
Abstract
Shikimic acid (SA) is an industrially important chiral compound used in diverse commercial applications, and the insufficient supply by isolation from plants and expensive chemical synthesis of SA has increased the importance of developing strategies for SA synthesis. In our previous studies, glycerol was observed to be an effective carbon source for SA accumulation in E. coli DHPYAAS-T7, where the PTS operon (ptsHIcrr) and aroL and aroK genes were inactivated, and the tktA, glk, aroE, aroF fbr, and aroB genes were overexpressed. For further investigation of the effects of glycerol aerobic fermentation on SA accumulation in E. coli BL21(DE3), the glpD, glpK genes and tktA, glk, aroE, aroF fbr, aroB genes were overexpressed simultaneously. The results indicated that SA production was increased 5.6-fold, while the yield was increased 5.3-fold over that of parental strain in shake flasks. It is demonstrated that the aerobic fermentation of glycerol associated with glpD and glpK gene overexpression increased glycerol flux, resulting in higher SA accumulation in E. coli BL21(DE3)-P-DK.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Shikimic acid (SA) is an important chiral compound used as a key chemical building block for the antiviral drug Oseltamivir (Tamiflu) (Johansson and Lidén 2006; Kancharla et al. 2009). SA is an intermediate of the aromatic amino acid biosynthesis pathway found in microbes and plants (Adachi et al. 2006). In recent years, SA has been used in the formulation of many industrially important chemical products, such as indole, aromatic amino acids and alkaloids (Bochkov et al. 2012). Isolation in insufficient quantities from plants and the high demand for SA during the pandemic of swine flu has increased the importance of this compound. Thus, fermentation using microorganisms gained our attention for large-scale production. Compared with extraction from plants, microbial fermentation has a variety of advantages, and E. coli is commonly applied in large-scale fermentation and industrial production. In order to improve shikimic acid’s availability, fermentative production processes from renewable resources like glucose and glycerol present an excellent and even more sustainable alternative to meet the current market volume at a competitive price level.
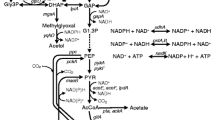
Microbial production of shikimic acid involves the shikimic acid pathway as shown in Fig. 1. Genetic manipulations and metabolic engineering strategies have been applied to E. coli for shikimic acid accumulation, and the four primary approaches are listed as follows. Firstly, the shikimic acid pathway begins with phosphoenolpyruvate (PEP) and erythrose-4-phosphate (E4P), which can be enhanced via metabolic engineering of the glycolytic and pentose phosphate pathway, respectively. Overexpression of the tktA gene (coding for transketolase) results in the increase of shikimic acid yield from 38 to 52 g/l, due to enhancing the concentration of E4P (Knop et al. 2001). Inactivation of the PTS operon (PTS−), expression of non-PTS glucose transporters genes such as glf (coding for glucose facilitators) and glk (coding for glucokinase) in combination with overexpression of tktA gene are reported to increase the production of shikimic acid to 71 g/l (Chandran et al. 2003; Gibson et al. 2001). Secondly, overexpression or integration of the genes for the key enzymes (e.g. aroH, aroF and aroG coding for DAHP (3-deoxy-d-arabinoheptulosonate-7-phosphate) synthase isoenzymes, aroB coding for dehydroquinate synthase, aroE coding for shikimate dehydrogenase) involved in the three rate-limiting step of shikimic acid pathway can increase its production. The expression of a six-gene group (aroG fbr, aroB, aroD, aroE, tktA and zwf), under the control of a synthetic operon constructed in a laboratory-evolved strain bearing simultaneous PTS− and pykF−, has been recently reported to increase the shikimic acid yield on glucose to 42 % mol/mol, which represents the highest reported yield (Rodriguez et al. 2013). Thirdly, preventing shikimic acid transformation to aromatic amino acids by blocking or down-regulating the downstream pathway after the SA production stage is an effective way. Deleting the aroK and aroL genes (coding for shikimate kinase I and II) can block aromatic amino acid pathway flux from central carbon metabolism. For example, in E. coli DHPYAAS-T7, where the PTS operon (ptsHIcrr), aroL and aroK genes are inactivated, and the tktA, glk, aroE, aroF fbr and aroB genes were overexpressed, the shikimic acid production increased to 1,850 mg/l (Chen et al. 2012). Finally, reducing the by-products from the three reversible reactions catalysed by shikimic acid dehydrogenase, DHQ dehydratase and quinic acid dehydrogenase accumulated in SA synthesis. Overexpression of feedback-resistant (fbr) DAHP synthase genes (e.g. aroF fbr and aroG fbr) can avoid the feedback inhibition in DADH transforming (Shinada et al. 1998; Rodriguez et al. 2013).

