
Abstract
Invertebrate iridescent virus 6 (IIV6) is a nucleocytoplasmic insect virus and a member of the family Iridoviridae. The IIV6 genome consists of 212,482 bp of linear dsDNA with 215 non-overlapping and putative protein-encoding ORFs. The IIV6 118L ORF is conserved in all sequenced members of the Iridoviridae and encodes a 515 amino acid protein with three predicted transmembrane domains and several N-glycosylation/N-myristoylation sites. In this study, we characterized the 118L ORF by both deleting it from the viral genome and silencing its expression with dsRNA in infected insect cells. The homologous recombination method was used to replace 118L ORF with the green fluorescent protein (gfp) gene. Virus mutants in which the 118L gene sequence had been replaced with gfp were identified by fluorescence microscopy but could not be propagated separately from the wild-type virus in insect cells. Unsuccessful attempts to isolate the mutant virus with the 118L gene deletion suggested that the protein is essential for virus replication. To support this result, we used dsRNA to target the 118L gene and showed that treatment resulted in a 99% reduction in virus titer. Subsequently, we demonstrated that 118L-specific antibodies produced against the 118L protein expressed in the baculovirus vector system were able to neutralize the virus infection. All these results indicate that 118L is a viral envelope protein that is required for the initiation of virus replication.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Iridoviridae, a family of nucleocytoplasmic viruses, is classified into two subfamilies Alphairidovirinae and Betairidovirinae. Iridovirids (members of the family Iridoviridae) in the Betairidovirinae subfamily are classified into three genera (Iridovirus, Chloriridovirus, and Decapodiridovirus) with members infecting invertebrates and thus named invertebrate iridescent viruses (IIVs) [1].
Invertebrate iridescent virus 6 (IIV6) is the type species of the Iridovirus genus. The virion structure comprises a central DNA/protein core surrounded in turn by an internal lipid membrane, an icosahedral protein capsid, and, in those viruses released by budding, an outer viral envelope that may also display a fringe of fibrils [1]. The IIV6 genome consists of 212,482 bp of linear, double-stranded DNA with 215 putative protein-encoding open reading frames (ORFs). Proteomic analysis has shown that IIV6 particles contain 54 structural, viral-encoded proteins [2]. These structural proteins constitute the capsid, and contribute to the envelope, and other components of the virion. Viral membrane proteins play important roles in regulating virus replication or virulence [3]. Some envelope or membrane proteins of iridovirids have been identified and functionally analyzed including 53R of Frog virus 3 (FV3) [4]; 53R, 2L, and 43R of Rana grylio virus (RGV) [5,6,7]; VP019, VP039, and VP088 of Singapore grouper iridovirus (SGIV) [8,9,10]; 007L, 056L, and 118L of Infectious spleen and kidney necrosis virus (ISKNV) [11]; and 168L of Decapod iridescent virus 1 (DIV1) [12].
IIV6 118L, one of the 54 structural protein coding ORFs, encodes a putative envelope protein. 118L protein consists of 515 amino acids, including three predicted transmembrane domains and several N-glycosylation/N-myristoylation sites. 118L has homologs in all other sequenced iridovirids and thus it is accepted as one of the 26 core proteins of the family Iridoviridae. Knockdown and knockout studies with its homologs, 53R of FV3, 53R of RGV, VP088 of SGIV, showed significant reductions in viral titers, suggesting that homologs are essential for viral infectivity, replication, and assembly [4, 13, 14]. Identifying the function of IIV6 118L protein will contribute significantly to clarifying the biology of the virus and its use as an experimental model.
