
Abstract
Accurate determination of emerging contaminants in drinking water constitutes a major environmental challenge for which highly sensitive analytical methods are needed. This work details the development of a novel highly sensitive solid-phase extraction-high performance liquid chromatography-tandem mass spectrometry (SPE-HPLC-MS/MS) method for simultaneous determination of a diverse panel of widely used trace contaminants, including two pharmaceuticals (fluoxetine and gemfibrozil), three pesticides (3-hydroxycarbofuran, azinphos-methyl, and chlorpyrifos), and two hormones (testosterone and progesterone) in water. The method is highly reproducible and sensitive with detection limits at subnanogram per liter level (0.05–0.5 ng/L). It was used to monitor the occurrence of these contaminants in source and drinking water across 18 drinking water treatment facilities in Missouri, USA in 1 year including cold winter and hot summer seasons. The experiment results indicated that all of the monitored contaminant concentrations are very low, lower than or close to the method detection limits, in the selected water treatment facilities. Pesticide concentrations were slightly elevated in some source waters during hot season, whereas slightly higher pharmaceuticals were observed during cold season. The concentrations of two hormones were lower than the limits of detection in all the water samples. These contaminants were present, if any, at below detection limits in all treated drinking water samples analyzed, suggesting that treatment processes effectively removed the contaminants studied herein.

Graphical Abstract
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
The growing need for access to clean drinking water in a populated world has been accentuated by the release of anthropogenic contaminants into the environment (Taheran et al. 2018). The widespread and pervasive release of contaminants from industry, agriculture, medicine, and household supplies have resulted in persistent environmental levels of many contaminants known to exert physiological effects at trace levels (Schwarzenbach et al. 2006; Tran et al. 2018). Common contaminants include pharmaceuticals, pesticides, endocrine disruptors, and so on, many of which have been extensively studied in wastewaters, surface waters, groundwater, and drinking water as regulated contaminants (de Alda and Barcelo 2001; Mauriz et al. 2006; Aznar et al. 2017; Castiglioni et al. 2017). In addition to these regulated compounds, many emerging environmental contaminants are considered potentially hazardous to humans, but currently not regulated by regulatory agencies due to either lack of sensitive enough analytical method or not enough occurrence data to make regulation decision. In the present study, we examine seven such compounds comprising a diverse panel of pharmaceuticals, pesticides, and hormones.
Fluoxetine (FLX) is a selective serotonin reuptake inhibitor, which has been used for over 25 years to treat major depression and other psychiatric disorders (Bedner and MacCrehan 2006; Andres-Costa et al. 2017). The prevalence of FLX in many aquatic environments has been attributed to its widespread use in numerous developing and developed countries (Alonso et al. 2010; Kolpin et al. 2002). The presence of FLX in environmental waters has been studied, where it has been shown to exert adverse behavioral effects on vertebrate fish (Sumpter et al. 2014). For example, 28 ng/L FLX could lead to reproductive complications in male fathead minnows (Schultz et al. 2011), and 0.3 μg/L affected swimming speed, schooling behavior, and response to predator alarm in the Arabian killifish (Barry 2013). Another common pharmaceutical, gemfibrozil (GEM), which is a widely prescribed lipid regulator and a member of the fibrate class of pharmaceuticals, is frequently detected in waters at concentrations up to 63.8 μg/L in USA (Bulloch et al. 2012). GEM is a highly persistent compound (Araujo et al. 2011) that is not readily susceptible to removal by oxidation or chlorination processes (Sharma 2008).
