 2005). These proteins also belong to the GH 36 family and demonstrate “possible activity of raffinose synthase” (http://www.cazy.org/; Cantarel et al. 2009). Significant structural differences between the C-terminal sequence of inducible α-galactosidase of Pa. flavescens and Q5K805 or Q55IG4 of Cr. neoformans were observed (Fig. 5b).
Finally, a tertial structure model of the C-terminal fragment of α-galactosidase from Pa. flavescens fitted perfectly (RMSD = 0.832) with the suggested template: the GH 36 family α-galactosidase (4fnq.pdb) of Geobacillus stearothermophilus (Fig. 6).

Homology model of the C-terminal sequence of the Pa. flavescens α-galactosidase: fragment position within the template structure, the GH 36 family alpha-galactosidase (4fnq.pdb) from Geobacillus stearothermophilus (a); detailed comparison of 210 amino acid long C-terminal of the Pa. flavescens α-galactosidase with the template structure (b)
Discussion
A recent study has indicated that the size and structure of the cryptococcal capsule is dependent on the ratio of its major polysaccharides: glucuronoxylomanan (GXM) and galactoxylomanan (GalXM). The absence of GalXM results in the overproduction of GXM and an increase in the capsule size (Moyrand et al. 2007). This finding was confirmed by other authors, who found that the strain Pa. laurentii, with a high production of GalXM (90% of all extracellular polysaccharides), was acapsular (Dudíková and Kolarova 2006). Pa. flavescens, grown on a medium with saccharides, in which galactose was bound into their structure, produced increased amounts of both surface and cytosole α-galactosidases. The highest induction was observed, using lactose as a carbon source, with the strain CCY 17-3-29 (Mészárosová et al. 2012). The induction of this enzyme was not observed with the acapsular strain. These findings resulted in the assumption that the inducible α-galactosidase may influence the GalXM/GXM ratio and, therefore, play a role in the composition and rearrangement of the capsule (Mészárosová et al. 2012).
Other results showed that the production of α-galactosidase was connected with poor growth of the strain, a decrease in the cell and capsule size, and a change in the composition of capsular polysaccharides of daughter cells during budding (Mészárosová et al. 2012). Moreover, the authors have hypothesized that the inducible α-galactosidase plays a key role in the rearrangement of the Cryptococcus capsules, which was also supported by the activity of the surface α-galactosidase from Cr. neoformans reported by Maceková et al. (2006). Nevertheless, our results show that the production of this enzyme was strain dependent (with the exception of the Pa. flavescens strains) and the pathogenic Cr. neoformans strains tested did not produce the lactose-inducible enzyme at all. The differences between these results can be explained using the data obtained from the genome analysis of Cr. neoformans (Loftus et al. 2005). It was found that Q5K805 and Q55IG4 (http://www.uniprot.org) are the only proteins in Cr. neoformans which may potentially participate in the galactose metabolism. They were found to exhibit “possible raffinose synthase activity” (http://www.cazy.org/; Cantarel et al. 2009) and thus, the low α-galactosidase activity of Cr. neoformans reported by Maceková et al. (2006) may be caused by the side activity of raffinose synthases.
The induction of some α-galactosidases by lactose in non-capsular yeasts was described in the early eighties (Church and Mayers 1980; Flórez et al. 1981) and later a similar phenomenon was observed within fungi (Zeilinger et al. 1993). Lactose consists of galactose and glucose, which are linked by β-bounds and, thus, it cannot be cleaved by α-galactosidases.
Although the induction is caused by the presence of bound galactose (Mészárosová et al. 2012), generally, it seems that microorganisms are not able to distinguish the character of the galactose binding. From an overview of currently available information (http://www.cazy.org/), it can be concluded that yeast enzymes belong to the GH 27 family, while fungal enzymes may also be members of the family 36.
β-Galactosidases are enzymes involved in the splitting of lactose and, as a result, in providing cells with galactose and glucose. However, a comparison of the growth and the production of β-galactosidase obtained by us showed that the link between the utilization of lactose from the medium and the enzyme production does not have a general character. Our results confirmed neither the expected impact of this enzyme on the growth of strains nor the anticipated induction of β-galactosidase by lactose.