To understand the contribution of a target viral gene in replication and pathogenesis in vivo, an efficient strategy is to construct a recombinant virus lacking the target gene [15]. Another widely used method to examine the gene function is to silence the expression of gene products [16,17,18]. In this study, we examined the function of IIV6 118L by both deleting and silencing it in the viral genome. Recombinant IIV6 (rIIV6Δ118-GFP) was generated by inserting a green fluorescent protein (gfp) gene in place of 118L ORF through homologous recombination. The significance of the 118L for IIV6 was demonstrated by the inhibition of virus replication when the gene was silenced by gene-specific dsRNAs. Specific antibodies were also used to block the 118L protein and to determine its function in virus infection.
Materials and methods
Cell line and viruses
Spodoptera frugiperda (Sf9) cells maintained at 28 °C in SF900II-SFM (Gibco) medium containing 5% fetal bovine serum (FBS, Sigma) were used for virus infections.
Invertebrate iridescent virus 6 (IIV6) and its recombinant (rCIVΔ157L-gfp) [19], that expressed the green fluorescent protein gene (gfp), were used in this study. The rCIVΔ157L-gfp displays similar characteristics to IIV6 except that it causes cells to fluoresce green when infected. rCIVΔ157L-gfp provides a sensitive and rapid quantitative assay for screening virus infections. Virus titers were determined by endpoint dilution assay (EPDA) and expressed as the tissue culture infectious dose 50 (TCID50 units/ml) [20].
Sequence analysis
So far, there are eight known IIV species whose complete genomes are available [1]. These are IIV6, IIV31, IIV3, IIV9, IIV22, IIV25, Anopheles minimus iridovirus (AMIV), and Decapod iridescent virus 1 (Shrimp hemocyte iridescent virus/SHIV). All of these have the homologs of IIV6 118L. The protein sequences of IIV6 118L and its homologs in other IIVs were downloaded from the NCBI database. Multiple amino acid sequence alignment was carried out using ClustalW Multiple alignment and edited with GeneDoc software. The prediction of myristoylation and glycosylation sites was performed using the ScanProsite tool. Transmembrane domains were searched using the transmembrane finder program (https://www.uniprot.org/help/transmem).
Generation of the recombinant virus with 118L deletion
To examine the importance of 118L for virus replication and infectivity, a recombinant virus, lacking the 118L gene, was generated through homologous recombination. A transfer vector was prepared carrying gfp under the control of the IIV6 major capsid protein gene (mcp; 274L) promoter between sequences, homologous to the left and right flanking regions of the 118L ORF. For this purpose, upstream (951 bp) and downstream (848 bp) sequences of the 118L ORF were amplified by polymerase chain reaction (PCR) from the viral genome and cloned into the pBlueScript/SK(-) vector (Stratagene). The gfp gene was amplified from the pJET1.2-mcp-pr-gfp vector [19] with the mcp promoter using mcp-Fw and gfp-2Rv primers and inserted between the flanking regions of 118L in the recombinant pBlueScript/SK(-). The primers used during the construction of the transfer vector are shown in Table 1.
The generated transfer vector was transfected into the Sf9 cells, 18 h after infection with IIV6 at a multiplicity of infection (MOI) of 5 TCID50 unit/cell in six-well plates. The transfection procedure was detailed in [19]. Transfections were incubated at 28 °C until the green fluorescent protein (GFP) was abundantly observed. Five days later, the supernatant containing progeny virus was collected and serial plaque assays were performed to purify the recombinant virus (rCIV-Δ118L/gfp) based on the green fluorescence of the plaques. Plaque purification was repeated several times. The purity of the recombinant virus was checked by PCR using 118L-specific primers (118L-Fw: 5’-GAGAATTCCAATGGACTG TGAGAGAAAT, 118L-Rv: 5’-GACTCGAGGCTGTTATTGTAGATGGTGGAC-3’) to detect the presence of wild-type IIV6 and specific primers (IIV6 MCP Fw: 5’-CCAAGCTTCAATACATAACAATCTTTCAT-3’, gfp-2Rv: 5’-CGGAATTCTCACTTGTA CAGCTCGTCCAT-3’), amplifying the cloned gfp and its promoter, to detect the recombinant IIV6.