Pesticides and their effects on the environment and human health have been extensively studied (Van Maele-Fabry et al. 2017); however, many pesticides and their degradation products are not currently regulated by government agencies. For example, 3-hydroxycarbofuran (3HCF) is a metabolite of the carbamate pesticides, which were first introduced as synthetic pesticides in the 1950s and remain in use today owing to their attractive performance and biological activities (Hsu et al. 2012). Remarkably, 3HCF has been found to have higher potency as an insecticide than its parent carbamate pesticides (Soler et al. 2007). For this reason and others, 3HCF is currently listed on the US Environmental Protection Agency (US EPA) Contaminant Candidate List 4 (CCL 4) as a possible compound requiring regulation in the near future. Another prominent pesticide that is not currently regulated is azinphos-methyl (AM). AM is one of the most widely used pesticides in the USA and other countries for a variety of fruits, nuts, and vegetables, with nearly 950,000 kg of the active ingredient being used during 1997 (Schulz et al. 2003). Nevertheless, AM is a highly toxic organophosphorus pesticide that affects many non-target organisms including beneficial insects, birds, small mammals, fish, aquatic invertebrates, and humans (US EPA, 2006). For example, AM was a potent inhibitor of muscular cholinesterase with an EC50 value of 1.05 ± 0.23 μg/L (Ferrari et al. 2007). Another commonly used organophosphate-based pesticide that merits attention is chlorpyrifos (CPF). CPF has been recently shown to be a potent acetylcholinesterase inhibitor that possesses considerable cytotoxicity in mammals (Goel et al. 2005; Mauriz et al. 2006).
Progesterone (PRO) and testosterone (TST) are the primary natural progestin and androgenic hormones, respectively. These compounds are routinely used in hormonal replacement therapy, contraception, and palliative care for cancer treatment (Méité et al. 2016). These hormones could function as endocrine disruptors after their inadvertent releasing into the environment, which can cause adverse reproductive changes in aquatic organisms, including feminization, hermaphroditism, and decrease in fertility (de Vidales et al. 2012). Although PRO and TST are not currently regulated as primary pollutants, their prevalence in US waterways is well documented, such as in a report by Koplin et al. (2002) that detected PRO and TST in 4.3%, and 2.8% of 139 US streams and a mean concentration of 0.11 μg/L (maximum 0.20 μg/L) and 0.12 μg/L (maximum 0.21 μg/L).
It is clear that these selected compounds represent important water contaminants with significant toxicities toward a range of organisms. However, analytical methodologies for their simultaneous determination are not currently available. While several methods for the analysis of individual compounds have been developed on various analytical platforms, including gas chromatography-mass spectrometry (GC-MS) (Araujo et al. 2011; Aznar et al. 2017; Planas et al. 2006), liquid chromatography-mass spectrometry (LC-MS) (Alonso et al. 2010; Salas et al. 2017), and liquid chromatography-tandem mass spectrometry (LC-MS/MS) (Kim et al. 2007; Tolgyesi et al. 2010), these methods often lack the sensitivity required for quantitation in real water samples. We therefore develop solid-phase extraction (SPE) as a sample preconcentration technique in combination with highly sensitive and selective HPLC-MS/MS method to enable sensitive quantification of these water contaminants. SPE is routinely used in conjunction with hyphenated analytical techniques (Anumol and Snyder 2015; Baker and Kasprzyk-Hordern 2011; Planas et al. 2006), which is one of the most commonly used preconcentration method for emerging contaminants in different environmental matrices.
The objectives of this study were to (1) develop a robust analytical methodology for simultaneous determination of FLX, GEM, 3HCF, AM, CPF, PRO, and TST in water matrices using SPE-HPLC-MS/MS; (2) monitor the occurrence of these environment contaminants in drinking water treatment facilities across the state of Missouri; (3) determine the seasonal effect of the targeted compounds in source water and treated water in selected treatment facilities to evaluate the removal efficiency of water treatment processes.
2 Materials and Methods
2.1 Chemicals
FLX, GEM, 3HCF, AM, CPF, PRO, and TST standards were purchased from Sigma-Aldrich (St. Louis, MO, USA). The general information of analytes including formula, CAS no., and molecular weight are shown in Table 1. Mass spectrometry grade formic acid was also purchased from Sigma-Aldrich. Ascorbic acid (AA), sodium thiosulfate (STS), 2-mercaptopyridine-1-oxide sodium salt (MOS), 2-chloroacetamide (CAM), disodium hydrogen phosphate (Na2HPO4), sodium phosphate monobasic (NaH2PO4), Optima-grade methanol, and methyl-tert-butylether (MtBE) were purchased from Fisher Scientific (Fair Lawn, NJ, USA). Ultra-high purity water (18.2 MΩ cm) was prepared by an Elix-3 water purification system (Millipore, Billerica, MA, USA). A Waters Oasis HLB SPE cartridge (Waters, Milford, MA, USA) was selected for analyte extraction from water matrices.
