
Abstract
Bacterial communities and denitrifiers in polluted paddy soils near e-waste processing centers were explored to investigate the effects of e-waste-derived heavy metals on soil bacteria. The abundance of denitrifying genes (narG, nirK, and nosZ), as well as the total bacterial community in soils, was slightly influenced by heavy metals. However, heavy metals, especially Ni and Cu, had a greater effect on the community structure of denitrifiers than other soil factors. Significant correlations were observed between heavy metals and the abundance of some dominant denitrifiers (P < 0.05). The Cu and Ni content had a significant positive effect on Methylobacterium based on the relative abundance of nitrite reducers to nitrous oxide reducers (P < 0.05). The exploration of the abundance and composition of denitrifiers in e-waste-contaminated paddy soils and their relationship with heavy metal content in soil offer an insight into the influence of heavy metals on denitrification in such soils.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
E-waste refers to the electronic products that have reached the end of their useful life. Recycling and disposal of e-waste are serious environmental problems in developing regions, including China (Awasthi et al. 2016; Schmidt 2006). Because these processes often lack pollution control measures, toxic pollutants, such as heavy metals and persistent organics, are released into the soil or to the river environment and cause serious health problems for people who live nearby (Kiddee et al. 2013; Tang et al. 2010; Xue et al. 2017; Wu et al. 2015, 2016, 2017, 2018). Paddy soil fields around e-waste recycling workshops, such as those in the town of Guiyu in Guangdong Province, China, have been extensively polluted by Cu, Cd, Ni, and other metals (Luo et al. 2011).
Soil microorganisms have a great impact on soil nutrient cycle and might influence the growth and metal uptake of plants; they are also sensitive to the changes in the surrounding environment (Rojas-Tapias et al. 2014; Sabadini-Santos et al. 2014; Zou et al. 2018). Heavy metal contaminants present substantial challenges to the maintenance of phylogenetically and functionally diverse soils (Subrahmanyam et al. 2016). For example, excessive Cu content impacts the urease activity and the community structure of soil microbes (Ge and Zhang 2011). The structures of bacterial communities and soil enzymes are also influenced by e-waste-derived heavy metal pollution (Tang et al. 2014; Zhang and Fan 2014). Soil enzymes are generally considered effective indicators of the activities of soil microorganisms and directly involved in element cycling in soils (Killham and Staddon 2002). Thus, the exploration of the functional genes of key enzymes of soil microbial communities in mechanisms such as nitrogen cycling may help to understand the role of heavy metals in microbial-mediated processes.
Microbial denitrification is a respiratory process that consists of four consecutive reaction steps, in which nitrate is reduced to dinitrogen gas, making it an essential component of nitrogen cycling (Liu et al. 2015; Zumft 1997). Among the established biomarkers, the narG gene encoding the alpha catalytic subunit of respiratory nitrate reductase, the nirK gene encoding the copper nitrite reductase (Braker et al. 2000; Henry et al. 2004), and the nosZ gene encoding the nitrous oxide reductase (Scala and Kerkhof 1998, 1999) have been most extensively applied in the assessment of the microbial diversity of the denitrification process in natural environments owing to their high conservation prosperity in gene sequences among microorganisms (Chon et al. 2011). Denitrification is sensitive to heavy metals, and the inhibition of denitrification is dependent on the amount of heavy metals in soils (Holtan-Hartwig et al. 2002). The inhibition of denitrification may have a close relationship with the changes in denitrifiers. The denaturing gradient gel electrophoresis (DGGE) analysis showed that metal pollution significantly influenced the composition of denitrifiers in paddy soils (Liu et al. 2016). However, there has been no detailed information on the relationship between heavy metal pollution and the structure of denitrifying communities in paddy soils around e-waste recycling workshops. The objective of this study was to identify the structure and diversity of denitrifying communities and total bacterial communities in paddy soils polluted by e-waste and to reveal the relationships between heavy metal content and the structure and diversity of denitrifiers.
2 Materials and Methods
2.1 Sample Collection
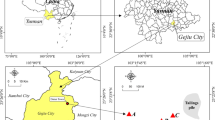
Field sampling was conducted in January 2013. Two polluted rice fields around a pickling field (N 116°37′, E 23°33′) and an e-waste recycling dump (N 116°35′, E 23°34′), hereafter referred to as LDTR and LGTR, respectively, along with a control site (CK) (N 116°22′, E 23°19′) in the town of Guiyu, Guangdong Province, were selected for this study. Ten soil samples from each site were randomly collected at the 0–20 cm soil layer. A portion of each soil sample was air-dried, passed through an 80-mesh sieve, and prepared for heavy metal and physicochemical properties analysis. The other portion of each soil sample was kept at − 20 °C for microflora analysis.
