
Abstract
A novel α-galactosidase gene (aga-F75) from Gibberella sp. F75 was cloned and expressed in Escherichia coli. The gene codes for a protein of 744 amino acids with a 24-residue putative signal peptide and a calculated molecular mass of 82.94 kDa. The native structure of the recombinant Aga-F75 was estimated to be a trimer or tetramer. The deduced amino acid sequence showed highest identity (69%) with an α-galactosidase from Hypocrea jecorina (Trichoderma reesei), a member of the glycoside hydrolase family 36. Purified recombinant Aga-F75 was optimally active at 60°C and pH 4.0 and was stable at pH 3.0–12.0. The enzyme exhibited broad substrate specificity and substantial resistance to neutral and alkaline proteases. The enzyme K m values using pNPG, melibiose, stachyose, and raffinose as substrates were 1.06, 1.75, 54.26, and 8.23 mM, respectively. Compared with the commercial α-galactosidase (Aga-A) from Aspergillus niger var. AETL and a protease-resistant α-galactosidase (Aga-F78) from Rhizopus sp. F78, Aga-F75 released 1.4- and 4.9-fold more galactose from soybean meal alone, respectively, and 292.5- and 8.6-fold more galactose from soybean meal in the presence of trypsin, respectively. The pH and thermal stability and hydrolytic activity of Aga-F75 make it potentially useful in the food and feed industries.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
α-Galactosidases (α-d-galactoside galactohydrolase; EC 3.2.1.22) catalyze the removal of α-linked terminal nonreducing galactose residues from different substrates (Dey and Pridham 1972), and some α-galactosidases catalyze transgalactosylation, especially at high substrate concentrations (Puchart and Biely 2005). Sources of α-galactosidase include microorganisms (Ademark et al. 2001; Baik et al. 2000; Brouns et al. 2006; Carrera-Silva et al. 2006; Duffaud et al. 1997), plants (Carmi et al. 2003; Fujimoto et al. 2003), and mammals (Hamers et al. 1977). On the basis of amino acid sequence similarity, α-galactosidases have been classified into the glycoside hydrolase (GH) families 4, 27, 36, and 57 in the CAZy database (http://www.cazy.org/fam/acc_GH.html). A majority of the known α-galactosidases belong to the GH-27 and GH-36 families and share a common conserved consensus sequence ([LIVMFY]-x(2)-[LIVMFY]-x-[LIVM]-D-[DS]-x-[WY]), implying a similarity of the reaction mechanism or active site (Fridjonsson et al. 1999). Most of the known eukaryotic α-galactosidases have been classified into the GH-27 family with an average molecular mass of about 50 kDa, which is smaller than that of most GH-36 α-galactosidases (about 80 kDa; Henrissat 1991; Henrissat and Bairoch 1993). GH-36 α-galactosidases are mainly from bacteria, although some are from fungi and plants, such as the α-galactosidases from Trichoderma reesei (Margolles-Clark et al. 1996), Penicillium sp. F63 (Mi et al. 2007), and Absidia corymbifera IFO 8084 (Baik et al. 2000). The latter α-galactosidases resemble those from bacteria; they form multimeric complexes comprised of subunits with high molecular mass. The biochemical and physical properties and substrate specificities of α-galactosidases from different organisms have been extensively studied and a presumed catalytic domain comprised of a (β/α)8 barrel topology has been proposed based on crystallographic data (Foucault et al. 2006; Fujimoto et al. 2003; Garman and Garboczi 2004; Golubev et al. 2004); however, the relationship between structure and function remains unknown.
α-Galactosidases are widely applied in many industries. In food and feed industries, α-galactosidases are added to eliminate α-d-galactosides (mainly raffinose and stachyose), which are antinutritional factors in legume seeds, and to improve digestibility alone or in combination with other enzymes (Ghazi et al. 2003; Graham et al. 2002; Igbasan et al. 1997; Puchal 1999). Furthermore, α-galactosidases are used in the sugar-making industry to improve the crystallization of sucrose (Linden 1982), in the pulp and paper industry to enhance pulp bleaching (Clarke et al. 2000), in medical treatments to catalyze erythrocyte conversion (Zhang et al. 2007) and to cure Fabry disease (Tsuboi 2007), and in scientific research as a selection marker using the substrate X-α-gal (Jeong et al. 2006).
The α-galactosidase activity from Gibberella fungus has been identified, but no detailed characterization or sequence data were reported (Thippeswamy and Mulimani 2002). Thus, this is the first report to describe the molecular cloning and expression of an α-galactosidase gene from Gibberella. The superior properties of the recombinant enzyme, such as strong resistance to protease and high hydrolytic activity, may be advantageous for many biotechnological applications.
Materials and methods
Microorganism isolation
The fungal strain F75 was isolated from the experimental plot soil, Beijing, China. The medium for isolation consisted of 0.4% (w/v) K2HPO4, 0.28% (w/v) (NH4)2SO4, 0.12% (w/v) CaCl2, 0.12% (w/v) urea, 0.12% (w/v) MgSO4, and 3% (w/v) of soybean meal extract. The taxon of strain F75 was identified based on rDNA sequence in the internal transcribed spacer (ITS) regions (Innis et al. 1990).