Gene silencing
dsRNA-mediated gene silencing was performed to temporarily suppress the expression of 118L ORF. A 320 bp region of 118L was amplified by PCR from viral DNA with 118L-Fw and 118L-Rv primers. A heterologous control of about 286 bp was also generated for the gfp gene from the pJET1.2-mcp-pr-gfp plasmid using gfp-Fw and gfp-Rv primers. The PCR products were gel purified and used as templates for amplifying the T7 RNA polymerase promoter sequence bearing fragments at both ends by a second PCR. dsRNAs were generated using the T7 RiboMAX Express RNAi system (Promega), by following the manufacturer’s instructions. The dsRNAs were then purified by isopropanol extraction, dissolved in RNase-free water, and quantified by Nanodrop 2000 (Thermo Scientific). The primers used for dsRNA production are shown in Table 2.
Sf9 cells (1.5 × 106 cells/well) were transfected with 100 μg dsRNA to silence the expression of 118L in six-well plate. At 24 h post-transfection, cells were infected with recombinant IIV6 (rCIV-Δ157L-gfp) at a moi of 3. Recombinant IIV6, generated in our previous study has the same replication kinetics as the wild-type virus, but includes gfp that makes it easy to trace. Treatment with dsRNA against gfp was used as a positive control for the silencing process, as it is expressed by the recombinant virus upon replication. Non-transfected but infected cells were used as a negative control. All cells were harvested at 24 hpi. Supernatants were used for the determination of progeny virus production by EPDA. Three replicates were performed for both 118L and gfp silencing experiments.
Production of 118L protein and antisera
The 118L gene of IIV6 was amplified by PCR using primers Fw 5’- GAGAATTCCAATGGACTGTGAGAGAAATATCT-3’ and Rv 5’- AAGCTTGCCTATAACCATTGTTTTTTACTTTCAA-3’ from the viral genome and cloned under the polyhedrin (polh) promoter in the baculovirus transfer vector pFastBacTMHT A. The recombinant transfer vector (pFastBacHT A-118L) was transformed into E. coli DH10Bac cells harboring helper plasmid to generate a recombinant Autographa californica multiple nucleopolyhedrovirus (AcMNPV) bacmid encoding the IIV6-118L protein. Recombinant bacmid DNA was first confirmed by using M13Fw and M13Rv universal primers, then transfected into Sf9 cells using Lipofectamine 2000 (Invitrogen) in a six-well plate following the manufacturer’s instructions. Transfected cells were incubated at 28 °C for 72 h. Subsequently, the medium with cells in wells was harvested and the supernatant containing the budded virus was obtained by centrifuging the cells at 600xg for 5 min. The obtained supernatant was saved as recombinant virus stock (Bac-AcMNPV-IIV6-118L). The recombinant virus was amplified, and the viral titer was determined by EPDA.
To produce a large amount of protein, Sf9 cells were infected at a moi of 5. At 72 h post-infection, cell suspension was centrifuged at 600 × g for 5 min and the cell pellet was used for protein purification. 118L protein fused to 6xHis-tag at its N-terminal was purified using MagneHis Protein Purification System (Promega) kit to collect His-tagged proteins. The concentration of the protein was estimated by the Bradford method [21]. 118L protein expression (0,45 μg) was observed by SDS-PAGE and western blot analysis using an anti-His monoclonal primary antibody produced in mouse at 1:1000 dilution. Following the first antibody, the alkaline phosphatase conjugated goat anti-mouse IgG antibody at 1:1000 dilution was used as the secondary antibody. Detection was performed using p-nitroblue tetrazolium chloride and 5-bromo-4-chloro-3-indolyl phosphate (NBT/BCIP) substrate for alkaline phosphatase.