2.2 Heavy Metal and Elementary Properties Analysis
The Cd, As, Pb, Cr, Cu, Zn, and Ni contents in soils were determined by inductively coupled plasma optical emission spectroscopy (ICP-OES) (PerkinElmer, Optima 2100DV) after digestion with a mixture of HNO3-HCl-HClO4 (4:1:1, v:v:v) (Hseu et al. 2002). The pH values were measured with a pH meter (Sartorius, 3C). Nitrite and nitrate in the soils were extracted with 2 mol/L KCl. Nitrite was determined spectrophotometrically at 540 nm with N-(1-naphthyl) ethylenediamine dihydrochloride, and nitrate was measured with a continuous flow analyzer (SAN++, Skalar, Breda, Holland) (Chen et al. 2011).
2.3 DNA Extraction and 16S rRNA Gene Analysis
DNA was extracted from 0.5 g soil samples with a Powersoil™ DNA extraction kit (Mo Bio Labs, USA) according to the manufacturer’s instructions and stored at − 20 °C for further molecular analysis. The V4 region of the 16S rRNA gene was amplified from each soil sample with primers and reaction conditions as outlined in Table S1. Polymerase Chain Reaction (PCR) products were purified with an E.Z.N.A.® Cycle-Pure Kit (OMEGA, USA), measured by a Qubit fluorometer (Life Technologies, Paisley, UK), and pooled in equimolar amounts. The 16S rRNA gene amplicon pool was then sequenced on the Illumina MiSeq system at the MACROGEN DNA Synthesis and Sequencing Facility (Seoul, Korea).
Quality trimming and high-quality set aligning for 16S rRNA sequences were performed using MOTHUR software package (Schloss et al. 2009). The alpha diversity of the bacterial communities, including Chao1, Shannon, and phylogenetic diversity (PD), was assessed from operational taxonomic unit (OTU) with an evolutionary distance of 0.03 (or 97% 16S rRNA gene sequence similarity) using Quantitative Insights Into Microbial Ecology (QIIME) software as the default parameters (Caporaso et al. 2010). The taxonomic composition analysis was conducted using a Ribosomal Database Project (RDP) classifier with an 80% threshold (Wang et al. 2007).
2.4 Quantitative Real-Time PCR Assay
Three denitrifying genes, narG, nirK, and nosZ, along with 16S rRNA were selected for the quantitative real-time PCR (qPCR) assay. The number of copies of the targeted genes was quantified in each sample using extracted DNA as a template for qPCR, and the primer sets are listed in Table S1. The qPCR was carried out in a MyiQ5 Cycler (BIO-RAD, USA). PCR reactions were performed following the qPCR kit manufacturer’s instructions (Zhang et al. 2009).
Linearized plasmids of ρGEM-T Easy Vector (Promega, USA) harboring PCR-amplified inserts of the targeted genes were used as template DNA to construct standard curves (Kidron et al. 2015). The specificity of each PCR assay was confirmed using both melting curve analysis and agarose gel electrophoresis. All the measurements were performed in triplicate.
2.5 Clone Library Construction of Denitrification Genes
Nine clone libraries of narG, nirK, and nosZ genes were constructed from mixed soil samples of LDTR, LGTR, and CK, respectively. The primer sets for the amplification of narG, nirK, and nosZ are listed in Table S1. PCR was conducted for each soil sample with a total volume of 50 μL (Wu et al. 2011). All PCR products were purified using an E.Z.N.A.® Gel Extraction Kit (OMEGA, USA) after being analyzed with 1.5% (w/v) agarose gels. The purified PCR products of the genes were mixed in equimolar amounts, ligated into the ρGEM-T Easy Vector (Promega, USA), and transformed into E. coli DH5α (Takara, Japan) cells. Transformants were selected on Luria-Bertani agar plates and verified using colony PCR. Approximately 120 positive recombinants from each of the nine libraries of narG, nirK, and nosZ were randomly selected for sequence analysis by BGI-Tech (BGI, China). The taxonomic information of clone sequences was acquired by comparing it to the National Center for Biotechnology Information-non-redundant (NCBI-nr) database using Basic Local Alignment Search Tool (BLAST) (Gołębiewski et al. 2014).