Pathways involved in glycerol dissimilation and biosynthesis of shikimic acid in E. coli BL21(DE3). Abbreviations: G3P, glycerol-3-phosphate; DHA, dihydroxyacetone; DHAP, dihydroxyacetone phosphate; PEP, phosphoenol pyruvate; DAHP, 3-deoxy-d-arabinoheptulosonate-7-P; DHQ, 3-dehydroquinic acid; DHS, 3-dehydroshikimic acid; SA, shikimic acid; E4P, erythrose-4-P; S3P, shikimate-3-phosphate; PPP, pentose phosphate pathway; EMP, glycolytic pathway; G6P, glucose-6-P; GlaP, galactose permease; Glc, glucose. Relevant reactions are represented by the names of the gene(s) coding for the enzymes: glpK, glycerol kinase; glpD, aerobic glycerol-3-phosphate dehydrogenase; glpABC, anaerobic glycerol-3-phosphate dehydrogenase; glpA, glycerol dehydrogenase; dhaKLM, dihydroxyacetone kinase; tktA, transketolase I; glk, glucokinase; aroF, aroG, aroH, DAHP synthase isoenzymes F, G and H, respectively; aroB, DHQ synthase; aroD, DHQ dehydratase; aroE, shikimic dehydrogenase; aroK, shikimate kinase I; aroL, shikimate kinase II. Broken lines or arrows represent two or more enzymatic reactions or incompletely characterized reactions. Continuous arrows represent unique reactions catalysed by one or more enzymes
In this work, we first investigated the effects of glycerol aerobic fermentation associated with the genes coding for glycerol-3-phosphate dehydrogenase (glpD) and glycerol kinase (glpK) on SA accumulation. The plasmid pET-28a-glpD–glpK was constructed to increase glycerol metabolic flux. Three engineered E. coli strains were constructed: E. coli BL21(DE3)-P-DK, in which the glpD and glpK genes of the glycerol aerobic metabolic pathway and the tktA, glk, aroE, aroF fbr and aroB genes of the aromatic amino acid pathway were overexpressed, E. coli BL21(DE3)-DK, in which the glpD and glpK genes were overexpressed and E. coli BL21(DE3)-P, in which the tktA, glk, aroE, aroF fbr and aroB genes were overexpressed. The data showed that glycerol aerobic fermentation associated with glpD and glpK genes overexpression increased glycerol metabolic flux, resulting in higher SA accumulation in E. coli BL21(DE3)-P-DK (5.6-fold more than E. coli BL21), suggesting that glycerol metabolic flux in E. coli BL21(DE3)-P-DK was very important for SA accumulation and glycerol.
Materials and methods
Strains, plasmids and genetic methods
T4 DNA ligase, restriction enzymes and Taq DNA polymerase were purchased from TaKaRa Biotechnology Co., Ltd. (Dalian, China). DNA molecular weight markers were purchased from Sangon Biotech Co., Ltd. (Shanghai, China). The glpD gene was amplified from E. coli genomic DNA using primers Pf-glpD/Pr-glpD. The PCR products were purified and cloned into the cloning vector pMD19-T. The DNA fragment digested by SacI/HindIII was ligated into the corresponding restriction sites of pET-28a to construct pET-28a-glpD. The ribosomal binding site (RBS AAGAAGGAG) and spacer (sequence between RBS and the starting codon, ATATACC) from the pET-28a vector were used to ligate glpD and glpK. The glpK fragment with the RBS and spacer sequence was amplified using primers Pf-glpK/Pr-glpK and E. coli genomic DNA. The PCR products were purified and inserted into the vector pMD19-T. After digested with HindIII/XhoI, the DNA fragment was cloned into the corresponding restriction sites of pET-28-glpD to construct pET-28a-glpD–glpK. PCR reactions were carried out in total 50 µl reaction solution, containing 5 U of Taq DNA polymerase, 20 ng E. coli genomic DNA, 1.0 μM of each primer, and 250 µM dNTP. The amplifications were performed for 30 cycles: 94 °C for 30 s, 55 °C for 30 s, 72 °C for 1.5 min, and plus an additional 10 min at 72 °C after the cycles. Oligonucleotide primers were synthesized by Genescript Co., Ltd. (Nanjing, Jiangsu, China). All resulting plasmids were sequenced by Genescript Co., Ltd. (Nanjing, Jiangsu, China). The genetically stable strains were stored in 25 % glycerol at −80 °C. Plates were prepared using LB medium containing 2 % agar, kanamycin (50 µg/ml) and chloramphenicol (25 µg/ml).