Strains, plasmids, enzymes, and reagents
Escherichia coli strain TOP10 and vector pEASY-T3 were purchased from TransGen Biotech (Beijing, China). E. coli strain BL21 and vector pET-22b(+) were purchased from Novagen (Darmstadt, Germany). Restriction endonucleases and pfu DNA polymerase were purchased from Takara (Kyotanabe, Japan) and T4 DNA ligase was purchased from Invitrogen (Carlsbad, CA, USA). Substrates p-nitrophenyl-α-d-galactopyranoside (pNPG), 6-bromo-2-naphthyl-α-d-galactopyranoside, melibiose and stachyose, and proteinases including trypsin, α-chymotrypsin, subtilisin A, collagenase, and proteinase K were purchased from Sigma (St. Louis, MO, USA). Proleather was obtained from Amano Enzyme Inc. (Nagoya, Japan) and alkaline protease (from Bacillus pumilus) was purified as Qiu and Cheng described (Qiu and Cheng 1984). d-Galactose and raffinose were purchased from Amresco (Solon, OH, USA), isopropyl-β-d-thiogalactopyranoside (IPTG) was from Calbiochem (Darmstadt, Germany), and low and high molecular weight calibration kits were from GE Healthcare (Piscataway, NJ, USA). All other chemicals were of analytical grade.
Cloning of the full-length genomic DNA of aga-F75
The fungal strain F75 was cultivated in potato dextrose broth medium at 30°C for 48 h. The cetyl trimethylammonium bromide (CTAB) method was used to extract genomic DNA from the mycelia (Graham et al. 1994). A pair of degenerate primers, P4 (5′-GAYGAYGGNTGGTTYGGN-3′) and Pr8 (5′-GACCATYTCNGGYTCNAMCC-3′; Y represents C or T, M represents A or C, and N represents A, T, C, or G), was designed based on the conserved motifs of fungal GH-36 α-galactosidases. The genomic DNA of strain F75 was used as a template for polymerase chain reaction (PCR) amplification. Touchdown PCR conditions were as follows: 5 min at 95°C, followed by 30 cycles of 94°C for 30 s, 58°C to 48°C (decreasing 1°C each cycle and holding at 48°C for remaining cycles ) for 30 s, and 72°C for 1 min, and a final extension at 72°C for 10 min. The PCR products were purified and ligated into pEASY-T3 for sequencing. The sequence of the DNA fragment was analyzed by BLAST (http://www.ncbi.nlm.nih.gov/BLAST) and the following six specific primers were designed based on the sequence: F78-sp1 (5′-GACGGTTGGTTCAAAGGACGAAAGTCAG-3′), F78-sp2 (5′-GATTGGTATCCAGACAAGTCAAAGTTTCCAC-3′), F78-sp3 (5′-TTGGGTATGAAATTTGGCTTGTGGGTGG-3′), F78-rsp1 (5′-CCACAAGCCAAATTTCATACCCAATTCATG-3′), F78-rsp2 (5′-AAGGGTCCTAAACCTTGTGGAAACTTTGAC-3′), and F78-rsp3 (5′-TGTCTGACTTTCGTCCTTTGAACCAACC-3′). The 5′ and 3′ flanking regions of the DNA fragment were obtained using a thermal asymmetric interlaced (TAIL)-PCR protocol (Liu and Whittier 1995) with an annealing temperature of 58°C and sequenced. These nucleotide sequences were assembled and analyzed using Vector NTI 7.0 software and Genomescan (http://genes.mit.edu/genomescan.html).
Cloning of the full-length complementary DNA of aga-F75
Strain F75 was grown in α-galactosidase-inducing medium containing 3% (w/v) soybean meal extract as a carbon source at 30°C on a rotary shaker at 250 rpm for 5 days (Cao et al. 2007). Mycelia were collected, frozen under liquid nitrogen, and immediately ground into fine powder. Total RNA was isolated and purified from mycelia using the RNeasy Plant Mini kit (Qiagen, Hilden, Germany). Full-length α-galactosidase cDNA was obtained from total RNA by reverse transcription (RT)-PCR amplification using a SuperScript III First-Strand Synthesis System for RT-PCR kit (Invitrogen); primer oligo(dT) was used to amplify the first strand and primers F75up1 (5′-AAGATGGTGCTTGTTACATTGAGGG-3′) and F75AG4 (5′-GAACTATTGCTTCTCAATCATCAAG-3′) were used to amplify the second strand. The cDNA containing the α-galactosidase gene, aga-F75, was analyzed using Vector NTI 7.0 software. The signal peptide sequence of the α-galactosidase from strain F75 was predicted using SignalP (http://www.cbs.dtu.dk/services/SignalP/). Homology searches in GenBank were performed using the BLAST server. Multiple alignments of protein sequences were accomplished using Vector NTI 7.0 software.
Expression of aga-F75 in E. coli
To construct the aga-F75 plasmid for expression in E. coli, a gene fragment containing aga-F75 was amplified by PCR using the primers F75AG-ES (5′-CTAGGATCCGATGGTGCTTGTTACATTGAG-3′, BamHI site underlined) and F78AG-YA (5′-CTAGCGGCCGCCTATTGCTTCTCAATCATC-3′, NotI site underlined). The PCR conditions were as follows: 95°C for 5 min, 30 cycles of 30 s at 94°C, 30 s at 62°C, and 2 min 30 s at 72°C, followed by a final extension at 72°C for 10 min. The PCR products were digested using BamHI and NotI and cloned into pET-22b(+) using T4 DNA ligase. The resulting construct, pET22b(+)/aga-F75, was then transformed into E. coli BL21 (DE3) cells. The strain harboring aga-F75 was grown to an OD600 of 0.6–0.8 in Luria–Bertani (LB) medium containing 100 μg/ml ampicillin at 37°C. The culture was then diluted 1:25 into fresh LB medium and cultivated at 37°C for 2–3 h until an OD600 of ∼0.6 was reached. After induction with 1 mM IPTG, the subculture was grown at 18°C for 12 h (Cao et al. 2008) with shaking at 180 rpm.