2.6 Terminal Restriction Fragment Length Polymorphism (T-RFLP) of Denitrifying Genes
The primers in the terminal restriction fragment length polymorphism (T-RFLP) analysis of narG, nirK, and nosZ were the same as those used in clone library construction (Table S1), and the forward primer was fluorescently labeled with 6-FAM. The PCR amplification was conducted in a total volume of 50 μL containing 5 μL of 10 × Ex Taq buffer, 4 μL dNTP mixture (2.5 mM each), 0.25 μL of 5 U μL−1 Ex Taq HS DNA polymerase (Takara, Shiga, Japan), 1 μL of purified soil DNA, and 1 μL of primer (10 μM each). The thermocycling steps were the same as those in clone library construction. Aliquots of purified PCR products were digested with the AluI restriction enzyme at 37 °C for 12 h and examined by electrophoresis on a 2% agarose gel. Fluorescently labeled T-RFs were detected using an ABI PRISM 310 genetic analyzer (Applied Biosystems, California, USA) after being separated by capillary electrophoresis. The fragment lengths and intensity were analyzed using Genescan software (Applied Biosystems). The data were subjected to quality control procedures, including T-RF alignment (clustering threshold = 2 base pairs (bp)) and noise filtering (peak area, standard deviation multiplier = 1). Besides, T-RFs with a proportion below 0.5% and size below 50 bp were excluded from subsequent analyses. The corresponding microorganism of a T-RF was accessed by matching the number of bp with the sequence from the clone libraries.
2.7 Statistical Analysis
One-way ANOVA (LSD) was conducted in SPSS 20.0 to assess the variance among the relative abundance of major microorganisms, soil properties, and heavy metal content at the LDTR, LGTR, and CK sites. The redundancy analysis (RDA) and virtual population analysis (VPA) of soil chemical and bacteria community structure properties were performed using Vegan package in R (http://vegan.r-forge.r-project.org). Moreover, Pearson’s correlation coefficients between bacterial community parameters and soil chemical properties were calculated using a two-tailed test implemented in SPSS 19.0. Statistical significance was determined at the 95% level (P < 0.05).
2.8 Sequence Accession
The 16S rRNA gene sequences reported in this study have been deposited in the NCBI BioProject under accession number SRP090656.
3 Results
3.1 Soil Characteristics and Heavy Metal Content
The soil samples collected in LDTR contained significantly higher Cu (378 mg/kg) and Ni (90 mg/kg) concentrations than those collected in LGTR and CK (P < 0.05); the Ni content in the LGTR soil was significantly higher than that in the CK soil (P < 0.05) (Table 1). Moreover, the average concentration of Ni and Cu in the soil of LDTR and LGTR exceeded the environmental quality standard for soils (the class II standard of National Soil Standard, China). However, the average concentrations of As and Cd in the soil of the three sites were very similar and far below the environmental quality standard for soils. In addition, pH (4.7~5.6) value and the concentrations of organic matter (OM) (38~59 mg/kg), NO2− (0.52~0.73 mg/kg), and NO3−(14~26 mg/kg) in soils from the three sites, especially from LDTR and LGTR, were close to each other (Table 1).
3.2 Composition and Diversity of e-Waste-Polluted Soil Microbial Communities
Chao1, PD, and the Shannon indices were determined to calculate the diversity of microbial communities (Table S2). Soil samples from LDTR had similar microbial richness and diversity (Chao1 38,861 ± 6409; PD 849 ± 75; Shannon 11 ± 0.45) to those from LGTR (Chao1 34,083 ± 4251; PD 802 ± 68; Shannon 11 ± 0.43) and CK (Chao1 36,757 ± 3579; PD 852 ± 62; Shannon 11 ± 0.35) at the same sequencing depth (normalized to 29,990 sequences per sample) (P > 0.05).
The overall microbial composition of the soil from LDTR, LGTR, and CK was similar (Fig. 1). Betaproteobacteria and Anaerolineae were the two most dominant orders, representing 16 and 9.7% of the LDTR samples, 15 and 8.6% of the LGTR samples, and 12 and 15% of the CK samples, respectively. At the family level, Hydrogenophilaceae, Enterobacteriaceae, and Gallionellaceae represented the most dominant lineages of all three sites and accounted for 5.0%, 2.5%, and 2.5% of all three sites, respectively (Fig. S1).

Relative abundance of dominant bacteria and archaea in LDTR, LGTR, and CK at the order level. Error bars indicate the standard errors of the relative abundance of dominant bacteria and archaea in soil samples from the same site
3.3 The Relationship Between Heavy Metal Content and Microbial Composition
RDA analysis showed that heavy metal concentrations and other physicochemical properties had a weak relationship with microbial diversity indices (Table S3). In contrast, more than 90% of the variance in soil bacterial composition could be explained by heavy metals and other soil properties investigated, and the permutation test indicated that both axes were significant (P < 0.05) (Table S3). Thus, the heavy metal content and other physicochemical properties might have a large impact on the soil bacterial composition at different taxa levels at the LDTR, LGTR, and CK sites (Table S3). The VPA analysis further illustrated that the heavy metals were the dominant factors that shaped the soil bacterial community structure (Fig. 2). Ni, followed by Cu, was the most significant dominant factor that shaped the soil bacterial composition (P < 0.05) (Table S4). Moreover, Bacteroidetes, Proteobacteria, and Chlorobi were significantly correlated with Ni and Cu (P < 0.05) (Table 2).