Due to the lack of data on the primary and tertiary structures of cryptococcal glycosidases, the next part of our work focused on purification and characterization of the main α-galactosidase from Pa. flavescens as well as determination of its partial structure.
During the purification of minor proteins from the material which contained slimy extracellular, intracellular and capsular polysaccharides, clogging of columns appeared (a well-known problem) and, therefore, the α-galactosidase was preferably purified from the CF rather than from the cell walls. The major part of neutral polysaccharides was removed at the first step of purification, using ion-exchange chromatography matrix, which bounded only charged proteins released later, due to a change in the eluent pH. The chromatography matrix was used only once for every time, which resulted in the protection of other columns used, and in further purification without complications. The use of affinity chromatography was based on the assumption that α-galactosidases migth be N-glycosylated (Ioannou et al. 1998). The Superose 12 column was employed due to its precise final purification of small amounts of proteins (Stratilová et al. 2005). The major CF proteome of Pa. flavescens was recognized in a crude protein extract relatively easily: by structural analysis, using MS, and by comparing known basic proteins derived from the complete genome of Cr. neoformans (Loftus et al. 2005). Although an intensive induction was observed, α-galactosidase as a minor protein was not identified by the MS analyses. After separation on CM-Sephadex C-50, and subsequent MS de-novo sequencing of tryptic digest of the weaker protein band on SDS-PAGE gel, three sequences corresponding to the fungal α-galactosidases were identified. A weak interaction of the major α-galactosidase with CM-Sephadex C-50 indicated the acidic character of this protein, with isoelectric point (pI) about 4.2. This result is in agreement with the data on the pIs of fungal α-galactosidases, which range from 3.7 to 7.0 (Zapater et al. 1990; Dey et al. 1993; Ríos et al. 1993; Koizumi et al. 1995; Puchart et al. 2000). The other biochemical features, such as the pH optimum, temperature optimum, pH stability and temperature stability (Fig. 2) were also comparable with features of fungal α-galactosidases.
Each additional purification step resulted in a dramatic loss of α-galactosidase due to the cumulative decreases in its solubility. This loss of soluble protein resulted in a concentration which was too low for further de-novo sequencing using MALDI-TOF/TOF MS. However, purification of the α-galactosidase on Concanavalin A-Sepharose column confirmed its glycoprotein character. The calibration of Superose 12 showed the molecular mass to be about 120 kDa for the protein with α-galactosidase activity and, together with the result from SDS-PAGE (110 kDa), confirmed its monomeric character. Nevertheless, the relationship between the cumulative decrease in α-galactosidase solubility and the possibility of its oligomerization, due to its increased concentration during purification, remains unclear.
Kinetic analysis of purified α-galactosidase showed a relatively high value of Km on PNP-α-galactopyranoside as a substrate (1.28 × 10−3 mol/l). This value is comparable with the values found for α-galactosidases isolated from fungi (Puchart et al. 2000). The transglycosylating activity of the surface α-galactosidase from Pa. laurentii has been described recently (Mastihuba et al. 2017). The authors reported a strict regioselectivity of this enzyme, resulting in di- and tri-α-(1–6)-galactosides as the only products. For this reason, only a simple experiment was carried out to determine transglycosylating activity in CF, using both PNP-α-galactopyranoside and galactose as substrates. The production of galactobiose, PNP-galactobiose, galactotriose and PNP-galactotriose showed that α-galactosidase from CF was able not only to release galactosyl groups from the substrate, but also to exhibit transglycosylation activities. A study on the practical exploitation of transglycosylating abilities of the lactose-inducible α-galactosidase from Pa. laurentii and Pa. flavescens, immobilized on the cell surface structures, is in progress.
Bioinformatic analysis of three peptide sequences, obtained by MS, positioned the “YPDVLWEGCASGGGR” sequence immediately after asparagine in the catalytic site of the GH 36 family α-galactosidases. This result was similar to that obtained with proteins from Phaeosphaeria nodorum (Q0U0J8_PHANO) and Leptosphaeria maculans (E4ZKD6_LEPMJ). Homology was found with a further 14 fungal α-galactosidases, which differed from the sequences we determined in only one amino acid. The sequences “TTPLEFR” and “AIPGLVALGER” were positioned closest to the C-terminus of proteins. For this reason, the “YPDVLWEGCASGGGR” sequence was selected to design a primer for the determination of the C-terminal sequence, using a molecular biology approach.