Enzyme assay
The enzyme assay methods varied according to the substrate. The pNPG method was used as described (Cao et al. 2007), in which 1 U of α-galactosidase activity was defined as the amount of enzyme that released 1 μmol of pNP from pNPG per min at 37°C. The dinitrosalicylic acid (DNS) method was performed according to Xu et al. (2004) with a little modification. McIlvaine buffer at pH 4.0 and reaction temperature at 37°C were used. One unit of α-galactosidase activity was defined as the amount of enzyme that released 1 μmol of reducing sugar per minute at 37°C. The glucose oxidase–peroxidase (GOD–POD) method was carried out using a GOD–POD kit (Biosino Bio-Techology & Science Inc, Beijing, China) according to the manufacturer’s instructions; 1 U of α-galactosidase activity was defined as the amount of enzyme that released 1 μmol of glucose per minute at 37°C.
Purification of the recombinant enzyme (r-Aga-F75)
To purify the recombinant His6-tagged enzyme, cells were harvested by centrifugation at 12,000×g for 5 min at 4°C, washed, and resuspended in sterilized ice-cold buffer A (20 mM Tris-HCl, pH 7.6 containing 500 mM NaCl) and stored at −20°C before use. After thawing, the cells were disrupted by 3 s sonication at 100 W on ice for several times and centrifuged at 12,000×g for 15 min at 4°C. Then 5 ml of the supernatant (crude enzyme) containing 151.62 mg protein was applied to a 1-ml Ni-NTA chelating column (Qiagen, Hilden, Germany) previously equilibrated with buffer A. The protein was eluted using an imidazole step gradient (0, 20, 40, 60, 80, 100, 200, and 300 mM) in 20 mM Tris-HCl (pH 7.6) containing 500 mM NaCl and 10% (w/v) glycerol. Fractions containing enzyme activity were determined using the pNPG method. The protein concentration at each step was determined using the Bradford method with bovine serum albumin as the standard (Bradford 1976).
Electrophoresis analysis
Protein induction and purification samples were subjected to 12% (w/v) sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) analysis in a vertical slab gel apparatus at pH 8.3. Protein bands were detected with Coomassie brilliant blue R-250. E. coli BL21 (DE3) containing pET-22b(+) was compared to that containing pET22b(+)/aga-F75 to verify α-galactosidase expression.
To determine the native molecular weight of r-Aga-F75, the purified protein subjected to nondenaturing gradient (4–12%, w/v) PAGE was analyzed by hydrolyzed 6-bromo-2-naphthyl-α-d-galactopyranoside and then stained with Fast blue B salt (Fluka, Buchs, Switzerland; Cao et al. 2007).
Biochemical characterization
The pH optimum of purified r-Aga-F75 α-galactosidase activity was determined at 37°C in 0.1 M McIlvaine buffers at pH 2.0–8.0 and in 0.1 M glycine-NaOH buffers at pH 9.0–11.0. The pH stability of r-Aga-F75 was determined by measuring the residual activity at the optimal pH and 37°C after pre-incubating the enzyme in the same buffers (α-galactosidase/buffer = 1:19, w/w) over a pH range of 2.0–11.0 at 37°C for 30 min.
The optimal temperature for α-galactosidase activity was determined in 0.1 M McIlvaine buffer at the optimal pH from 0°C to 70°C. Thermal stability of the purified recombinant enzyme was determined by measuring the residual enzyme activity under standard conditions (pH 4.0, 37°C for 5 min) after incubation of the enzyme at 50°C or 60°C for 2, 5, 10, 15, 20, and 30 min.
The effect of metal ions and chemical reagents on the activity of purified r-Aga-F75 was determined by adding 1 or 5 mM of various metal salts (NaCl, KCl, CaCl2, LiCl, CoCl2, CrCl3, NiSO4, CuSO4, MgSO4, FeCl3, MnSO4, ZnCl2, PbCl2, AgNO3, or HgCl2) or reagents (β-Met, SDS, ethylenediaminetetraacetic acid (EDTA), CTAB, or Triton X-100) to the assay system. The system without any additive was used as a control.
Resistance of Aga-F75 to proteases including trypsin, α-chymotrypsin, proteinase K, subtilisin A, collagenase, proleather, and alkaline protease was determined as described (Cao et al. 2007). All enzyme activity assays in biochemical characterization were verified in triplicate using the pNPG method.
Substrate specificity and kinetic parameters
The substrate specificity and kinetic parameters of r-Aga-F75 were determined by measuring the enzyme activity after incubation in 0.1 M McIlvaine buffer (pH 4.0) containing pNPG (0.0125–1 mM), melibiose (1–10 mM), raffinose (1–10 mM), and stachyose (1–10 mM) at 37°C for 5, 15, 20, and 20 min (within the first-order reaction period). Enzyme activity was assayed using the pNPG method (pNPG), the GOD–POD method (melibiose), and the DNS method (raffinose and stachyose). The apparent Michaelis constant (K m) and maximal reaction rate (V max) were calculated from the Lineweaver–Burk plot. Each value represents an average of three independent triplicates.