Purified fusion protein (2 mg) was mixed with an equal volume of Freund’s Complete Adjuvant and used to immunize mice by hypodermal injection (first injection). The inoculations were performed a total of 4 times at intervals of 2 weeks. Two weeks after the fourth immunization, blood samples, including the first and last injections, were titrated against the protein used in inoculations by checkerboard ELISA. Three days before removing the spleen of mice, 118L fusion protein (without adjuvant) was inoculated intraperitoneally. After the 5th inoculation, anti-IIV6 118L serum was collected, and its specificity was validated.
Neutralization assay
To determine whether 118L encodes an envelope protein that is important in virus entry, a neutralization assay was performed as described previously [14]. Virus neutralization assay was carried out on 96-well cell culture plate, where about 2 × 104 cells were inoculated into each well. Recombinant IIV6 (rCIVΔ157L-gfp), including gfp, was incubated with anti-118L at a final concentration of 8 μg /ml for 1 h at room temperature. As a control, the same amount of recombinant IIV6 was incubated with the irrelevant antibody anti-AMV248 of Amsacta moorei entomopoxvirus (AMEV). After incubation, a 100 μl volume of mixtures containing virus and antibody was added to cells in each well. The inocula were removed 2 h later and 200 μl of culture medium was added to each well. The infected cells together with supernatant were collected at regular intervals and stored at −80 °C until titer determination by TCID50 analysis.
Results
IIV6 118L sequence is conserved among known IIVs
Amino acid sequence alignment revealed several conserved features, including three transmembrane domains, several N-glycine myristoylation and N-Glycosylation sites, and 5 invariant cysteines (Fig. 1). The N-terminal region of the IIV6 118L homologs is more conserved than the C-terminal region. Besides, species in each genus (IIV6 and IIV31 belong to Iridovirus genus, IIV3, IIV9, IIV22, IIV25 and Anopheles minimus iridovirus (AMIV) belong to Chloriridovirus genus, and Shrimp hemocyte iridescent virus (SHIV) belongs to the Decapod iridovirus genus) show higher homology with one another than with species in other genera (Fig. 1).

Multiple sequence alignment of IIV6-118L homologs in invertebrate iridescent viruses (IIVs). IIV6 118L amino acid sequence and all homologs in IIVs are included in the alignment. IIV6 and IIV31 belong to the Iridovirus genus, IIV3, IIV9, IIV22, IIV25 and Anopheles minimus iridovirus (AMIV) belong to the Chloriridovirus genus, and Shrimp hemocyte iridescent virus (SHIV) belongs to the Decapodiridovirus genus. Open reading frame numbers of IIV6 118L homologs are written after abbreviated names. Conserved columns are shaded (black, 100%; dark gray, 80%; light gray, 80–60%). The myristylation and glycosylation sites are indicated as M and G, respectively, and a short line under these letters shows the responsible amino acid positions. The predicted transmembrane (TM) domains are shown with long lines indicating the amino acids constituting TM domain. The conserved cysteines are indicated by an arrowhead
Construction of 118L deleted IIV6
To determine whether 118L is necessary for viral replication and infectivity, a recombinant virus lacking the 118L ORF and harboring gfp was generated (Fig. 2A). GFP fluorescence was observed in cells as a result of homologous recombination between the transfer vector and the IIV6 genome (Fig. 2B). Transfection liquid was harvested 5 days later, and plaque assay was performed to purify the recombinant virus (rIIV6Δ118L-gfp) from the transfection mixture containing wild-type virus (Fig. 2C). The purity of the recombinant virus was verified by PCR once after the 5th cycle of plaque purification and then after every five successive plaque purification cycles. The plaque procedure was repeated twenty times, but still the PCR product of viral DNA obtained from the final plaque assay showed a 559 nt fragment indicating the presence of the wild-type virus along with a 1032 nt fragment of the recombinant virus (Fig. 2D). The inability to isolate the mutant virus with the 118L gene deletion indicates that protein is necessary for virus replication.