Bioinformatic analysis of a 210 amino acid-long C-terminal sequence showed more than 70% matrix identity with seven fungal long-chain α-galactosidases belonging to the GH 36 family. Subsequent alignment of the sequence with those of three α-galactosidases from T. reseii (Margolles-Clark et al. 1996), two of the GH 27 (G0RQ44_HYPJQ and G0RA73_HYPJQ with matrix identity 20.81% and 22.45%, respectively) and one of the GH 36 family (G0RVT1_HYPJQ with matrix identity 70.95%) confirmed the inclusion of α-galactosidase from Pa. flavescens, as the first yeast α-galactosidase, into the GH 36 family. Interestingly, one of these α-galactosidases from T. reseii was also induced by lactose (Flórez et al. 1981), even though it was a member of the GH 27 family (Margolles-Clark et al. 1996). In the GH 36 family of proteins, Q5K805 (matrix identity 20.99%) and Q55IG4 (matrix identity 21.55%) of Cr. neoformans (Loftus et al. 2005) showed major structural variations, which indicated functional differences between the enzymes of Pa. flavescens and Cr. neoformans, which are involved in the galactose metabolism.
The C-terminus sequence was long enough for its homology model to be created, which confirmed assignment of the Pa. flavescens α-galactosidase to the GH family. The value of 0.832 for RMSD between the C-terminal of Pa. flavescens α-galactosidase and the model structure (crystal structure of the GH 36 family α-galactosidase AgaB from Geobacillus stearothermophilus, 4fnq.pdb) showed that the choice of model structure was correct and confirmed the identification of the first yeast α-galactosidase as belonging to the GH 36 family.
References
Ankel H, Ankel E, Schutzbach JS, Garancis JC (1970) Mannosyl Transfer in Cryptococcus laurentii. J Biol Chem 245:3945–3955
Brumer H III, Sims PFG, Sinnott ML (1999) Lignocellulose degradation by Phanerochaete chrysosporium: purification and characterization of the main α-galactosidase. Biochem J 339:43–53
Cantarel BL, Coutinho PM, Rancurel C, Bernard T, Lombard V, Henrissat B (2009) The Carbohydrate-Active EnZymes database (CAZy): an expert resource for Glycogenomics. Nucleic Acids Res 37:233–238
Cao Y, Wang Y, Meng K, Bai Y, Shi P, Luo H, Yang P, Zhou Z, Zhang Z, Yao B (2009) A novel protease-resistant alpha-galactosidase with high hydrolytic activity from Gibberella sp. F75: gene cloning, expression, and enzymatic characterization. Appl Microbiol Biotechnol 83:875–884
Church FC, Mayers SP (1980) Alpha-galactosidase from Pichia guillermondii. Mycologia 72:278–287
Comfort DA, Bobrov KS, Ivanen DR, Shabalin KA, Harris JM, Kulminskaya AA, Brumer H, Kelly RM (2007) Biochemical analysis of Thermotoga maritima GH36 alpha-galactosidase (TmGalA) confirms the mechanistic commonality of clan GH-D glycoside hydrolases. Biochemistry 46:3319–3330
David M, Gabriel M, Kopecká M (2007) Cytoskeletal structures, ultrastructural characteristics and the capsule of the basidiomycetous yeast Cryptococcus laurentii. Antonie Van Leeuwenhoek 92:29–36
Dey PM, Patel S, Brownleader MD (1993) Induction of α-galactosidase in Penicillum ochrochloron by guar (Cyamopsis tetragonobola) gum. Biotechnol Appl Biochem 17:361–371
Dudíková J, Kolarova N (2006) Extracellular polysaccharides produced by acapsular mutant of Cryptococcus laurentii. Chem Pap 60:69–70
Eneyskaya EV, Golubev AM, Kachurin AM, Saveljev AN, Neustroev KN (1998) Transglycosylation activity of α-D-galactosidase from Trichoderma reesei. An investigation of the active site. Carbohydr Res 305:83–91