Assay of hydrolytic activity
The hydrolytic activity of purified r-Aga-F75 was determined by measuring the amount of galactose released from oligosaccharides including melibiose, raffinose, and stachyose. Oligosaccharides were dissolved in 0.1 M McIlvaine buffer (pH 4.0) to 1 mg/ml and incubated in the presence of 1 or 3 U/ml r-Aga-F75 at 37°C for 24 h (Luonteri et al. 1998b). The amount of released galactose in the samples was detected using a Dionex system (Dionex, Sunnyvale, CA, USA) containing a high-performance anion-exchange chromatography column (4 × 250 mm) equipped with a CARBOPAC PA10 (HPAEC-PAD) and a 25-μl sample volume in 18 mM NaOH and 50 mM borate solution. The buffer flow rate was 1 ml/min. Two controls were performed under the same conditions without addition of the enzyme or with boiling inactivated enzyme. The results of the controls were almost the same.
The synergistic effect of Aga-F75 (1 U/ml) and trypsin (10:1 (w/w) ratio of α-galactosidase/trypsin) on hydrolysis of 5% (w/v) soybean powder was performed in 0.1 M McIlvaine buffer (pH 4.0) at 37°C for 24 h (Eneyskaya et al. 1999). The amount of released galactose was determined using HPAEC-PAD as described above. Aga-F75 activity was compared to two α-galactosidases: protease-resistant Aga-F78 from Rhizopus sp. F78 ACCC 30795 (Cao et al. 2007) and commercial Aga-A from Aspergillus niger var. AETL (Maharashtra, India).
Nucleotide sequence accession numbers
The nucleotide sequence for the Gibberella sp. F75 ITS region and the α-galactosidase gene (aga-F75) were deposited in the GenBank database under accession numbers FJ360899 and FJ392036, respectively.
Results
Identification of the fungal strain
The rDNA sequence of ITS1-5.8S rRNA–IST2 (536 bp) amplified by PCR from strain F75 had the highest nucleotide identity (100%) with that from Fusarium proliferatum (Gibberella fujikuroi complex; Genbank accession no. FJ040179) and Gibberella moniliformis strain Fm-X.1.7-030527-31 (Genbank accession no. EU364864). Based on the ITS sequence analysis, F75 was identified as a member of Gibberella and deposited in the China General Microbiological Culture Collection Center (CGMCC) under CGMCC 2499.
Molecular cloning and sequence analysis
A partial α-galactosidase gene (160–180 bp) was amplified by PCR using degenerate primers (P4 and Pr8) derived from the conserved amino acid sequences of the fungal GH-36 α-galactosidases, and the PCR product was sequenced and analyzed. The sequence of this fragment exhibited 74% identity with α-galactosidase C from Aspergillus clavatus NRRL 1 (Genbank accession no. XP_001275168) and 69% identity with Agl1 from Penicillium sp. F63 (Genbank accession no. ABC70181), suggesting the fragment was derived from the α-galactosidase coding sequence. Many PCR products from the 5′ and 3′ flanking regions amplified by TAIL-PCR were isolated, sequenced, and assembled with the partial gene fragment. The resulting assembled sequence was ∼3,000 bp, and one open reading frame (ORF) consisting of 2,339 bp was identified within it.
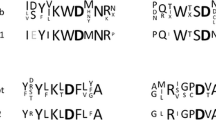
The full-length cDNA sequence obtained via RT-PCR contained one open reading frame of 2,232 bp. Compared to the ORF, two introns, 86–138 and 746–796 bp, were identified to interrupt the α-galactosidase coding sequence. The putative protein was 744 residues in length with a calculated pI of 5.10 and a calculated molecular mass of 82.94 kDa. SignalP analysis indicated the presence of an N-terminal signal peptide at residues 1–24. The deduced amino acid sequence of Aga-F75 had higher identities with fungal GH-36 α-galactosidases from Hypocrea jecorina (T. reesei; Genbank accession no. CAA93245; 69%), Penicillium sp. F63 (Genbank accession no. ABC70181; 63%), and A. niger (Genbank accession no. CAB63901; 63% identity) than with the bacterial GH-36 α-galactosidases from G. stearothermophilus (Genbank accession no. AAG49420; 43% identity) and E. coli (Genbank accession no. AAA24497; 22% identity; Fig. 1) and had lower identities (no more than 10%) with GH-27 α-galactosidases (data not shown). This result suggests that Aga-F75 is a novel fungal α-galactosidase belonging to GH-36 family.

Amino acid sequence alignment of Aga-F75 with other GH-36 α-galactosidases. All sequences are indicated with species initials, including G. sp. F75 (Gibberella sp. F75, FJ392036), H. jecorina (Hypocrea jecorina RUTC-30, CAA93245), P. sp F63 (Penicillium sp. F63, ABC70181), A. niger (Aspergillus niger CBS 120.49/N400, CAB63901), G. stearothermophilus (Geobacillus stearothermophilus KVE39, AAG49420), and E. coli (Escherichia coli K12, AAA24497). Hyphens indicate gaps. Identical amino acid residues at the same position are shaded
Expression and purification of Aga-F75
The recombinant gene encoding the Gibberella α-galactosidase containing a C-terminal His6-tag was expressed in E. coli BL21 (DE3) cells. The crude enzyme activity in cells was 1.42 U/ml. The recombinant Aga-F75 was purified using single-step affinity chromatography on a Ni-NTA column. After 1,037-fold purification (47.61% recovery), the specific activity of r-Aga-F75 was 48.33, 3.17, 0.26, and 11.90 U/mg for the substrates pNPG, melibiose, raffinose, and stachyose, respectively.