E. coli BL21(DE3) and E. coli DH5α-T7 were used as expression hosts. The pET-28a was used as expression vectors. The plasmid pAOC-TGEFB was kindly provided by Dr. Fang (Zou et al. 2004). E. coli BL21(DE3)-P and E. coli BL21(DE3)-DK were constructed by transforming plasmid pAOC-TGEFB and pET-28a-glpD–glpK, respectively, into the parental strain E. coli BL21(DE3). The plasmid pAOC-TGEFB and pET-28a-glpD–glpK were transformed into E. coli BL21(DE3) to form E. coli BL21(DE3)-P-DK. E. coli DH5α-T7-P and E. coli DH5α-T7-DK were obtained by transforming plasmid pAOC-TGEFB and pET-28a-glpD–glpK, respectively, into E. coli DH5α-T7. The plasmid pAOC-TGEFB and pET-28a-glpD–glpK were transformed into E. coli DH5α-T7 to form E. coli DH5α-T7-P-DK. E. coli strains were transformed by the calcium chloride methods. All strains, along with the primers and plasmids used in this study are listed in Table 1.
Cultivation conditions
Prior to fermentations, the strains of interest were streaked from their frozen glycerol stocks onto appropriate LB plates consisting of 5 g/l yeast extract, 10 g/l peptone and 5 g/l NaCl and incubated overnight at 37 °C. Three colonies were used to inoculate into 2 ml of LB medium supplemented (when needed) with 25 µg/ml chloramphenicol and 50 µg/ml kanamycin. The culture was incubated overnight at 37 °C. 1 ml of this culture was transferred into 50 ml of modified M9 medium consisting of (/l) 7 g K2HPO4·3H2O, 3 g NaH2PO4·3H2O, 2.5 g·NaCl, 0.5 g MgSO4·7H2O, 10 g peptone, 15 g yeast extract and 20 g glycerol. The filled volume was 50 ml in 250-ml erlenmeyer flask with agitation at 200 rev/min and the modified M9 medium was adjusted to pH 7.8 with 1 M NaOH. IPTG was used with a final concentration of 1 mM to induce the overexpression of genes in the E. coli BL21(DE3) derivative strains and l-arabinose (l-ara) was used with a final concentration of 3 g/l to induce the overexpression of genes in the E. coli DH5α-T7 derivative strains. All of the fermentation experiments were performed in triplicate. The data was averaged and presented as the mean standard deviation, and statistical significance was defined as P < 0.05.
Analysis of gene amplification by RT-PCR
RNA extraction from E. coli was performed using Bacterial Total RNA Isolation Kit (Sangon Biotech Co., Ltd, Shanghai, China). First-strand cDNA synthesis was according to Su et al. (2012). glpD was amplified using primers RTf-glpD/RTr-glpD, glpK was amplified using primers RTf-glpK-RTr-glpK, and the internal control 16S rDNA was amplified using primers RTf-16s/RTr-16s. The amplification was performed for 30 cycles: 94 °C for 30 s, 51 °C for 30 s, and 72 °C for 30 s; plus an additional 10 min at 72 °C after the cycles. 2 % (w/v) agarose gel electrophoresis was performed to analyse PCR product and Bio-Imaging System with the Quantity One 1-D analysis software (Bio-Rad, CA, USA) was used to analyse the band intensity.
Glycerol-3-phosphate dehydrogenase and glycerol kinase activity assay
The glycerol-3-phosphate dehydrogenase was assayed as described previously (Peres et al. 2010). Briefly, the reaction mixture was prepared as follows: 2.6 mM NAD+, 11 mM glycerol-phosphate, 2.5 mM MgCl2·6H2O and 50 mM glycine/NaOH buffer at pH 9.5. The reaction was started by adding cell lysate to the assay mixture with the final reaction volume of 3 ml and ended by adding 2 % SDS after incubating at 25 °C for 3 min. 2 % SDS was added before adding cell lysate in blank control. NADH formation was detected at 340 nm. One enzymatic unit was defined as the amount of enzyme required to form 1 μM of NADH (molar absorption coefficient of 6,200 M−1 cm−1) per min at 25 °C under the conditions specified. The glycerol kinase activity was assayed as described previously (Hayashi and Lin 1967). The phosphorylation of glycerol was coupled to the reduction of NAD+ by the action of l-α-glycerol-P dehydrogenase in glycine/NaOH buffer at pH 9.5. The reaction mixture contained the following: 10 mM glycerol, 5 mM ATP, 2.5 mM MgCl2·6H2O, 3 mM NAD+, 500 μM sodium carbonate, 20 U of l-α-glycerol-P dehydrogenase, and cell lysate in a final volume of 3.0 ml. The l-α-glycerol-P dehydrogenase was omitted from the blank. The UV detection wavelength was 340 nm as above. The unit of enzyme activity was expressed as 1 μM of l-α-glycerol-P formed per min at 25 °C under the conditions specified. Protein concentration was determined by the method of Lowry with bovine serum albumin as a standard.
SDS-PAGE
Protein molecular weight markers were purchased from Sangon Biotech Co., Ltd. (Shanghai, China). The supernatant from the cell lysate harboring expression proteins was used for SDS-PAGE. The expression of recombinant plasmids was detected by SDS-PAGE. The concentration of resolving gel and stacking gel were 12 and 5 %, respectively. Protein concentration was determined by the same method mentioned above.