Generation and characterization of recombinant Invertebrate iridescent virus 6, expressing gfp, with 118L ORF deletion (rIIV6Δ118L-gfp). A Schematic diagram of the construction of the recombinant virus. B Bright-field and fluorescence images of transfection result, produced by the recombinant virus as a result of homologous recombination between the transfer vector and the wild-type IIV6 genome. C Bright-field and fluorescence images of plaques, produced by the recombinant IIV6, viewed under fluorescent and halogen light. D PCR products of viral DNAs were obtained from four different plaques at the final plaque assay. 1–4: PCR results with 118L-specific primers showing the presence of wild-type IIV6-specific fragment, 5–7: PCR results of the same DNA with primers, specific to placed gfp and its promoter, showing the presence of the recombinant IIV6-specific fragment. M: Thermo Scientific GeneRuler 1 kb Plus DNA Ladder
RNA interference-mediated gene silencing
To evaluate the effect of 118L silencing on virus titers, the expression of 118L was silenced by the dsRNA-mediated RNAi assay. Supernatants obtained from 118L-specific dsRNA-treated cells were used to determine the production of infectious virions by EPDA. The average virus titers produced in cells treated with 118L- and gfp-specific dsRNAs were recorded as 6.53 × 104 and 6.01 × 107, respectively. Results showed that 118L-specific dsRNA treatment resulted in a 99.8% decrease in progeny virus production compared to the gfp-specific dsRNA-treated cells (Fig. 3).

Virus yield of Sf9 cells treated with dsRNAs. Cells were treated with dsRNA-gfp and dsRNA-118L 24 h before recombinant IIV6 (rCIV-Δ157L-gfp) infection. Non-transfected but infected cells were also used as control. At 24 h p.i., cells were harvested. Supernatants were used for the determination of progeny virus production by EPDA. Results are the averages and standard deviations of three replicates from dsRNA-treated cell supernatants
Expression of 118L protein
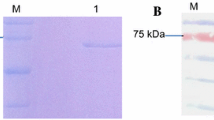
The recombinant 118L protein expressed in the baculovirus vector system with a 6xHis-tag fused to its N-terminus was confirmed by SDS-PAGE (10%) and western blot analysis before the antibody production. SDS-PAGE result (Fig. 4A) and western analysis (Fig. 4B) with an anti-6 × His-tag-specific antibody showed the presence of the 415R fusion protein expressed at approximately 34 kDa in size.

Confirmation of the purified 118L protein sample by SDS-PAGE (0.45 μg protein) (A) and western blot (B) analyses. Arrows show the protein sample locations (~ 34 kDa). Marker; Prestained Protein Ladder (Fisher BioReagents)
Neutralization assay
A virus neutralization assay was performed to determine whether the 118L protein was involved in virus infection. IIV6 was incubated with anti-118L prior to inoculation into Sf9 cells. A time course analysis revealed a decrease in virus titer at all intervals examined, which became clear from 36 hpi onwards when IIV6 was neutralized by incubation with 8 μl/ml of anti-118L prior to inoculation. As a control, pre-incubation with anti-AMEV248 as an irrelevant antibody had no effect on viral titer (Fig. 5). IIV6 neutralization with 118L antibody markedly reduced the virus titer. This result indicates that 118L is likely to be located on the surface of the virus particle and may be involved in virus entry.

Neutralization of IIV6 infection in vitro on Sf9 cells by mouse anti-IIV6 118L antibody. IIV6 neutralization with anti-118L serum markedly reduced the virus titer at 36 hpi to 96 hpi. Each data point represents the average titer of three independent infections. Error bars indicate standard deviations
Discussion
IIV6 118L is a highly conserved, virion-associated protein that is present in all five genera within the family Iridoviridae [2, 22]. Studies with 118L homologs, 53R of FV3, 53R of RGV, VP088 of SGIV, showed its importance in virus infectivity, replication, and assembly [4, 13, 14]. The detailed function of 118L in IIV6 replication remained to be determined. In this study, we have characterized the function of 118L by deleting the gene from the viral genome, silencing its expression during viral infection, and neutralizing the protein on virion structure.