Electrophoresis analysis
Purified r-Aga-F75 migrated as a single band of ∼82 kDa on a 12% (w/v) SDS-PAGE, corresponding to the calculated molecular weight (Fig. 2a). Native gradient gel electrophoresis of the purified r-Aga-F75 (Fig. 2 b) demonstrated that the purified protein migrated as two bands of ∼220 and ∼320 kDa; this was verified using two staining methods and suggested that native r-Aga-F75 might be a trimer or tetramer.

Electrophoretic analysis of recombinant Aga-F75. a SDS-PAGE analysis of Aga-F75 expressed in E. coli BL21 (DE3). Lanes: M low molecular weight markers, 1 E. coli pET-22b(+) cell lysate following IPTG induction (negative control), 2 pET-22b(+)/aga-F75 cell lysate following IPTG induction, 3 purified Aga-F75 after filtration through Ni-NTA chelating column. b Nondenaturing gradient PAGE of purified Aga-F75. Lanes: M high molecular weight markers, 1 purified Aga-F75 stained with Coomassie brilliant blue, 2 the purified Aga-F75 hydrolyzed 6-bromo-2-naphthyl-α-d-galactopyranoside was stained by Fast blue B salt. The arrows indicate the two multimeric proteins migrating at ∼220 and ∼320 kDa
Properties of purified r-Aga-F75
The optimal pH of r-Aga-F75 was 4.0 (Fig. 3a), and the enzyme retained ≥80% activity at pH 3.0–5.0. Purified r-Aga-F75 was stable over the pH range of 3.0–12.0, maintaining ≥80% activity. The optimal temperature of Aga-F75 activity was 60°C at pH 4.0, and the enzyme exhibited about 60% of its activity at 37°C (Fig. 3b). In the thermostability assay, no enzyme activity was lost after pretreatment at 50°C for 30 min, while 98.95, 83.30, 53.29, 30.53, 10.79, and 4.61% activity was retained after pretreatment at 60°C under the same conditions for 2, 5, 10, 15, 20, and 30 min, respectively.

Characterization of purified r-Aga-F75. a Effect of pH on r-Aga-F75 activity. The enzyme activity assay was performed at 37°C in 0.1 M McIlvaine buffer at pH 2.0–8.0 and in 0.1 M glycine-NaOH at pH 9.0–11.0. b Effect of temperature on Aga-F75 activity. The enzyme activity was measured in 0.1 M McIlvaine buffer (pH 4.0) at 0–70°C. Data represent the means ± standard deviation of triplicate measurements
The activity of r-Aga-F75 was evaluated in the presence of 1 and 5 mM of various metal ions and chemical reagents. The activity of r-Aga-F75 was significantly enhanced (P < 0.05) in the presence of 5 mM Pb2+ and strongly inhibited by 1 and 5 mM Cu2+, Fe3+, Hg2+, and SDS. Partial inhibition was observed in the presence of 1 mM Triton or 1 and 5 mM CTAB. Certain metal ions, such as Ca2+, Co2+, Cr3+, Ni2+, Mn2+, and Zn2+, were partially inhibitory at the higher concentration (5 mM). The addition of Na+, K+, Li+, Mg2+, Ag+, EDTA, and β-Met had little or no effect on the activity.
r-Aga-F75 was strongly resistant to trypsin, α-chymotrypsin, subtilisin A, and collagenase digestion. After treatment with these proteases at 37°C for 30 min, the enzyme retained almost 100% activity. Proteinase K, proleather, and alkaline protease partially inhibited Aga-F75 by less than 50%.
Substrate specificity and kinetic parameters
Purified r-Aga-F75 exhibited broad substrate specificity. Calculated from the Lineweaver-Burk plot, the K m values of Aga-F75 using the substrates pNPG, melibiose, raffinose, and stachyose were 1.06, 1.75, 54.26, and 8.23 mM, respectively, and the V max values were 0.19, 0.21, 0.85, and 0.70 U/mg, respectively. These results indicated that Aga-F75 had higher affinity for the synthetic substrate pNPG and the natural substrate melibiose, but the reaction rates were higher in the presence of the substrates raffinose and stachyose.
Hydrolytic activity of Aga-F75
Purified r-Aga-F75 efficiently catalyzed the hydrolysis of natural galacto-oligosaccharides. After treatment with both 1 and 3 U/ml of Aga-F75, 23.06 and 24.75%, 32.36 and 46.06%, and 25.40 and 32.60% of galactose was released from the substrates melibiose, raffinose, and stachyose, respectively.
The hydrolytic activity of Aga-F75 on soybean meal was compared with that of Aga-F78 and Aga-A in the presence and absence of trypsin (Table 1). When soybean meal was treated with enzyme alone, Aga-F75 released 1.4- and 4.9-fold more galactose than Aga-A and Aga-F78, respectively. When combined with trypsin, galactose release by Aga-F75 was significantly enhanced by 37% and was 292.5- and 8.6-fold more than that released by Aga-A and Aga-F78, respectively.
Discussion
In this study, a novel GH-36 α-galactosidase gene, aga-F75, from Gibberella sp. F75 was obtained using degenerate PCR and TAIL-PCR techniques. Comparison of the chromosomal and complementary DNA of aga-F75 indicated that the gene contains two introns, which are common to many GH-36 α-galactosidases, such as AglC from A. niger and α-galactosidase of A. corymbifera IFO 8084 (Ademark et al. 2001; Baik et al. 2000). However, some fungal GH-36 α-galactosidases lack introns, such as Agl1 from Penicillium sp. F63 (Mi et al. 2007). The presence or absence of introns in fungal GH-36 α-galactosidase genes might be related to the enzyme’s evolutionary history.