2.2 Instrumentation
A Shimadzu ultra-fast liquid chromatography (UFLC) system (Columbia, MD, USA) including two pumps (LC-20 AD XR), an autosampler (SIL-20 AC XR), an online degasser (DGU-30A3), and a column oven (CTO-20A) with a Synergi Polar-RP column (150 × 2.0 mm, I.D. 4-μm particle size, Phenomenex, Torrance, CA, USA) was used for separation. Analyst 1.5 software was used for data acquisition and quantification. Samples were eluted with a flow rate set to 0.30 mL/min under a gradient elution program with eluent A (ultra-pure water with 0.1% (v/v) formic acid) and eluent B (methanol with 0.1% (v/v) formic acid) that was defined as the following: 50% B for 2 min, linear increase to 100% B over 2 min and maintained at 100% B for 5 min, then decreased to 50% B over 0.1 min and maintained for 6 min prior to next injection. The sample injection volume was 20 μL.
A 4000Q Trap tandem mass spectrometer (AB Sciex, Foster City, CA, USA) was operated under positive electrospray ionization (ESI) mode and scheduled multiple reaction monitoring (MRM) was used for quantitation. Nitrogen gas was used for curtain and collision gases. Specifically, declustering potentials (DP), collision energies (CE), and collision cell exit potentials (CXP) were optimized for the two most sensitive ion transitions for each analyte: one for quantification ion pair and one for confirmation ion pair. Flow injection analysis (FIA) was performed to optimize ion source conditions: ion source temperature at 550 ̊C, ion spray voltage at 4000 V, curtain gas pressure at 10 psi, ion source gas 1 pressure at 40 psi, and ion source gas 2 pressure at 55 psi. The entrance potential was 10 V for all compounds.
2.3 Water Sample Collection and Pretreatment
A total of 18 drinking water treatment facilities across Missouri were sampled for the occurrence study. The source water used for selected facilities include groundwater (GW) and surface water (SW). Treatment facilities employed various disinfection treatment processes, including chlorination with gaseous chlorine and alkali hypochlorite solution, chloramination, and chlorine dioxide disinfection. Paired source water and treated drinking water samples were collected during consecutive winter and summer seasons.
Sample collection and preparation were in accordance with the US Environmental Protection Agency (US EPA) Standard Method 540 (US EPA, 2013). Briefly, samples were collected with precleaned 500-mL amber glass bottles fitted with Teflon liner screw caps. Sample preservation to minimize analyte degradation included the addition of 0.1 g/L AA as a quenching agent and 2 g/L CAM as an antimicrobial agent prior to collection. Thereafter, water samples were placed in iced coolers and transported to the laboratory overnight.
The linear range of calibration for each compound, method detection limits, reproducibility, and spike recoveries for each compound in ultra-pure water and in matched water sample matrices were performed in accordance with the quality control (QC) guidelines recommended by US EPA. Additional QC measures included matrix blanks, sample duplicates, and spiked sample recoveries throughout the SPE-HPLC-MS/MS method. QC sample sets were analyzed for every ten water samples.