Flórez IG, Lazo PS, Ochoa AG, Gascón S (1981) The specificity of induction of α-galactosidase from Saccharomyces carlsbergensis. Biochim Biophys Acta 674:71–77
Franc V, Šebela M, Řehulka P, Končitíková R, Lenobel R, Madzak C, Kopečný D (2012) Analysis of N-glycosylation in maize cytokinin oxidase/dehydrogenase 1 using a manual microgradient chromatographic separation coupled offline to MALDI-TOF/TOF mass spectrometry. J Proteomics 75:4027–4037
Frohman MA, Dush MK, Martin GR (1988) Rapid production of full-lenght cDNAs from rare transcripts: Amplification using a single gene-specific oligonucleotide primer. Proc Natl Acad Sci USA 85:8998–9002
Golubev WI (1991) Capsules. In: Rose AH, Harrison JS (eds) The yeasts, vol 4, 2nd edn. Academic, London, pp 175–197
Hashimoto H, Katayama C, Goto M, Kitahata S (1993) Purification and some properties of α-galactosidase from Candida guilliermondii H-404. Biosci Biotech Biochem 57:372–378
Hashimoto H, Katayama C, Goto M, Okinaga T, Kitahata S (1995) Transgalactosylation catalyzed by α-galactosidase from Candida guilliermondii H-404. Biosci Biotech Biochem 59:619–623
Ioannou YA, Zeidner KM, Grace ME, Desnick RJ (1998) Human α-galactosidase A: glycosylation site 3 is essential for enzyme solubility. Biochem J 332:789–797
Koizumi K, Tanimoto T, Okada Y, Hara K, Hashimoto H, Kitahata S (1995) Isolation and characterization of novel heterogeneous branched cyclomalto-oligosaccharides (cyclodextrins) produced by transgalactosylation with α-galactosidase from coffee bean. Carbohydr Res 278:129–142
Kurakake M, Moriyama Y, Sunouchi R, Katani S (2011) Enzymatic properties and transglycosylation of α-galactosidase from Penicillium oxalicum SO. Food Chem 126:177–182
Laemmli UK (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685
Liu XZ, Wang QM, Göker M, Groenewald M, Kachalkin AV, Lumbsch HT, Millanes AM, Wedin M, Yurkov AM, Boekhout T, Bai FY (2015) Towards and integrated phylogenetic classification of the Tremellomycetes. Stud Mycol 81:85–147
Loftus BJ, Fung E, Roncaglia P, Rowley D, Amedeo P, Bruno D, Vamathevan J, Miranda M, Anderson IJ, Fraser JA et al (2005) The genome of the basidiomycetous yeast and human pathogen Cryptococcus neoformans. Science 307:1321–1324
Maceková D, Farkaš V, Kishida E, Takeo K (2006) Ecto-glycanases and metabolic stability of the capsule in Cryptococcus neoformans. J Basic Microbiol 46:470–479
Margolles-Clark E, Tenkanen M, Luonteri E, Pentillä M (1996) Three α-galactosidase genes of Trichoderma reesei cloned by expression in yeasts. Eur J Biochem 240:104–111
Mastihuba V, Mastihubová M, Belák M, Dudíková J, Karnišová-Potocká E, Petruš L (2017) Preparation of α-galactooligoglycosides by cell walls from Cryptococcus laurentii using a novel α-galactosyl donor. Tetrahedron: Asymmetry. https://doi.org/10.1016/j.tetasy.2017.07.007
Merceron R, Foucault M, Haser R, Mattes R, Watzlawick H, Gouet P (2012) The molecular mechanism of thermostable α-galactosidases AgaA and AgaB explained by X-ray crystallography and mutational studies. J Biol Chem 287:39642–39652
Mészárosová Cs, Zaragoza O (2012) Correlation between cryptococcal capsule size and structure to activities of surface glycosidases of Cryptococcus laurentii. Bull ČSSM 1:5–12
Mészárosová Cs, Kolarova N, Vadkertiová R, Stratilová E (2012) An induction of Cryptococcus laurentii α-galactosidase. Chem Pap 66:806–813
Moyrand F, Fontaine T, Janbon G (2007) Systematic capsule gene disruption reveals the central role of galactose metabolism on Cryptococcus neoformance virulence. Mol Microbiol 64:771–781