Many microorganisms, plants, and animals produce α-galactosidases, often in multiple forms (Dey and Pridham 1972). Most GH-36 α-galactosidases have a molecular mass of ∼80 kDa, calculated from the protein sequence or determined from electrophoretic mobility, and are large proteins comprised of many subunits (often trimers or tetramers) in their native state (Table 2; Baik et al. 2000; Cao et al. 2007; Mi et al. 2007; van den Broek et al. 1999). Heterologous expression does not influence this multimer property. The α-galactosidase of A. corymbifera IFO 8084 expressed in E. coli and Agl1 of Penicillium sp. F63 expressed in Pichia pastoris display a similar multimeric structures property as they do when isolated from their native host (Baik et al. 2000; Mi et al. 2007). In this study, r-Aga-F75 had a relative molecular mass of ∼82 kDa based on SDS-PAGE, corresponding to its calculated molecular mass, and zymogram staining indicated that r-Aga-F75 exists as a trimer or tetramer in the native structure (Fig. 2).
The optimal conditions for activity of some GH-36 α-galactosidases are compared in Table 2. α-Galactosidases from plants are significantly different than those from microorganisms. The α-galactosidase from Cucumis melo (melon) exhibits maximal activity at 30–37°C and pH 7.5–8.5 and is defined as an alkaline α-galactosidase (Carmi et al. 2003). Most α-galactosidases from fungi have pH optima of 4.0–6.0, whereas bacterial α-galactosidases have somewhat higher pH optima between pH 5.0 and 7.5 (Luonteri 1998a). In the present study, Aga-F75 exhibited a pH optimum of 4.0 and was more adaptable to weak acid conditions than other fungal α-galactosidases (Table 2). Some α-galactosidases display maximal activity at high temperature, but are inactive at 37°C. For example, α-galactosidase from Thermomyces lanuginosus CBS 395.62/b has an optimal temperature of 65°C and retains less than 15% activity at 37°C (Rezessy-Szabo et al. 2007); the most thermostable α-galactosidase from Thermotoga neapolitana 5068 has a temperature optimum of 100–105°C and is almost completely inactivated at 50°C (Duffaud et al. 1997). Compared with other known α-galactosidases, Aga-F75 showed high activity under acidic conditions and physiological temperatures, suggesting that it has potential in food or feed applications.
The metal ions Cu2+, Fe3+, Ag+, and Hg2+ inhibit most known α-galactosidases to some degree (Brouns et al. 2006; Fridjonsson et al. 1999; Mi et al. 2007; Rezessy-Szabo et al. 2007; Sripuan et al. 2003; Wong et al. 1986). Ag+ and Hg2+ often severely inactivate α-galactosidases, because they may attack cysteine residues in the active site and interfere with substrate interaction by binding in the catalytic pocket (Fujimoto et al. 2003). In our study, Ag+ did not affect Aga-F75 activity, implying that some distinct catalytic mechanism exists, but further study is needed.
The raffinose family oligosaccharides (RFOs), mainly raffinose and stachyose, are widely distributed in the seeds of many plants, especially in legumes (Vila and Mascarell 1999). RFOs are an antinutritional factor in food and feed, leading to abdominal distension in monogastric animals caused by intestinal microorganism metabolism. If all the RFOs in soybean meal were hydrolyzed, the available energy would increase by 8.0% (Puchal 1999). The addition of exogenous α-galactosidase has been proven to be more economical and efficient for eliminating the raffinose type antinutritional factors (Graham et al. 2002). In this study, Aga-F75 can efficiently hydrolyze many natural substrates, such as melibiose, raffinose, and stachyose, suggesting that it will be a good candidate to eliminate the raffinose type antinutritional factors from soybean meal.
In food and feed industries, food products are often supplemented with proteases and glycoside hydrolases (such as α-galactosidase and xylanase) to make protein-rich materials (for example, soybean meal, lean meat, and fish) more edible and increase the nutritional value (Gupta et al. 2002) and elevate the nutritional value of many materials such as polysaccharides and oligosaccharides (Fontes et al. 1995; Ghazi et al. 2003). Thus, the positive synergic effect of both types of enzymes is very important in nutrition utilization. In this study, three α-galactosidases were added to soybean meal alone or in combination with trypsin. As shown in Table 1, all the tested α-galactosidases hydrolyzed soybean meal efficiently alone, and Aga-F75 produced more galactose compared with Aga-A and Aga-F78. When combined with trypsin, the hydrolytic activity of Aga-F75 was promoted by 37%, whereas that of Aga-F78 and Aga-A were decreased by 21.54% and 99.33%, respectively (Table 1). On the other hand, soybean oligosaccharides are likely responsible for increasing the viscosity of digesta, thereby decreasing their interaction with digestive enzymes (such as trypsin) and interfering with nutrient digestion (Smits and Annison 1996). Dietary supplementation with α-galactosidase may therefore reduce viscosity and improve nutrient digestion (Smiricky et al. 2002). Thus, in the case of Aga-F75, its high hydrolytic activity when combined with trypsin likely reduces the viscosity of the reaction system and makes the trypsin substrates more accessible to trypsin. Furthermore, the action of trypsin likely helps liberate more α-galactosidase substrates. Consequently, more galactose was released by Aga-F75 when was combined with trypsin.
In summary, a novel protease-resistant α-galactosidase, Aga-F75, was cloned, expressed, and characterized in this study. This enzyme efficiently hydrolyzed the raffinose type antinutritional factors in soybean meal, and the hydrolysis was enhanced in the presence of trypsin, suggesting its potential value in the feed or food industry.