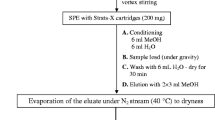
2.4 Water Sample Solid-Phase Extraction
Water samples (500 mL) were vacuum-filtered through 0.45-μm nylon filters, followed by adjusting pH to 7.00 ± 0.05 using phosphate buffer. Following pretreatments, samples were stored under dark conditions at 4 °C for no longer than 48 h prior to analysis. Solid-phase extraction of the target compounds was performed using Oasis HLB cartridges (6 mL, 200 mg, Waters, Milford, MA, USA). Cartridges were conditioned sequentially with 6.0 mL of methanol and 12.0 mL of ultra-pure water. Then, 500 mL sample was passed through the cartridge at a flow rate of 10–15 mL/min. The HLB cartridges were subsequently rinsed with 6.0 mL of ultra-pure water, and then air dried for 5 min. Thereafter, each sample cartridge was eluted with 6.0 mL methanol. The eluate was collected in a 10-mL glass vial and concentrated to 1 mL by a gentle nitrogen stream using a Zymark Turbovap LV evaporator (LabX, Midland, Ontario, Canada) at 32 ± 1 °C. The concentrated eluate was adjusted to 1.0 mL with methanol, which was then subjected to analysis.
3 Results and Discussion
3.1 Optimization of MS/MS Analysis Conditions
Individual compounds were infused at a concentration of 100 μg/L in mobile phase A (0.1% (v/v) formic acid in ultra-pure water) at a flow rate of 0.5 mL/h. The MS/MS parameters and the MRM transitions for each selected compound were optimized to achieve the maximum instrumental sensitivity. Two MRM transitions between the precursor ion and the two most abundant product ions were monitored for each compound: the first ion pair (generally most sensitive one) was used for quantitation, whereas the second was used for confirmation, which have been summarized in Table 2. The MS/MS parameters that were optimized for each transition included declustering potential (DP), collision energy (CE), and collision cell exit potential (CXP). Ion source parameters were subsequently optimized with the following values: ion source temperature 550 °C, ion spray voltage 4000 V, curtain gas pressure 10 psi, GS1 40 psi, and GS2 55 psi. The entrance potential was 10 V for all transitions.
3.2 Optimization and Validation of HPLC Conditions
A representative chromatogram of standard mixture is provided in Fig. 1. All the analytes eluted between 2.77 and 6.51 min. The reproducibility of retention time was tested by injections of standard mixture spiked in ultra-high purity water and in Missouri river water sample (Table 1). Excellent reproducibility of retention time was observed with percent relative standard deviation (%RSD) for six injections ranging from 0.18 to 0.53%.

A representative chromatogram of HPLC-MS/MS method with injection of standard mixture
The performance of this HPLC-MS/MS method was validated in different water matrices including Milli-Q water, river water, and drinking water (Table 3). The instrument limits of detection (LOD) without SPE, defined as having a signal-to-noise ratio (S/N) between 3 and 5, ranged from 0.005 to 0.2 μg/L, whereas limits of instrument quantification (LOQ, S/N=9-10) ranged from 0.01 to 0.5 μg/L. Linear regression analyses were performed using an external calibration curve that ranged from instrument quantification limits to 100 μg/L with coefficients of determination (R2) all higher than 0.99 for all the analytes.
3.3 SPE-HPLC-MS/MS Method Performance
SPE cartridge selection is dependent on the physicochemical properties of the desired analytes as well as that of the matrix. In this study, a Waters Oasis HLB cartridge was selected based on an exhaustive review of the literature on the extraction and preconcentration of the individual compounds from water matrices of our experiments. All the influencing factors were optimized, including flow rate of extraction, nitrogen pressure for solvent evaporation, preservation agents used at water sample collection to prevent from microbial degradation, and chemical reaction to the analytes. Two different quenching agents (0.1 g/L sodium thiosulfate and 0.1 g/L ascorbic acid) and two antimicrobial agents (65 mg/L MOS and 2 g/L 2-chloroacetamide) were tested. Each additive was added to water samples at the sample collection time and analyzed to make sure they have no interference with the analysis of water samples. Certain amount of quenching agent and antimicrobial was added into 500 mL drinking water and surface water (river water) samples with the spiked analytes, and processed through SPE-HPLC-MS/MS method to evaluate the spike recoveries. From Fig. 2, results demonstrate that sodium thiosulfate is a better quenching agent with more reproducible recoveries. Comparing the two antimicrobial agents MOS and CAM, the recoveries of GEM, TST, AM, CPH, and PRO had no significant difference but CAM results a better performance than MOS for 3HCF and FLX. Therefore, STS and CAM were chosen as preservation agents for water sample analysis in this developed method.