Analytical methods
Biomass concentrations were determined every few hours until the end of the culture period. Samples (2 ml) were collected, and the dry cell weight (DCW) was monitored. For the determination of SA accumulation in the fermentation broth, samples were prepared by centrifugation at 4,500g for 3 min (TGL-16G, Anting Scientific Instrument Factory, Shanghai, China), and the supernatant was filtered through 0.45 µm nylon membranes. Glycerol concentration and HPLC analysis were carried out according to Chen et al. (2012). HPLC was performed with Shimadzu LC-10A instrument with a SPD-20A UV–visible diode-array detector. The mobile phase was deionized water adjusted to pH 2.0 with H3PO4. Filtered samples of SA were chromatographed on a reverse-phase Hedera ODS-2 column (4 × 150 mm; 5 µm; Hanbon Science and Technology, Huaian, Jiangsu, China) and the flow rate was 0.8 ml min−1 at 30 °C. The detection wavelength was 217 nm.
Results
Construction of pET-28a-glpD–glpK and overexpression of glpK and glpD genes
To investigate the effects of glycerol aerobic fermentation associated with glpK and glpD genes on SA accumulation, glpK and glpD genes were cloned into pET-28a under the control of the T7 promoter, while five genes of the SA pathway, containing tktA, glk, aroE, aroF fbr and aroB, were cloned into the plasmid pAOC-TGEFB (provided by Dr. Fang, Zou et al. 2004) (Fig. 2a). As shown in Fig. 2b, 55 kDa GlpD/GlpK were notably overexpressed (lane 2), the same as 72 kDa TktA, 43 kDa AroF/AroB, 35 kDa Glk and 29 kDa AroE (lane 3). Seven proteins, GlpD, GlpK, TktA, AroF, AroB, Glk and AroE, were observed as distinct bands (lane 4), indicating that plasmids pET-28a-glpD–glpK and pAOC-TGEFB could be compatible and expressed in E. coli BL21(DE3)-P-DK. As shown in Fig. 2c, the glpD and glpK genes were not expressed in E. coli DH5α-T7-DK and E. coli DH5α-T7-P-DK (lanes 2 and 4), suggesting that the expression of pET-28a-glpD–glpK in the PTS− system background of E. coli DH5α-T7 was not well regulated. To further investigate the transcriptional level of glpK and glpD genes, SqRT-PCR was performed among the strains as above (Fig. 3a–d). As shown in Fig. 3c, d, the transcription ratio of glpD in E. coli BL21 (DE3)-P-DK was 2.3-fold than E. coli BL21(DE3), while the ratios in E. coli DH5α-T7-DK and E. coli DH5α-T7-P-DK were no obvious differences with E. coli BL21(DE3). In E. coli BL21(DE3)-P-DK, the transcription ratio of glpK was 2.0-fold higher than E. coli BL21(DE3), and the ratios in E. coli DH5α-T7-DK and E. coli DH5α-T7-P-DK showed no obvious differences with E. coli BL21(DE3). To further investigate translational level of glpD and glpK, the activities of glycerol-3-phosphate dehydrogenase and glycerol kinase were detected (Fig. 3e, f). The activities of glycerol-3-phosphate dehydrogenase in E. coli BL21 (DE3), E. coli BL21 (DE3)-P-DK, E. coli DH5α-T7-DK and E. coli DH5α-T7-P-DK were 0.023, 0.057, 0.025, and 0.024 U/mg, respectively. The activities of glycerol kinase in the strains above were 0.019, 0.045, 0.018, and 0.02 U/mg, respectively. It was shown that the enzyme activity in E. coli BL21(DE3)-P-DK was obviously higher than that of the other three strains. The date of RT-PCR and enzyme activity indicated that glpD and glpK genes were co-expressed effectively in E. coli BL21(DE3), but not in E. coli DH5α-T7-DK or E. coli DH5α-T7-P-DK. Due to the apparent poor expression of glpD and glpK, we had to change from past host E. coli DH5α-T7 to a BL21 (DE3), three engineered E. coli strains were constructed: E. coli BL21(DE3)-DK, in which the glpD and glpK genes of the glycerol aerobic metabolic pathway were overexpressed, E. coli BL21(DE3)-P, in which the tktA, glk, aroE, aroF fbr and aroB of the aromatic amino acid pathway genes were overexpressed and in E. coli BL21(DE3)-P-DK, the genes above were co-expressed.