Virtual population analysis (VPA) of heavy metals, pH, and OM as well as NO2− and NO3− on the community structure of bacteria at the phylum level
3.4 Characteristics of Denitrifying Microbial Communities under e-Waste Pollution
The qPCR assays showed that all four genes were similar in copy numbers in soils from LDTR, LGTR, and CK; their differences did not reach significant levels (P > 0.05) (Table S5). In regard to the 16S rRNA gene, the copy number per gram soil of LDTR, LGTR, and CK was 5.7 × 109 ± 3.1 × 109, 4.4 × 109 ± 4.1 × 109, and 3.7 × 109 ± 2.7 × 109 (mean ± SE), respectively. Meanwhile, per gram soil of LDTR, LGTR, and CK contained 4.2 × 105 ± 6.5 × 105, 3.6 × 105 ± 3.2 × 105, and 1.5 × 105 ± 1.4 × 105 copies of narG, 3.1 × 105 ± 3.7 × 105, 4.4 × 105 ± 4.6 × 105, and 5.4 × 105 ± 5.8 × 105 copies of nirK, and 6.5 × 106 ± 6.2 × 106, 8.2 × 106 ± 7.0 × 106, and 7.7 × 106 ± 5.9 × 106 copies of nosZ, respectively.
Approximately 120 clones were randomly selected from the libraries of LDTR, LGTR, and CK soil that targeted narG, nirK, or nosZ as marker genes, providing an insight into the taxonomic composition of denitrifying communities in e-waste polluted soils. Figure 2 shows that there are some differences in the phylogenetic distribution of the communities with different denitrifying genes, while the difference among the three sites was also obvious. Deinococci was the most dominant order containing the narG gene, while Alphaproteobacteria was the most dominant order containing the nirK or the nosZ gene. Moreover, Betaproteobacteria represented another most dominant order of clones with the nosZ gene, accounting for 18%, 25%, and 60% of the clones in LDTR, LGTR, and CK, respectively. In contrast, only less than 9% of the clones containing nirK belonged to Betaproteobacteria. Likewise, Azoarcus genus in Betaproteobacteria family was the most dominant genus of clones with the nosZ gene, but not the nirK gene, and represented 22% of the nosZ clone library. Bradyrhizobium was the dominant genus both in nirK and nosZ libraries and accounted for 8% of both clone libraries (Fig. 3). Furthermore, the relative abundance of some orders was much higher in polluted sites than in the control site, suggesting that these orders might be metal resistant. For example, Deltaproteobacteria, a dominant order in the narG library accounted for 20 and 16% of the clones in LDTR and LGTR, respectively, while it only accounted for 4% of the clones in CK.

Community structure of denitrifying bacteria at the order level in soils of LDTR, LGTR, and CK based on the clone libraries of narG (a), nirK(b), and nosZ (c)
To further investigate the diversity of the denitrifiers, the PCR-amplified narG, nirK, and nosZ fragments from soils in LDTR, LGTR, and CK were subjected to AluI digestion and analyzed by T-RFLP (Table S6-S8). For all three genes, the major T-RFs and the genera they belonged to were varied among the three study sites, especially between polluted (LDTR and LGTR) and unpolluted (CK) sites. The predominant T-RFs of narG fragments in LDTR, LGTR, and CK belonged to Achromobacter (358 bp, with 11.4% ± 13.7% relative abundance), Achromobacter (136 bp, 5.09% ± 6.34%), and Thauera (568 bp, 11.6% ± 16.4%). Moreover, the T-RF of 287 bp assigned to Meiothermus frequently occurred both in LDTR (5.12% ± 9.00%) and LGTR (3.49% ± 9.60%) (Table S6). For nirK, the dominant T-RFs of 174 bp and 350 bp in LDTR and LGTR, as well as the T-RF of 238 bp in CK, were related to Bradyrhizobium (Table S7). The T-RFs for the nosZ gene assigned to Bradyrhizobium were also found in LDTR (237 bp, 11.5% ± 10.7%), LGTR (237 bp, 5.33% ± 6.67%), and CK (193 bp, 3.24% ± 2.63%) (Table S8).
3.5 The Correlation Between Soil Properties and Denitrifying Microbial Communities
In order to determine the extent of the soil properties that affect denitrifier communities, the gene copy numbers and the relative abundance of the major genera with narG, nirK, and nosZ were analyzed using RDA (Table S9). The copy number of narG, nirK, and nosZ was only slightly impacted by the soil properties. Furthermore, the soil properties might be important in explaining the relative abundance of major genera with narG, nirK, and nosZ genes, indicated by the P value (< 0.01) of the permutation test on three corresponding RDA analyses (Table S9). Similar to the total bacterial community, heavy metals were also the dominant factors that shaped the community structure of denitrifying bacteria. Heavy metals could explain 50.6%, 46.1%, and 48.0% of the variation in the relative abundance of the major denitrifier genera with narG, nirK, and nosZ genes, respectively (Fig. 3). Ni was also the most important factor that determined the community structure of denitrifying bacteria (P < 0.05) (Table S10).