Recoveries of target compounds with different antimicrobial agents (MOS, CAM) in Missouri river water and quenching agents (STS, AA) in tap water
Method sensitivity was determined through the new SPE-HPLC-MS/MS method with method quantification limits (MQL) ranged from 0.1 to 1.0 ng/L (S/N = 9-10). The compounds provided a linear detector response from their MQL to 200 ng/L with good linearity (R2 > 0.99). The spiked recoveries at a concentration of 10 ng/L ranged from 66 to 97% with acceptable reproducibility (< 15% RSD), as shown in Table 4. The spike recoveries should be improved more if isotope-labeled internal standards are available. These results indicated that the newly developed method may be used to screen the selected contaminants in source and municipal water matrices.
3.4 Occurrence Screening of Contaminants in Missouri Drinking Water System
A total of 18 drinking water treatment facilities across the state of Missouri were selected for screening study of the selected water contaminants. These facilities used a variety of source waters, including river, lake, reservoir, and well waters. Free chlorine, monochloramine, and chlorine dioxide are commonly used for water disinfection in Missouri state water treatment facilities. Source and finished drinking water samples were collected from these facilities during cold winter season (December) and hot summer season (July). The analyses results are summarized in Table 5, and the detailed data for individual water treatment facility are shown in Tables S1 and S2 of the supplementary information. All the selected contaminant concentrations were low, with levels close or below the method detection limits for all facilities selected. The pharmaceutical (i.e., FLX and GEM) concentrations tended to be higher during the cold season than hot season. Specifically, FLX was detected in source water samples in 2 of 20 (10%) summer season–collected water samples, and in 5 of 21 (24%) of winter season–collected water samples. No detectable level of GEM was found in all water samples analyzed except one river water sample containing 1.85 ng/L GEM, above the method quantification limit of 1.0 ng/L, during winter time. A study by Kim et al. (2007) did not find any samples at higher than detection limit of GEM in South Korean surface water. Another survey conducted by Benotti et al. (2009) reported that FLX and GEM were found in 3 (15.8%) and 11 (57.9%) samples, respectively, in 19 selected source water samples across USA during 2006 to 2007. The median concentrations for FLX and GEM in the study of Benotti et al. were 0.80 and 2.2 ng/L, respectively, in the source water (Benotti et al. 2009), which were matched with our study. The seasonal variations observed in our study may be attributed to seasonality of pharmaceutical usage/discharge and weather conditions, such as less rainfall during cold season resulting less dilution of the contaminants, as well as less degradation from less microbial activity at lower temperature. All pharmaceuticals at detectable levels were from surface water samples, and none of them was found in any well water sample. On the other hand, pesticide (3HCF) was detected only in source water samples during the hot season with the highest concentration of 0.10 ng/L, which should be attributed to increased pesticide agricultural application during the spring and summer months. During hot season, there were no detectable AM founded in the raw and drinking water samples. However, during cold season, AM was founded in 2 out of 21 source samples with the maximum concentration at 0.56 ng/L. In the Ria Formosa Lagoom, Portugal, there was no significant seasonal variation of AM, which had a level from 8.3 to 12.5 ng/L for the whole year (Cruzeiro et al. 2015). The concentrations of AM in Ebro River, Spain, were lower than 0.5 ng/L and around 2.3 ng/L for 2010 and 2011, respectively (Ccanccapaa et al. 2016). The two hormones (i.e., PRO and TST) were at non-detectable level in all the selected water samples during this study. In the survey conducted by Benotti et al. (2009), PRO and TST were found in 4 (21.1%) and 2 (10.5%), respectively, out of 19 selected source water samples of drinking water facilities with the median concentration ad 2.2 and 1.1 ng/L. In the City Lake and City River of India, TST concentrations were below 0.55 ng/L, and PRO concentrations were below 0.88 ng/L and 16.64 ng/L, respectively (Appa et al. 2018). The results of this occurrence study demonstrated that all the selected contaminant concentrations present at the level below detection limits or in the subnanogram per liter range, and changed seasonally. Thus, these contaminants may not rise serious health concern in the Missouri drinking water systems selected, at least in the time period of this study (2015–2016). This occurrence study results are not only very useful to the water facilities and customers for understanding their water quality, it is also necessary for regulation agencies to establish public health guidelines.