The construction of pET-28a-glpD–glpK and expression of enzymes. a The maps of polycistronic plasmids. b Overexpression of the vector pET-28a-glpD–glpK and pAOC-TGEFB in E. coli BL21(DE3) derivative strains. The lane names are denoted as follows: M, protein marker; 1, E. coli BL21(DE3) wild type; 2, E. coli BL21(DE3)-DK; 3, E. coli BL21(DE3)-P; 4, E. coli BL21(DE3)-P-DK. c Overexpression of the vector pET-28a-glpD–glpK and pAOC-TGEFB in E. coli DH5α-T7 derivative strains. The lane names are denoted as follows: M, protein marker; 1, E. coli DH5α-T7; 2, E. coli DH5α-T7-DK; 3, E. coli DH5α-T7-P; 4, E. coli DH5α-T7-P-DK; (TktA, 72 kDa; GlpK/GlpD, 55 kDa; AroF/AroB, 43 kDa; Glk, 35 kDa; AroE, 29 kDa)

RT-PCR analysis of glpD and glpK genes and the enzyme activity assay of glycerol-3-phosphate dehydrogenase and glycerol kinase. a RT-PCR products of glpD and16S rDNA, b RT-PCR products of glpK and16S rDNA, c the ratios of glpD transcription level in the recombinant groups to that of the E. coli BL21(DE3) wild type, d the ratios of glpK transcription level in the recombinant groups to that of the E. coli BL21(DE3), e the relative activity of glycerol-3-phosphate dehydrogenase compared to E. coli BL21(DE3), f the relative activity of glycerol kinase compared to E. coli BL21(DE3). Housekeeping gene 16S rDNA was an internal reference. Lanes 1, 2, 3 and 4 stood for BL21(DE3), Bl21(DE3)-P-DK, DH5α-T7-DK and DH5α-T7-P-DK respectively. The ratios of glpD, glpK transcription level in the recombinant group compared to that of the wild type group were calculated after the BL21 (DE3) normalized to 1. *P < 0.05, **P < 0.01 and compared with BL21 (DE3) group
Aerobic fermentation of glycerol associated with glpK and glpD genes enhanced SA accumulation
SA accumulation in E. coli BL21 (DE3), E. coli BL21 (DE3)-DK, E. coli BL21 (DE3)-P, E. coli BL21 (DE3)-P-DK and E. coli DH5α-T7-P-DK were evaluated in flask cultures with modified M9 medium. SA production in E. coli BL21 (DE3)-DK was 72.4 mg/l, which was 2.0-fold higher than that in E. coli BL21 (DE3). Moreover, SA accumulation in E. coli BL21(DE3)-P-DK reached 200.2 mg/l, which was 2.8- and 2.1-fold higher than that in E. coli BL21(DE3)-DK and E. coli BL21(DE3)-P, respectively (Fig. 4a, b). While, SA accumulation in E. coli DH5α-T7-P-DK was 146.6 mg/l, which was lower than 200.2 mg/l in E. coli BL21(DE3)-P-DK. After 48 h of cultivation, glycerol was mostly consumed, with no obvious differences among the samples (Fig. 4c). Therefore, the SA yield of E. coli BL21(DE3)-P-DK reached 7.4 mmol SA/mol Gly, which was higher than that of E. coli BL21(DE3)-DK and E. coli BL21(DE3)-P (Table 2). The culture of E. coli BL21 (DE3)-P-DK showed a lower DCW at 3.6 g/l than the 5.3 g/l of E. coli BL21 (DE3) (Fig. 4d). Namely, the presence of the plasmids pET-28a-glpD–glpK and pAOC-TGEFB enhanced the yield of SA, but reduced the biomass of E. coli BL21 (DE3)-P-DK to some degree.

Shikimic acid accumulation in E. coli BL21(DE3) derivative strains and E. coli DH5α-T7-P-DK. a Shikimic acid accumulation, c glycerol consumption and d growth curves. (open circle) E. coli BL21(DE3); (open square) E. coli BL21(DE3)-P; (inverted triangle) E. coli BL21(DE3)-DK; (triangle) E. coli BL21(DE3)-P-DK; (diamond symbols) E. coli DH5α-T7-P-DK. b Relative SA accumulation in E. coli BL21(DE3) derivative strains and E. coli DH5α-T7-P-DK: DCW (open symbols); SA accumulation (closed symbols). Error bars represent the standard deviations from triplicate measurements. *P < 0.05, **P < 0.01 and ***P < 0.001 compared with BL21(DE3) group
Glycerol effectively improved SA production in E. coli BL21 (DE3)-P-DK
To investigate the effects of glycerol flux on SA accumulation further, glycerol, glucose, maltose, sucrose, and sorbitol were utilized as different carbon sources. E. coli BL21(DE3) and E. coli BL21(DE3)-P-DK grown aerobically in modified M9 complex medium containing carbon source of glycerol (20 g/l) or other carbon sources (20 g/l). The data showed that the highest SA production in E. coli BL21(DE3)-P-DK was 200.2 ± 10.9 mg/l in glycerol culture, followed by 142.8 ± 1.2 mg/l in glucose culture, 119.1 ± 5.8 mg/l in maltose culture, 89.9 ± 4.5 mg/l in sucrose culture and 132.9 ± 3.2 in sorbitol (Fig. 5a). In contrast, E. coli BL21(DE3) exhibited similar SA production with different carbon sources (Fig. 5b). The biomass of E. coli BL21(DE3)-P-DK with different carbon sources was similar (Fig. 5c) but lower than that of E. coli BL21(DE3) (Fig. 5d). The SA yield per biomass for E. coli BL21(DE3)-P-DK with different carbon sources was then calculated: the highest SA yield was 61.1 mg SA/g glycerol, followed by 42.8 mg SA/g glucose, 34.1 mg SA/g maltose, 23.9 mg SA/g sucrose and 38.1 mg SA/g in sorbitol. It is important to emphasize that E. coli BL21 (DE3)-P-DK displayed higher carbon flux toward SA, which was 5-fold higher than that of E. coli BL21 (DE3) in glycerol culture. The SA accumulation in E. coli BL21(DE3)-P-DK in glucose culture, maltose culture, sucrose and sorbitol culture were 3.0-, 2.2-, 1.7- and 2.8-fold, respectively, higher than that in E. coli BL21(DE3) (Fig. 5e, f).