References
Ademark P, Vries RP, Hagglund P, Stalbrand H, Visser J (2001) Cloning and characterization of Aspergillus niger encoding an α-galactosidase and a β-mannosidase involved in galactomannan degradation. Eur J Biochem 268:2982–2990
Baik SH, Saito K, Yokota A, Asano K, Tomita F (2000) Molecular cloning and high-level expression in Escherichia coli of fungal α-galactosidase from Abisidia Corymbifera IFO 8084. J Biosci Bioeng 90:168–173
Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72:248–254
Brouns SJ, Smits N, Wu H, Snijders AP, Wright PC, de Vos WM, van der Oost J (2006) Identification of a novel α-galactosidase from the hyperthermophilic archaeon Sulfolobus solfataricus. J Bacteriol 88:2392–2399
Cao Y, Yang P, Shi P, Wang Y, Luo H, Meng K, Zhang Z, Wu N, Yao B, Fan Y (2007) Purification and characterization of a novel protease-resistant α-galactosidase from Rhizopus sp. F78 ACCC 30795. Enzyme Microb Tech 41:835–841
Cao Y, Huang H, Meng K, Yang P, Shi P, Wang Y, Luo H, Zhang Z, Wu N, Yao B (2008) Cloning and functional expression of an α-galactosidase from Yersinia pestis biovar Microtus str. 91001. Biosci Biotech Bioch 72:2203–2205
Carmi N, Zhang G, Petreikov M, Gao Z, Eyal Y, Granot D, Schaffer AA (2003) Cloning and functional expression of alkaline α-galactosidase from melon fruit: similarity to plant SIP proteins uncovers a novel family of plant glycosyl hydrolases. Plant J 33:97–106
Carrera-Silva EA, Silvestroni A, LeBlanc JG, Piard JC, Savoy de Giori G, Sesma F (2006) A thermostable α-galactosidase from Lactobacillus fermentum CRL722: genetic characterization and main properties. Curr Microbiol 53:374–378
Clarke JH, Davidson K, Rixon JE, Halstead JR, Fransen MP, Gilbert HJ, Hazlewood GP (2000) A comparison of enzyme-aided bleaching of softwood paper pulp using combinations of xylanase, mannanase and α-galactosidase. Appl Microbiol Biotechnol 53:661–667
Dey PM, Pridham JB (1972) Biochemistry of α-galactosidases. Adv Enzymol 36:91–130
Duffaud GD, McCutchen CM, Leduc P, Parker KN, Kelly RM (1997) Purification and characterization of extremely thermostable β-mannanase, β-mannosidase, and α-galactosidase from the hyperthermophilic eubacterium Thermotoga neapolitana 5068. Appl Environ Microb 63:169–177
Eneyskaya EV, Kulminskaya AA, Savel’ev AN, Savel’eva NV, Shabalin KA, Neustroev KN (1999) Acid protease from Trichoderma reesei: limited proteolysis of fungal carbohydrases. Appl Microbiol Biotechnol 52:226–231
Fontes CM, Hall J, Hirst BH, Hazlewood GP, Gilbert HJ (1995) The resistance of cellulases and xylanases to proteolytic inactivation. Appl Environ Microb 43:52–57
Foucault M, Watzlawick H, Mattes R, Haser R, Gouet P (2006) Crystallization and preliminary X-ray diffraction studies of two thermostable α-galactosidases from glycoside hydrolase family 36. Acta Cryst 62:100–103
Fridjonsson O, Watzlawick H, Gehweiler A, Rohrhirsch T, Mattes R (1999) Cloning of the gene encoding a novel thermostable α-galactosidase from Thermus brockianus ITI360. Appl Environ Microb 65:3955–3963
Fujimoto Z, Kaneko S, Momma M, Kobayashi H, Mizuno H (2003) Crystal structure of rice α-galactosidase complexed with D-galactose. J Biol Chem 278:20313–20318
Garman SC, Garboczi DN (2004) The molecular defect leading to Fabry disease: structure of human α-galactosidase. J Mol Biol 337:319–335
Ghazi S, Rooke JA, Galbraith H (2003) Improvement of the nutritive value of soybean meal by protease and α-galactosidase treatment in broiler cockerels and broiler chicks. Br Poult Sci 44:410–418
Golubev AM, Nagem RAP, Brandao Neto JR, Neustroev KN, Enryskaya EV, Kulminskaya AA, Shablin KA, Savel’ev AN, Polikarpov I (2004) Crystal structure of α-galactosidase from Trichoderma reesei and its complex with galactose: implications for catalytic mechanism. J Mol Biol 339:413–422
Graham GC, Mayers P, Henry RJ (1994) A simplified method for the preparation of fungal genomic DNA for PCR and RAPD analysis. Biotechniques 16:48–50
Graham KK, Kerley MS, Firman JD, Allee GL (2002) The effect of enzyme treatment of soybean meal on oligosaccharide disappearance and chick growth performance. Poult Sci 81:1014–1019
Gupta R, Beg QK, Lorenz P (2002) Bacterial alkaline proteases: molecular approaches and industrial applications. Appl Environ Microb 59:15–32
Hamers MN, Westerveld A, Khan M, Tager JM (1977) Characterization of α-galactosidase isoenzymes in normal and Fabry human–Chinese hamster somatic cell hybrids. Hum Genet 36:289–297
Henrissat B (1991) A classification of glycosyl hydrolases based on amino acid sequence similarities. Biochem J 280:309–316
Henrissat B, Bairoch A (1993) New families in the classification of glycosyl hydrolases based on amino acid sequence similarities. Biochem J 293:781–788