4 Conclusions
A rapid and sensitive SPE-HPLC-MS/MS method was developed for the simultaneous determination of seven environmental contaminants including pharmaceutical, pesticides, and hormones in source and drinking waters. This novel method was well suitable for various source and treated drinking water sample analyses. The method is highly reproducible and sensitive. The method calibration linearity is excellent for every compound (R2 > 0.99). This method is suitable for different source and treated drinking water matrix with acceptable spike recoveries, ranged mostly higher than 70%, except TST with an average spike recovery of 66.3% in source water sample. The method had been applied to monitor the occurrence of these contaminants in 18 drinking water treatment facilities across the state of Missouri, USA. The seven contaminants were detected at near or below the method detection limits and exhibited slight seasonal variations, although more sampling times should be more conclusive to determine the generality of these trends. Furthermore, all the monitored contaminants were lower than their detection limits in treated drinking water samples, indicating that the treatment procedures used by the drinking water facilities could remove the selected contaminants effectively.
References
de Alda, M. J. L., & Barcelo, D. (2001). Determination of steroid sex hormones and related synthetic compounds considered as endocrine disrupters in water by fully automated on-line solid-phase extraction-liquid chromatography-diode array detection. Journal of Chromatography A, 911(2), 203–210.
Alonso, S. G., Catala, M., Maroto, R. R., Gil, J. L. R., de Miguel, A. G., & Valcarcel, Y. (2010). Pollution by psychoactive pharmaceuticals in the Rivers of Madrid metropolitan area (Spain). Environmental International, 36(2), 195–201.
Andres-Costa, M. J., Proctor, K., Sabatini, M. T., Gee, A. P., Lewis, S. E., Pico, Y., & Kasprzyk-Hordern, B. (2017). Enantioselective transformation of fluoxetine in water and its ecotoxicological relevance. Scientific Reports, 7, 157–177.
Anumol, T., & Snyder, S. A. (2015). Rapid analysis of trace organic compounds in water by automated online solid-phase extraction coupled to liquid chromatography-tandem mass spectrometry. Talanta, 132(15), 77–86.
Appa, R., Mhaisalkar, V. A., Bafana, A., Devi, S. S., Krishnamurthi, K., Chakrabarti, T., & Naoghare, P. K. (2018). Simultaneous quantitative monitoring of four indicator contaminants of emerging concern (CEC) in different water sources of Central India using SPE/LC (ESI)MS-MS. Environmental Monitoring and Assessment, 190, 489.
Araujo, L., Villa, N., Camargo, N., Bustos, M., Garcia, T., & Prieto, A. D. (2011). Persistence of gemfibrozil, naproxen and mefenamic acid in natural waters. Environmental Chemistry Letters, 9(1), 13–18.
Aznar, R., Albero, B., Sanchez-Brunete, C., Miguel, E., Martin-Girela, I., & Tadeo, J. L. (2017). Simultaneous determination of multiclass emerging contaminants in aquatic plants by ultrasound-assisted matrix solid-phase dispersion and GC-MS. Environmental Science and Pollution Research, 24(9), 7911–7920.
Baker, D. R., & Kasprzyk-Hordern, B. (2011). Multi-residue analysis of drugs of abuse in wastewater and surface water by solid-phase extraction and liquid chromatography-positive electrospray ionisation tandem mass spectrometry. Journal of Chromatography A, 1218(12), 1620–1631.