Based on the correlation analysis between the soil properties and relative abundance of denitrifying bacteria with the narG gene, Ni and Cu positively influenced Meiothermus (P < 0.05) (Table 3). With regard to the species with the nirK gene, Methylobacterium and Bradyrhizobium were positively correlated with Ni and Cu (P < 0.05), while significant negative correlations were detected between Nocardia and Ni and Cu (P < 0.05). Caulobacter, Anaeromyxobacter, and Mesorhizobium were also negatively correlated with Ni content in soil (P < 0.05) (Table 3). Among the bacteria containing the nosZ gene, Anaeromyxobacter and Oligotropha (P < 0.05) were negatively affected by Ni and Cu, while Mesorhizobium was positively affected by Ni and Cu (Table 3). Ni also positively affected the relative abundance of Bradyrhizobium (P < 0.05) (Table 3).
4 Discussion
4.1 Effects of Heavy Metals on the Diversity and Structure of Bacterial Communities
Zn, Hg, and Cu might lead to a simultaneous decrease in microbial community diversity in soil (Garcia-Sanchez et al. 2015; Li et al. 2015). It was also reported that the contents of residual Cd and Zn could explain 89% and 43% of the variations in the abundance of total bacteria and metal-resistant bacteria in paddy soil contaminated by e-waste (Zhang and Fan 2014). However, previous DGGE analysis results showed that there was little correlation between the microbial community diversity and slightly elevated contents of Cu and Cd in e-waste-polluted paddy soils (Tang et al. 2014). Few correlations were observed between heavy metals and the diversity index of soil microbial communities in this study (Table S3), likely caused by the moderate metal content in the soil. Moreover, bacteria in soil, for example, Cytophagaceae, Chitinophagaceae, Acidobacteriaceae, KD4-96, Sphingomonadaceae, and Nitrosomonadaceae, might have adapted to heavy metals such as Zn (Gołębiewski et al. 2014). While, in large-scale studies, the local variability of soil properties may obscure the effect of heavy metals on the compositions of the soil bacteria (Chodak et al. 2013). However, in this study, the RDA analysis indicated that heavy metals, especially Ni and Cu, had a greater influence on the structure of soil bacterial communities than other soil properties such as soil pH and NO3− (Table S4, Fig. 3). The pH and NO3− of the investigated soils were somewhat similar at the study sites; however, Ni and Cu concentrations were rather different. In addition, Bacteroidetes and Proteobacteria were positively correlated with Cu and Ni (P < 0.05) (Table 2), suggesting that these bacteria might resist to Cu and Ni and their stress-response mechanism to Cu and Ni needs further research.
4.2 Effects of Heavy Metals on the Abundance and Structure of Denitrifying Microbial Communities
Denitrification process is sensitive to heavy metals (Liu et al. 2016; Sobolev and Begonia 2008). Thus, the copy number of denitrifying genes such as narG, nirK, and nosZ might be significantly impacted by heavy metal concentrations. However, the RDA analysis suggested that there was only a slight correlation between the copy number of narG, nirK, and nosZ genes and the heavy metal content in polluted soil (Table S9). The abundance of nirK genes, representing the nitrite-reducing bacteria, was not affected by elevated Cu, Zn, and Cd levels in paddy soils (Liu et al. 2014). The copy number of nirK could temporarily be inhibited under the stress of Cu, Zn, and Cd (Holtan-Hartwig et al. 2002). Thus, the adaptation of denitrifiers to heavy metals might lead to the poor relationship between the copy number of denitrifying genes and heavy metals.
Species such as Bradyrhizobium sp. (Torres et al. 2011), Marinithermus sp. (Sikorski et al. 2010), and Hyphomicrobium sp. (Venkatramanan et al. 2013), which play an important role in denitrification, were also identified in nirK and nosZ clone libraries in this study. Some of the denitrifying bacteria identified in our study were resistant to heavy metals. The abundance of Meiothermus genus was tightly associated with Cu in the soil of copper mines (Pereira et al. 2015). Methylobacterium was also identified in the rhizosphere and endosphere of the Ni hyperaccumulating plant Thlaspi goesingense and exhibited characteristics of Ni resistance (Idris et al. 2006). In our study, the relative abundance of soil denitrifying communities largely depended on the concentrations of heavy metals, especially Ni and Cu (Fig. 4, Table S10). Ni and Cu were both significantly related to major denitrifying genera, including Meiothermus as nitrate-reducers, Bradyrhizobium, Methylobacterium, and Nocardia as nitrite reducers, and Mesorhizobium, Anaeromyxobacter, and Oligotropha as nitrous oxide reducers (P < 0.05) (Table 3). The likely reason for the tight relationship between Cu and denitrifying bacteria is that the expression of the gene encoding the nitrous oxide reductase (nosZ), which converts N2O to N2, is regulated by extracellular Cu concentrations (Sullivan et al. 2013). Our research suggests that Ni might be related to denitrification via the change in the relative abundance of major denitrifying bacteria. Further research is needed to illustrate the relationship between the changes in major denitrifying bacteria and their interactions under the stress of heavy metals.