Igbasan FA, Guenter W, Slominski BA (1997) The effects of pectinase and α-galactosidase supplementation on the nutritive value of peas for broiler chickens. Can J Anim Sci 77:537–539
Innis MA, Gelfand DH, Sninsky JJ, White TJ (1990) PCR protocols: a guide to methods and applications. Academic, New York
Jeong DW, Lee JH, Kim KH, Lee HJ (2006) A food-grade expression/secretion vector for Lactococcus lactis that uses an α-galactosidase gene as a selection marker. Food Microbiol 23:468–475
Linden JC (1982) Immobilized α-D-galactosidase in the sugar beet industry. Enzyme Microb Tech 4:130–136
Liu YG, Whittier RF (1995) Thermal asymmetric interlaced PCR: automatable amplification and sequencing of insert end fragment from PI and YAC clones for chromosome walking. Genomics 25:674–681
Luonteri E (1998a) Fungal α-arabinofuranosidase and α-galactosidase acting on polysaccharides. VTT Biotechnology and Food Research. VTT Publication 371
Luonteri E, Tenkanen M, Viikari L (1998b) Substrate specificities of Penicillium simplicissimum α-galactosidases. Enzyme Microb Tech 22:192–198
Margolles-Clark E, Tenkanen M, Luonteri E, Penttila M (1996) Three α-galactosidase genes of Trichoderma reesei cloned by expression in yeast. Eur J Biochem 240:104–111
Mi S, Meng K, Wang Y, Bai Y, Yuan T, Luo H, Yao B (2007) Molecular cloning and characterization of a novel α-galactosidase gene from Penicillium sp. F63 CGMCC 1669 and expression in Pichia pastoris. Enzyme Microb Tech 40:1373–1380
Puchal F (1999) Role of feed enzyme in poultry nutrition examined. Feedstuffs 11:12–14
Puchart V, Biely P (2005) Glycosylation of internal sugar residues of oligosaccharides catalyzed by α-galactosidase from Aspergillus fumigatus. Biochim Biophys Acta 1726:206–216
Qiu X, Cheng X (1984) A study on purification and some properties of alkaline protease from Bacillus pumilum. Acta Microbiol Sin 24:66–73
Rezessy-Szabo JM, Nguyen QD, HoschkeA Braet C, Hajos G, Claeyssens M (2007) A novel thermostable α-galactosidase from the thermophilic fungus Thermomyces lanuginosus CBS 395.62/b: purification and characterization. Biochim Biophys Acta 1770:55–62
Smiricky MR, Grieshop CM, Albin DM, Wubben JE, Gabert VM, Fahey GC Jr (2002) The influence of soy oligosaccharides on apparent and true ileal amino acid digestibilities and fecal consistency in growing pigs. J Anim Sci 80:2433–2441
Smits CHM, Annison G (1996) Non-starch plant polysaccharides in broiler nutrition—towards a physiologically valid approach to their determination. Worlds Poult Sci J 52:204–221
Sripuan T, Aoki K, Yamamoto K, Tongkao D, Kumagai H (2003) Purification and characterization of thermostable α-galactosidase from Ganoderma lucidum. Biosci Biotechnol Biochem 67:1485–1491
Talbot G, Sygusch J (1990) Purification and characterization of thermostable β-mannanase and α-galactosidase from Bacillus stearothermophilus. Appl Environ Microbiol 56:3505–3510
Thippeswamy S, Mulimani VH (2002) Enzymic degradation of raffinose family oligosaccharides in soymilk by immobilized α-galactosidase from Gibberella fujikuroi. Process Biochem 38:635–640
Tsuboi K (2007) Enzyme replacement therapy in patients with Fabry’s disease. J Int Med Res 35:574–581
van den Broek LAM, Ton J, Verdoes JC, van Laere KMJ, Voragen AGJ, Beldman G (1999) Synthesis of α-galacto-oligosaccharides by a cloned α-galactosidase from Bifidobacterium adolescentis. Biotechnol Lett 21:441–445
Vila B, Mascarell J (1999) α-Galactosides in soybean meal: can enzyme help. Feed Inter 6:24–29
Wong HC, Hu CA, Yeh HL, Su W, Lu HC, Lin CF (1986) Production, purification, and characterization of α-galactosidase from Monascus pilosus. Appl Environ Microb 52:1147–1152
Xu Y, Yao X, Xu S, Li X, Fan Y (2004) An evaluation of two methods for determination of activity in α-galactosidase used in feed. Acta Agri Zhejiangensis 16:349–353
Zhang Y, Gong F, Bao G, Gao H, Ji S, Tan Y, Li S, Li L, Wang Y, Xu H, Xu L, Tian S, Zhang Z, Lv Q, Qiu Y, Bai J, Chen J (2007) B to O erythrocyte conversion by the recombinant α-galactosidase. Chin Med J (Engl) 120:1145–1150
Acknowledgments
This work was supported by the Chinese National High Technology Research and Development Program (863 Program, grant no. 2007AA100601 and no. 2006AA02Z220) and the 948 program of the Ministry of Agriculture (grant no. 2007-Z3).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Cao, Y., Wang, Y., Meng, K. et al. A novel protease-resistant α-galactosidase with high hydrolytic activity from Gibberella sp. F75: gene cloning, expression, and enzymatic characterization. Appl Microbiol Biotechnol 83, 875–884 (2009). https://doi.org/10.1007/s00253-009-1939-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-009-1939-2