Glycerol-enhanced shikimic acid accumulation in strain E. coli BL21(DE3)-P-DK. a, b SA accumulation in E. coli BL21(DE3)-P-DK and E. coli BL21(DE3). c, d DCW of E. coli BL21(DE3)-P-DK and E. coli BL21(DE3). (open circle) glycerol; (open square) glucose; (triangle) maltose; (inverted triangle) sucrose; (diamond symbols) sorbitol. e, f The relative values of SA accumulation and DCW using different carbon sources after 24 h of cultivation are shown: E. coli BL21(DE3) (closed symbols); E. coli BL21(DE3)-P-DK) (open symbols). Error bars represent the standard deviations from triplicate measurements. *P < 0.05, **P < 0.01 and ***P < 0.001 compared with glycerol of BL21(DE3) group
Discussion
Previous studies have reported successful expression of key enzymes’ genes (aroG fbr, aroB, aroE and gene) in E. coli that can produce 8.9-fold more SA than the parental strain E. coli in which ΔaroK and ΔaroL genes were knocked out (Cui et al. 2014). In the present study, a co-expression plasmid system was constructed with plasmids pAOC-TGEFB, where the tktA, glk, aroE, aroF fbr and aroB genes of the aromatic amino acid pathway were overexpressed, and pET-28a-glpD–glpK. In addition, previous studies have mainly focused on the inactivation of the PTS system to channel part of the phosphoenolpyruvate (PEP) not consumed in glucose transport to the aromatic pathway (Rodriguez et al. 2013; Cortés-Tolalpa et al. 2014). Although a PTS− strain accumulated more aromatic compounds in glycerol–glucose culture than in glucose culture, the productivity of aromatic compounds in glycerol culture was higher than that in glucose culture (Martínez et al. 2008; Chen et al. 2012). The strategy of overexpressing glpK and glpD genes in E. coli production strains growing on glycerol has been reported previously (Mazumdar et al. 2010, 2013; Wong et al. 2014), none of these are applied in SA biosynthesis. To investigate the effects of glycerol aerobic fermentation on SA accumulation, increasing glycerol metabolic flux by glpD and glpK genes overexpressed was carried out to obtain higher SA accumulation. Meanwhile carbon source is one of the main components of culture medium which is a crucial factor for SA production. The effects of various carbon sources on the growth and SA accumulation in E. coli strains were explored (Liu et al. 2014). The effect of different carbon source on the SA production was performed in this study. From the data obtained, the glpD and glpK genes were not successfully expressed in E. coli DH5α-T7 (Fig. 2c). The protein IIIGlc is involved in regulation by inducer exclusion in enteric bacteria and GlpK is inactivated by unphosphorylated IIIGlc protein. In the ptsHIcrr mutant, E. coli DH5α-T7, IIIGlc is present inside the cell and inactivates GlpK (Dean et al. 1990), which could explain why the glpD and glpK genes were not expressed well in E. coli DH5α-T7. Due to the apparent poor expression of glpD and glpK, we had to change from past host E. coli DH5α-T7 to a BL21 (DE3).
In E. coli BL21(DE3)-DK, the glpD and glpK genes were expressed compatibly and SA accumulation reached 72.4 mg/l (Fig. 4a), a 2.0-fold increase over that in E. coli BL21(DE3) (Fig. 4b), suggesting that expression of the glpK and glpD genes could increase glycerol metabolic flux and result in higher SA accumulation. Moreover, SA accumulation in E. coli BL21(DE3)-P-DK reached 200.2 mg/l (Fig. 4a), which was 5.6-fold higher than that of E. coli BL21(DE3) (Fig. 4b), suggesting increased glycerol metabolic flux toward SA due to the overexpression of glpD and glpK genes in glycerol dissimilation and aroE, aroB, glk, tktA, and aroF fbr genes in SA pathway. Previous studies reported that increasing the availability of PEP and erythrose-4-phosphate (E4P) effectively contributes to the accumulation of SA in recombinant E. coli (Krämer et al. 2003). Glycerol kinase, encoded by glpK, catalyses the conversion of glycerol into glycerol-3-phosphate (G3P). G3P is then converted to dihydroxyacetone phosphate (DHAP) by aerobic glycerol-3-phosphate dehydrogenase, encoded by glpD. Thus, the overexpression of the glpK and glpD genes increases the availability of DHAP, which ultimately enters the Embden–Meyerhof–Parnas pathway and is converted to PEP and E4P (Ahn et al. 2008). Hence, the flux toward SA was increased.