Virtual population analysis (VPA) of soil properties on the community structure of denitrifying bacteria with narG (a), nirK (b), and nosZ (c) genes
5 Conclusion
Our study elucidates the major genera of denitrifying bacteria, as well as the whole microorganism community in two e-waste-polluted paddy soil sites containing elevated Ni and Cu and a control site. The possible relationship between heavy metals and bacteria in soils was also explored. Statistical analysis showed that Ni was the most important factor, followed by Cu, that controls the relative abundance of major denitrifying bacteria or total bacteria in two e-waste-polluted paddy soil sites. However, the influence of Ni and Cu on the abundance of the microorganism community was not obvious. Significant correlations (P < 0.05) were found between the metal concentrations and the abundance of specific denitrifying bacteria, such as Bradyrhizobium, Meiothermus, and Methylobacterium.
References
Awasthi, A. K., Zeng, X., & Li, J. (2016). Environmental pollution of electronic waste recycling in India: a critical review. Environmental Pollution, 211, 259–270.
Braker, G., Zhou, J., Wu, L., Devol, A. H., & Tiedje, J. M. (2000). Nitrite reductase genes (nirK and nirS) as functional markers to investigate diversity of denitrifying bacteria in pacific northwest marine sediment communities. Applied and Environmental Microbiology, 66, 2096–2104.
Caporaso, J. G., Kuczynski, J., Stombaugh, J., Bittinger, K., Bushman, F. D., Costello, E. K., et al. (2010). QIIME allows analysis of high-throughput community sequencing data. Nature Methods, 7, 335–336.
Chen, Y. L., Xu, Z. W., Hu, H. W., Hu, Y. J., Hao, Z. P., Jiang, Y., et al. (2011). Responses of ammonia-oxidizing bacteria and archaea to nitrogen fertilization and precipitation increment in a typical temperate steppe in Inner Mongolia. Applied Soil Ecology, 68, 36–45.
Chodak, M., Gołębiewski, M., Morawska-Płoskonka, J., Kuduk, K., & Niklińska, M. (2013). Diversity of microorganisms from forest soils differently polluted with heavy metals. Applied Soil Ecology, 64, 7–14.
Chon, K., Chang, J. S., Lee, E., Lee, J., Ryu, J., & Cho, J. (2011). Abundance of denitrifying genes coding for nitrate (narG), nitrite (nirS), and nitrous oxide (nosZ) reductases in estuarine versus wastewater effluent-fed constructed wetlands. Ecological Engineering, 37, 64–69.
Garcia-Sanchez, M., Garcia-Romera, I., Cajthaml, T., Tlustos, P., & Szakova, J. (2015). Changes in soil microbial community functionality and structure in a metal-polluted site: the effect of digestate and fly ash applications. Journal of Environmental Management, 162, 63–73.
Ge, C. R., & Zhang, Q. C. (2011). Microbial community structure and enzyme activities in a sequence of copper-polluted soils. Pedosphere, 21(2), 164–169.
Gołębiewski, M., Deja-Sikora, E., Cichosz, M., Tretyn, A., & Wróbel, B. (2014). 16S rDNA pyrosequencing analysis of bacterial community in heavy metals polluted soils. Microbial Ecology, 67(3), 635–647.
Henry, S., Baudoin, E., Lopez-Gutierrez, J. C., Martin-Laurent, F., Brauman, A., & Philippot, L. (2004). Quantification of denitrifying bacteria in soils by nirK gene targeted real-time PCR. Journal of Microbiological Methods, 59(3), 327–335.
Holtan-Hartwig, L., Bechmann, M., Hoyas, T. R., Linjordet, R., & Bakken, L. R. (2002). Heavy metal tolerance of soil denitrifying communities: N2O dynamics. Soil Biology and Biochemistry, 34, 1181–1190.
Hseu, Z. Y., Chen, Z. S., Tsai, C. C., Tsui, C. C., Cheng, S. F., Liu, C. L., et al. (2002). Digestion methods for total heavy metals in sediments and soils. Water, Air, and Soil Pollution, 141, 189–205.
Idris, R., Kuffner, M., Bodrossy, L., Puschenreiter, M., Monchy, S., Wenzel, W. W., et al. (2006). Characterization of Ni-tolerant methylobacteria associated with the hyperaccumulating plant Thlaspi goesingense and description of Methylobacterium goesingense sp. nov. Systematic and Applied Microbiology, 29, 634–644.