Barry, M. J. (2013). Effects of fluoxetine on the swimming and behavioural responses of the Arabian killifish. Ecotoxicology, 22(2), 425–432.
Bedner, M., & MacCrehan, W. A. (2006). Reactions of the amine-containing drugs fluoxetine and metoprolol during chlorination and dechlorination processes used in wastewater treatment. Chemosphere, 65(11), 2130–2137.
Benotti, M. J., Trenholm, R. A., Vanderford, B. J., Holady, J. C., Stanford, B. D., & Snyder, S. A. (2009). Pharmaceuticals and endocrine disrupting compounds in US drinking water. Environmental Science & Technology, 43(3), 597–603.
Bulloch, D. N., Lavado, R., Forsgren, K. L., Beni, S., Schlenk, D., & Larive, C. K. (2012). Analytical and biological characterization of halogenated gemfibrozil produced through chlorination of wastewater. Environmental Science & Technology, 46(10), 5583–5589.
Castiglioni, S., Davoli, E., Riva, F., Palmiotto, M., Camporini, P., Manenti, A., & Zuccato, E. (2017). Mass balance of emerging contaminants in the water cycle of a highly urbanized and industrialized area of Italy. Water Research, 131, 287–298.
Ccanccapaa, A., Masiáa, A., Navarro-Ortegab, A., Picóa, Y., & Barcelóbc, D. (2016). Pesticides in the Ebro River basin: occurrence and risk assessment. Environmental Pollution, 211, 414–424.
Cruzeiro, C., Pardal, A. M., Rocha, E., & Rocha, M. J. (2015). Occurrence and seasonal loads of pesticides in surface water and suspended particulate matter from a wetland of worldwide interest—the Ria Formosa Lagoon, Portugal. Environmental Monitoring Assessment, 187, 669.
Ferrari, A., Venturino, A., & de D’Angelo, A. M. P. (2007). Muscular and brain cholinesterase sensitivities to azinphos methyl and carbaryl in the juvenile rainbow trout Oncorhynchus mykiss. Comparative Biochemistry and Physiology Part C: Toxicology & Pharmacology, 146(3), 308–313.
Goel, A., Dani, V., & Dhawan, D. K. (2005). Protective effects of zinc on lipid peroxidation, antioxidant enzymes and hepatic histoarchitecture in chlorpyrifos-induced toxicity. Chemico-Biological Interactions, 156(2–3), 131–140.
Hsu, C. H., Hu, C. C., & Chiu, T. C. (2012). Analysis of carbofuran, carbosulfan, isoprocarb, 3-hydroxycarbofuran, and 3-ketocarbofuran by micellar electrokinetic chromatography. Journal of Separation Science, 35, 1359–1364.
Kim, S. D., Cho, J., Kim, I. S., Vanderford, B. J., & Snyder, S. A. (2007). Occurrence and removal of pharmaceuticals and endocrine disruptors in south Korean surface, drinking, and waste waters. Water Research, 41, 1013–1021.
Kolpin, D. W., Furlong, E. T., Meyer, M. T., Thurman, E. M., Zaugg, S. D., Barber, L. B., & Buxton, H. T. (2002). Pharmaceuticals, hormones, and other organic wastewater contaminants in US streams, 1999-2000: a national reconnaissance. Environmental Science & Technology, 36(6), 1202–1211.
Mauriz, E., Calle, A., Lechuga, L. M., Quintana, J., Montoya, A., & Manclus, J. J. (2006). Real-time detection of chlorpyrifos at part per trillion levels in ground, surface and drinking water samples by a portable surface plasmon resonance immunosensor. Analytica Chimica Acta, 561(1–2), 40–47.
Méité, L., Soro, B. D., Aboua, N. K., Mambo, V., Traoré, K. S., Mazellier, P., De Laat, J. (2016). Qualitative determination of photodegradation products of progesterone and testosterone in aqueous solution. American Journal of Analytical Chemistry, 07(01), 22–33.