SA accumulation in E. coli BL21(DE3)-P-DK reached 200.2 ± 10.9 mg/l in glycerol culture (Fig. 5a), which was the highest in comparison to glucose, maltose and sucrose cultures, suggesting that glycerol is an effective carbon source for SA accumulation. E. coli BL21 (DE3)-P-DK displayed higher carbon flux toward SA production, which was 5.0-fold higher than that of E. coli BL21 (DE3) in glycerol culture (Fig. 5e). Glucose, maltose and sucrose can be transported or regulated by the PTS system, which consumes PEP (Ahn et al. 2008). In comparison to other carbon sources, however, glycerol enters the glycolytic pathway without consuming PEP. Therefore, part of the PEP not consumed in sugar transport could be converted to DHAP by glycerol kinase and glycerol-3-phosphate dehydrogenase, which are encoded by glpK and glpD, and then isomerized to glyceraldehyde 3-phosphate (GA3P). GA3P is the substrate of transketolase I (coded by tktA), which interconnects the non-oxidative branch of the pentose phosphate pathway and glycolysis (Zou et al. 2004). For these reasons, increasing glycerol metabolic flux can enhance SA accumulation in E. coli BL21 (DE3)-P-DK. Previous studies reported that many organisms carry out the fermentative metabolism of glycerol in a 1,3-propanediol-dependent manner under anaerobic conditions. E. coli is able to ferment glycerol when appropriate conditions are maintained in the absence of 1,3-propanediol synthesis under aerobic conditions (Yazdani and Gonzalez 2007). These conditions include avoiding the accumulation of the fermentation gas hydrogen, an acidic condition and appropriate medium composition (Dharmadi et al. 2006). Therefore, the accumulation of SA in the cultures simultaneously facilitated the utilization of glycerol.
In the present study, although the overexpression of glpK and glpD genes led to a notable increase in SA accumulation, it was not high enough to reach the level of previous research (Chen et al. 2012). Further metabolic modifications need to be studied in E. coli BL21(DE3)-DK-P, such as blocking SA from the next step in the SA pathway and overexpression of the galP gene, which encodes GalP in glycerol transport (Martínez et al. 2008).
References
Adachi O, Ano Y, Toyama H, Matsushita K (2006) High shikimate production from quinate with two enzymatic systems of acetic acid bacteria. Biosci Biotechnol Biochem 70:2579–2582
Ahn JO, Lee HW, Saha R, Park MS, Jung JK, Lee DY (2008) Exploring the effects of carbon sources on the metabolic capacity for shikimic acid production in Escherichia coli using in silico metabolic predictions. J Microbiol Biotechnol 18:1773–1784
Bochkov DV, Sysolyatin SV, Kalashnikov AI, Surmacheva IA (2012) Shikimic acid: review of its analytical, isolation and purification techniques from plant and microbial sources. J Chem Biol 5:5–17
Chandran SS, Yi J, Draths KM, Von Daeniken R, Weber W, Frost JW (2003) Phosphoenolpyruvate availability and the biosynthesis of shikimic acid. Biotechnol Prog 19:808–814
Chen K, Dou J, Tang S, Yang Y, Wang H, Fang H, Zhou C (2012) Deletion of the aroK gene is essential for high shikimic acid accumulation through the shikimate pathway in E. coli. Bioresour Technol 119:141–147
Cortés-Tolalpa L, Gutiérrez-Ríos RM, Martínez LM, de Anda R, Gosset G, Bolivar F, Escalante A (2014) Global transcriptomic analysis of an engineered Escherichia coli strain lacking the phosphoenolpyruvate: carbohydrate phosphotransferase system during shikimic acid production in rich culture medium. Micro Cell Fact 13:28
Cui YY, Ling C, Zhang YY, Huang J, Liu JZ (2014) Production of shikimic acid from Escherichia coli through chemically inducible chromosomal evolution and cofactor metabolic engineering. Microb Cell Fact 13:21
Dean DA, Reizer J, Nikaido H, Saier MH Jr (1990) Regulation of the maltose transport system of Escherichia coli by the glucose-specific enzyme III of the phosphoenolpyruvate-sugar phosphotransferase system. J Biol Chem 265:21005–21010
Dharmadi Y, Murarka A, Gonzalez R (2006) Anaerobic fermentation of glycerol by Escherichia coli: a new platform for metabolic engineering. Biotechnol Bioeng 94:821–829