Kiddee, P., Naidu, R., & Wong, M. H. (2013). Metals and polybrominated diphenyl ethers leaching from electronic waste in simulated landfills. Journal of Hazardous Materials, 252-253, 243–249.
Kidron, G. J., Posmanik, R., Brunner, T., & Nejidat, A. (2015). Spatial abundance of microbial nitrogen-transforming genes and inorganic nitrogen in biocrusts along a transect of an arid sand dune in the Negev Desert. Soil Biology and Biochemistry, 83, 150–159.
Killham, K., & Staddon, W. J. (2002). Bioindicators and sensors of soil health and the application of geostatistics. In R. G. Burns & R. P. Dick (Eds.), Enzymes in the environment (pp. 391–405). New York: Marcel Dekker Inc.
Li, X., Bond, P. L., Van Nostrand, J. D., Zhou, J., & Huang, L. (2015). From lithotroph- to organotroph-dominant: directional shift of microbial community in sulphidic tailings during phytostabilization. Scientific Reports, 5, 12978.
Liu, Y., Ding, Y., Zheng, J., Zhou, T., Pan, G., Crowley, D., et al. (2014). Abundance, composition and activity of ammonia oxidizer and denitrifier communities in metal polluted rice paddies from South China. PLoS One, 9, e102000.
Liu, X., Chen, C., Wang, W., Hughes, J. M., Lewis, T., Hou, E., et al. (2015). Vertical distribution of soil denitrifying communities in a wet sclerophyll forest under long-term repeated burning. Microbial Ecology, 70(4), 993–1003.
Liu, Y., Zhou, H., Li, L., Zheng, J., Zhang, X., & Pan, G. (2016). Abundance, composition and activity of denitrifier communities in metal polluted paddy soils. Scientific Reports, 6, 19086.
Luo, C., Liu, C., Wang, Y., Liu, X., Li, F., Zhang, G., et al. (2011). Heavy metal contamination in soils and vegetables near an e-waste processing site, South China. Journal of Hazardous Materials, 186(1), 481–490.
Pereira, L. B., Vicentini, R., & Ottoboni, L. M. (2015). Characterization of the core microbiota of the drainage and surrounding soil of a Brazilian copper mine. Genetics and Molecular Biology, 38(4), 484–489.
Rojas-Tapias, D. F., Bonilla, R., & Dussán, J. (2014). Effect of inoculation and co-inoculation of Acinetobacter sp. RG30 and Pseudomonas putida GN04 on growth, fitness, and copper accumulation of maize (Zea mays). Water, Air, and Soil Pollution, 225(12), 1–13.
Sabadini-Santos, E., Silva, T. S. D., Lopes-Rosa, T. D., Mendonça-Filho, J. G., & Santelli, R. E. (2014). Microbial activities and bioavailable concentrations of Cu, Zn, and Pb in sediments from a tropic and Eutrothicated Bay. Water, Air, and Soil Pollution, 225(5), 1–11.
Scala, D. J., & Kerkhof, L. J. (1998). Nitrous oxide reductase (nosZ) gene-specific PCR primers for detection of denitrifiers and three nosZ genes from marine sediments. FEMS Microbiology Letters, 162(1), 61–68.
Scala, D. J., & Kerkhof, L. J. (1999). Diversity of nitrous oxide reductase (nosZ) genes in continental shelf sediments. Applied and Environmental Microbiology, 65(4), 1681–1687.
Schloss, P. D., Westcott, S. L., Ryabin, T., Hall, J. R., Hartmann, M., Hollister, E. B., et al. (2009). Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Applied and Environmental Microbiology, 75(23), 7537–7341.
Schmidt, C. W. (2006). Unfair trade: e-waste in Africa. Environmental Health Perspectives, 114(4), 232–235.
Sikorski, J., Tindall, B. J., Lowry, S., Lucas, S., Nolan, M., Copeland, A., et al. (2010). Complete genome sequence of Meiothermus silvanus type strain (VI-R2). Standards in Genomic Sciences, 3(1), 37–46.
Sobolev, D., & Begonia, M. F. (2008). Effects of heavy metal contamination upon soil microbes: lead-induced changes in general and denitrifying microbial communities as evidenced by molecular markers. International Journal of Environmental Research and Public Health, 5(5), 450–456.
Subrahmanyam, G., Shen, J. P., Liu, Y. R., Archana, G., & Zhang, L. M. (2016). Effect of long-term industrial waste effluent pollution on soil enzyme activities and bacterial community composition. Environmental Monitoring and Assessment, 188(2), 112.
Sullivan, M. J. G., Appia-Ayme, C., Rowley, G., & Richardson, D. (2013). Copper control of bacterial nitrous oxide emission and its impact on vitamin B12-dependent metabolism. PNAS, 110, 199926–119931.