Pirofski L, Casadevall A (1996) Antibody immunity to Cryptococcus neoformans: paradigm for antibody immunity to the fungi? Zentralbl Bacteriol 284:475–495
Puchart V, Biely P (2005) Glycosylation of internal sugar residues of oligosacharides catalyzed by α-galactosidase from Aspergillus fumigatus. Biochim Biophys Acta 1726:206–216
Puchart V, Vršanská M, Bhat MK, Biely P (2000) Purification and characterisation of α-galactosidase from a thermophilic fungus Thermomyces lanuginosus. Biochim Biophys Acta 1524:27–37
Radola BJ (1980) Ultrathin-layer isoelectric focusing in 50–100 μm polyacrylamide gels on silanized plates or polyester films. Electrophoresis 1:43–56
Rappsilber J, Ishihama Y, Mann M (2003) Stop and go extraction tips for matrix-assisted laser desorption/ionization, nanoelectrospray, and LC/MS sample pretreatment in proteomics. Anal Chem 75:663–670
Ríos S, Pedregosa AM, Monistrol IF, Laborda F (1993) Purification and molecular properties of an α-galactosidase synthesized and secreted by Aspergillus nidulans. FEMS Microbiol Lett 112:35–42
Rose TM, Henikoff JG, Henikoff S (2003) CODEHOP (COnsensus-DEgenerate Hybrid Oligonucleotide Primer) PCR primer design. Nucleic Acids Res 31:3763–3766
Saveľev AN, Ibatyllin FM, Eneyskaya EV, Kachurin AM, Neustroev KN (1996) Enzymatic properties of α-galactosidase from Trichoderma reesei. Carbohydr Res 296:261–273
Scigelová M, Crout DHG (2000) Purification of α-galactosidase from Aspergillus niger for application in the synthesis of complex oligosaccharides. J Mol Catal B: Enzym 8:175–181
Shevchenko A, Tomas H, Havlis J, Olsen JV, Mann M (2006) In-gel digestion for mass spectrometric characterization of proteins and proteomes. Nat Protocols 1:2856–2860
Spangenberg P, André C, Dion M, Rabiller C, Mattes R (2000) Comparative study of new α-galactosidases in transglycosylation reactions. Carbohydr Res 329:65–73
Spangenberg P, André C, Dion M, Langlois V, Rabiller C (2002) α-Galactosyl fluoride in transfer reactions mediated by the green coffee beans α-galactosidase in ice. Carbohydr Res 337:221–228
Stratilová E, Dzúrová M, Malovíková A, Omelková J (2005) Oligogalacturonate hydrolase from carrot roots. Z Naturforsch 60:899–905
Van Laere KMJ, Hartemink R, Beldman G, Pitson S, Dijkema C, Schols HA, Voragen AGJ (1999) Transglycosidase activity of Bifidobacterium adolescentis DSM 20083 α-galactosidase. Appl Microbiol Biotechnol 52:681–688
Zapater IG, Ullah AH, Wodzinski RJ (1990) Extracellular α-galactosidase (E.C.3.2.1.22) from Aspergillus ficuum NRRL 3135, purification and characterisation. Prep Biochem 20:263–296
Zeilinger S, Kristufek D, Arisan-Atak I, Hodits R, Kubicek CP (1993) Conditions of formation, purification and characterization of an α-galactosidase of Trichoderma reseii RUT C-30. Appl Environm Microbiol 59:1347–1353
Acknowledgements
This work was supported by the Scientific Grant Agency of Ministry of Education, Science, Research and Sport of Slovak Republic and Slovak Academy of Sciences (VEGA 2/0023/14 and VEGA 2/0058/16), and by the Ministry of Defence of the Czech Republic - long-term organization development plan of the Faculty of Military Health Sciences, University of Defence.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Stratilová, B., Klaudiny, J., Řehulka, P. et al. Characterization of a long-chain α-galactosidase from Papiliotrema flavescens. World J Microbiol Biotechnol 34, 19 (2018). https://doi.org/10.1007/s11274-017-2403-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11274-017-2403-6