Gibson JM, Thomas PS, Thomas JD, Barker JL, Chandran SS, Harrup MK, Draths KM, Frost JW (2001) Benzene-free synthesis of phenol. Angew Chem Int Ed 40:1945–1948
Hayashi SI, Lin EC (1967) purification and properties of glycerol kinase from Escherichia coli. J Biol Chem 242:1030–1035
Johansson L, Lidén G (2006) Transcriptome analysis of a shikimic acid producing strain of Escherichia coli W3110 grown under carbon- and phosphate-limited conditions. J Biotechnol 126(4):528–545
Kancharla PK, Doddi VR, Kokatla H, Vankar YD (2009) A concise route to (–)-shikimic acid and (–)-5-epishikimic acid, and their enantiomers via Barbier reaction and ring-closing metathesis. Tetrahedron Lett 50:6951–6954
Knop DR, Draths KM, Chandran SS, Barker JL, von Daeniken R, Weber W, Frost JW (2001) Hydroaromatic equilibration during biosynthesis of shikimic acid. J Am Chem Soc 123:10173–10182
Krämer M, Bongaerts J, Bovenberg R, Kremer S, Müller U, Orf S, Wubbolts M, Raeven L (2003) Metabolic engineering for microbial production of shikimic acid. Metab Eng 5:277–283
Liu X, Lin J, Hu H, Zhou B, Zhu BQ (2014) Metabolic engineering of Escherichia coli enhance shikimic acid production from sorbitol. World J Microbiol Biotechnol 30:2543–2550
Martínez K, de Anda R, Hernández G, Escalante A, Gosset G, Ramírez OT, Bolívar FG (2008) Coutilization of glucose and glycerol enhances the production of aromatic compounds in an Escherichia coli strain lacking the phosphoenolpyruvate: carbohydrate phosphotransferase system. Microb Cell Fact 7:1–12
Mazumdar S, Clomburg JM, Gonzalez R (2010) Escherichia coli strains engineered for homofermentative production of d-lactic acid from glycerol. Appl Environ Microbiol 76:4327–4336
Mazumdar S, Blankschien MD, Clomburg JM, Gonzalez R (2013) Efficient synthesis of l-lactic acid from glycerol by metabolically engineered Escherichia coli. Microb Cell Fact 12:1–7
Peres MDS, Solra VC, Valentini SR, Gattas EAD (2010) Recombinant expression of glycerol-3-phosphate dehydrogenase using the Pichia pastorissystem. J Mol Catal B Enzym 65:128–132
Rodriguez A, Martínez JA, Báez-Viveros JL, Flores N, Hernández-Chávez G, Ramírez OT, Gosset G, Bolivar F (2013) Constitutive expression of selected genes from the pentose phosphate and aromatic pathways increases the shikimic acid yield in high-glucose batch cultures of an Escherichia coli strain lacking PTS and pykF. Microb Cell Fact 12:86
Shinada T, Yoshida Y, Ohfune Y (1998) Direct conversion of 1,2-diol into allyl sulfide. Regioselective transformation of (–)-quinic acid to (–)-shikimic acid. Tetrahedron Lett 39:6027–6028
Su ZZ, Dou J, Xu ZP, Guo QL, Zhou CL (2012) A novel inhibitory mechanism of baicalein on influenza A/FM1/1/47 (H1N1) virus: interference with mid-late mRNA synthesis in cell culture. Chin J Nat Med 6:0415–0420
Wong MS, Li M, Black RW, Le TQ, Puthli S, Campbell P, Monticello DJ (2014) Microaerobic conversion of glycerol to ethanol in Escherichia coli. Appl Environ Microbiol AEM 80:3276–3282
Yazdani SS, Gonzalez R (2007) Anaerobic fermentation of glycerol: a path to economic viability for the biofuels industry. Curr Opin Biotechnol 18:213–219
Zou YK, Zhou JZ, Sun X, Cai YF, Dai HM, Li SL, Zhou CL, Fang HQ (2004) Construction of shikimic acid-producing engineered Escherichia coli strains based on ptsHIcrr mutants. Microbiol China 38:1186–1192
Acknowledgments
This research was financially supported by the Fundamental Research Funds for the Central Universities (ZL2014SK0035), and the Priority Academic Program Development of Jiangsu Higher Education Institution (PAPD).
Author information
Authors and Affiliations
Corresponding author
Additional information
Yang Yang and Chao Yuan have contributed equally to this work.
Rights and permissions
About this article

Cite this article
Yang, Y., Yuan, C., Dou, J. et al. Recombinant expression of glpK and glpD genes improves the accumulation of shikimic acid in E. coli grown on glycerol. World J Microbiol Biotechnol 30, 3263–3272 (2014). https://doi.org/10.1007/s11274-014-1753-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11274-014-1753-6