Tang, X., Shen, C., Shi, D., Cheema, S. A., Khan, M. I., Zhang, C., et al. (2010). Heavy metal and persistent organic compound contamination in soil from Wenling: an emerging e-waste recycling city in Taizhou area, China. Journal of Hazardous Materials, 173(1–3), 653–660.
Tang, X., Hashmi, M. Z., Long, D., Chen, L., Khan, M. I., & Shen, C. (2014). Influence of heavy metals and PCBs pollution on the enzyme activity and microbial community of paddy soils around an e-waste recycling workshop. International Journal of Environmental Research and Public Health, 11(3), 3118–3131.
Torres, M. J., Rubia, M. I., Bedmar, E. J., & Delgado, M. J. (2011). Denitrification in Sinorhizobium meliloti. Biochemical Society Transactions, 39(6), 1886–1889.
Venkatramanan, R., Prakash, O., Woyke, T., Chain, P., Goodwin, L. A., Watson, D., et al. (2013). Genome sequences for three denitrifying bacterial strains isolated from a uranium- and nitrate-contaminated subsurface environment. Genome Announcements, 1(4), e00449–e00413.
Wang, Q., Garrity, G. M., Tiedje, J. M., & Cole, J. R. (2007). Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Applied and Environmental Microbiology, 73(16), 5261–5267.
Wu, Z., Zou, L., Lu, D., & Liu, Z. (2011). Restoration of taxonomic and functional genes after bioaugmentation of petroleum contaminated soil. Journal of Environmental Monitoring, 13(10), 2904–2913.
Wu, C., Zou, Q., Xue, S. G., Mo, J. Y., Pan, W. S., Lou, L. Q., & Wong, M. H. (2015). Effects of silicon (Si) on arsenic (As) accumulation and speciation in rice (Oryza sativa L.) genotypes with different radial oxygen loss (ROL). Chemosphere, 138, 447–453.
Wu, C., Zou, Q., Xue, S. G., Pan, W. S., Yue, X., Hartley, W., Huang, L., & Mo, J. Y. (2016). Effect of silicate on arsenic fractionation in soils and its accumulation in rice plants. Chemosphere, 165, 478–486.
Wu, C., Huang, L., Xue, S. G., Shi, L. Z., Hartley, W., Cui, M. Q., & Wong, M. H. (2017). Arsenic sorption by red mud-modified biochar produced from rice straw. Environmental Science and Pollution Research, 24(22), 18168–18178.
Wu, C., Wang, Q. L., Xue, S. G., Pan, W. S., Lou, L. Q., Li, D. J., & Hartley, W. (2018). Do aeration conditions affect arsenic and phosphate accumulation and phosphate transporter expression in rice (Oryza sativa L.)? Environmental Science and Pollution Research, 25, 43–51.
Xue, S. G., Shi, L. Z., Wu, C., Wu, H., Qin, Y. Q., Pan, W. S., Hartley, W., & Cui, M. Q. (2017). Cadmium, lead, and arsenic contamination in paddy soils of a mining area and their exposure effects on human HEPG2 and keratinocyte cell-lines. Environmental Research, 156, 23–30.
Zhang, J. H., & Fan, W. W. (2014). Metal partitioning and relationships to soil microbial properties of submerged paddy soil contaminated by electronic waste recycling. Chemistry and Ecology, 31(2), 147–159.
Zhang, B., Sun, B., Ji, M., & Liu, H. (2009). Population dynamic succession and quantification of ammonia-oxidizing bacteria in a membrane bioreactor treating municipal wastewater. Journal of Hazardous Materials, 165(1–3), 796–803.
Zou, Q., An, W. H., Wu, C., Li, W. C., Fu, A. Q., Xiao, R. Y., Chen, H. K., & Xue, S. G. (2018). Red mud-modified biochar reduces soil arsenic availability and changes bacterial composition. Environmental Chemistry Letters, 16(2), 615–622.
Zumft, W. G. (1997). Cell biology and molecular basis of denitrification. Microbiology and Molecular Biology Reviews, 61(4), 533–616.
Funding
This work was supported by grants from the National Science Foundation of China (Grant No. 41330857, 31700470), Natural Science Foundation of Guangdong Province (2017A030313218), Natural Science Foundation of Guangxi Province (2017GXNSFBA198099), and Guangxi Key Laboratory project (YB2014201, 2016TZYKF10).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Electronic Supplementary Material
ESM 1
(DOCX 177 kb)
Rights and permissions
About this article

Cite this article
Deng, D., Hu, M., Li, L. et al. Denitrifying Microbial Communities in Heavy-Metal-Contaminated Paddy Soils near Electronic-Waste Processing Centers. Water Air Soil Pollut 229, 318 (2018). https://doi.org/10.1007/s11270-018-3974-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11270-018-3974-z