Planas, C., Puig, A., Rivera, J., & Caixach, J. (2006). Analysis of pesticides and metabolites in Spanish surface waters by isotope dilution gas chromatography/mass spectrometry with previous automated solid-phase extraction—estimation of the uncertainty of the analytical results. Journal of Chromatography A, 1131(1–2), 242–252.
Salas, D., Borrull, F., Fontanals, N., & Marce, R. M. (2017). Hydrophilic interaction liquid chromatography coupled to mass spectrometry-based detection to determine emerging organic contaminants in environmental samples. TrAC Trends in Analytical Chemistry, 94, 141–149.
Schultz, M. M., Painter, M. M., Bartell, S. E., Logue, A., Furlong, E. T., Werner, S. L., & Schoenfuss, H. L. (2011). Selective uptake and biological consequences of environmentally relevant antidepressant pharmaceutical exposures on male fathead minnows. Aquatic Toxicology, 104(1–2), 38–47.
Schulz, R., Hahn, C., Bennett, E. R., Dabrowski, J. M., Thiere, G., & Peall, S. K. C. (2003). Fate and effects of azinphos-methyl in a flow-through wetland in South Africa. Environmental Science & Technology, 37(10), 2139–2144.
Schwarzenbach, R. P., Escher, B. I., Fenner, K., Hofstetter, T. B., Johnson, C. A., von Gunten, U., & Wehrli, B. (2006). The challenge of micropollutants in aquatic systems. Science, 313(5790), 1072–1077.
Sharma, V. K. (2008). Oxidative transformations of environmental pharmaceuticals by Cl2, ClO2, O3, and Fe (VI): kinetics assessment. Chemosphere, 73(9), 1379–1386.
Soler, C., Hamilton, B., Furey, A., James, K. J., Manes, J., & Pico, Y. (2007). Liquid chromatography quadrupole time-of-flight mass spectrometry analysis of carbosulfan, carbofuran, 3-hydroxycarbofuran, and other metabolites in food. Analytical Chemistry, 79(4), 1492–1501.
Sumpter, J. P., Donnachie, R. L., & Johnson, A. C. (2014). The apparently very variable potency of the anti-depressant fluoxetine. Aquatic Toxicology, 151, 57–60.
Taheran, M., Naghdi, M., Brar, S. K., Verma, M., & Surampalli, R. Y. (2018). Emerging contaminants: here today, there tomorrow! Environmental Nanotechnology, Monitoring & Management, 10, 122–126.
Tolgyesi, A., Verebey, Z., Sharma, V. K., Kovacsics, L., & Fekete, J. (2010). Simultaneous determination of corticosteroids, androgens, and progesterone in river water by liquid chromatography-tandem mass spectrometry. Chemosphere, 78(8), 972–979.
Tran, N. H., Reinhard, M., & Gin, K. Y. (2018). Occurrence and fate of emerging contaminants in municipal wastewater treatment plants from different geographical regions-a review. Water Research, 133, 182–207.
Van Maele-Fabry, G., Gamet-Payrastre, L., & Lison, D. (2017). Residential exposure to pesticides as risk factor for childhood and young adult brain tumors: a systematic review and meta-analysis. Environment International, 106, 69–90.
de Vidales, M. J. M., Saez, C., Canizares, P., & Rodrigo, M. A. (2012). Electrolysis of progesterone with conductive-diamond electrodes. Journal of Chemical Technology and Biotechnology, 87, 1173–1178.
Acknowledgments
The authors would like to also thank Dr. Casey Burton and Charles Roberts for their editing assistance on this manuscript.
Funding
This study received funding from the Missouri Department of Natural Resources.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic Supplementary Material
ESM 1
(DOCX 49 kb)
Rights and permissions
About this article

Cite this article
Zhang, H., West, D., Shi, H. et al. Simultaneous Determination of Selected Trace Contaminants in Drinking Water Using Solid-Phase Extraction-High Performance Liquid Chromatography-Tandem Mass Spectrometry. Water Air Soil Pollut 230, 28 (2019). https://doi.org/10.1007/s11270-018-4066-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11270-018-4066-9