The 118L protein that is conserved among all sequenced iridovirids contains three transmembrane (TM) domains and several N-myristoylation/N-glycosylation sites. One of the TM domains is at the C-terminus of the protein, while the other two are located in the central section of the amino acid sequence. Previous reports have suggested that virus-encoded transmembrane (TM) domain-containing proteins invariably participate in the process of membrane fusion between the virus and host cells [23, 24]. 118L contains thirteen potential N-myristoylation sites. Myristoylated viral proteins play important roles in assembly, structure, budding, intracellular host interactions, and viral entry [25]. These proteins establish associations with cellular membranes through hydrophobic myristoyl groups, enabling them to integrate into lipid bilayers [26]. The conserved cysteines found in 118L may be involved in the formation of disulfide bonds. For vaccinia virus, A16 protein that is required for the entry of poxviruses into cells, the conserved cysteine residues were disulfide bonded via the poxvirus cytoplasmic redox system [27]. In addition, 118L contains a total of ten potential N-glycosylation sites. Glycosylation can have significant effects on the interactions between viruses and cell surface attachment factors and may also have effect on virion stability and virus replication. Envelope protein glycosylation has been identified as a virulence determinant for multiple flaviviruses [28]. As such, 118L appears to be a significant virus protein that plays roles during several steps of virus replication including virus attachment, fusion to host, particle formation, and release from the cell.
A recombinant virus was generated to test the gene functionality. The 118L deletion mutant of IIV6 was constructed in this study using a conventional homologous recombination technique. The 118L coding sequence was deleted and replaced with sequence encoding green fluorescent protein (GFP). The recombinant virus in which 118L had been replaced with gfp was identified by fluorescence microscopy but could not to be propagated separately from the wild-type virus despite multiple plaque purification steps. No fluorescence was observed in plaques prepared from the extended dilutions of the previous plaques. This result showed that the 118L-negative virus was generated only in the presence of 118L protein provided by the wild-type virus during mixed infection but failed when the mutant and the wild-type virus were separated at higher dilutions. Our inability to isolate the pure IIV6 118L deletion mutant strongly indicates that 118L protein is essential for virus replication.
Genes essential for the viral lifecycle cannot be deleted from the viral genome without loss of replication. For example, Cao and McFadden (2001) attempted to generate a recombinant myxoma virus carrying M118L deletion but failed to segregate the pure M118L deletion mutant from wild-type parental virus [29]. They concluded that M118L is an essential poxvirus structural component of intracellular mature virus (IMV) particles. Similarly, Armat et al., (2022) deleted glycoproteins E and I from the genome of Infectious laryngotracheitis virus (ILTV) but were unable to purify the recombinant virus from the wild-type virus [30]. They noticed that deletion produced an in vivo phenotype of single-infected cells with no cell-to-cell spread. Recombinant viruses, lacking essential genes or have protein deficiency, can be created using different solutions. He et al. (2013) generated a conditional lethal mutant of Rana grylio virus (RGV) in which the expression of the 53R protein, which is homologous to IIV6 118L, was regulated by Isopropyl-beta-D-thiogalactoside (IPTG) [31]. They observed that in the absence of IPTG, 53R expression was strongly reduced, and that caused a sharp decrease in plaque formation and virus titers. The result from Rana virus 53R is consistent with our findings for IIV6 118L and suggests that these proteins are functionally similar.
The RNA silencing method was used to investigate the effects of inhibiting the IIV6 118L gene in the viral genome on virus replication. Results showed that virus production decreased sharply in cells in which 118L was silenced. Frog virus 3 ORF 53R, an ortholog of IIV6 118L, was studied using antisense morpholine oligonucleotides (asMOs) technology [4]. Those results showed that the knockdown of 53R resulted in a decrease in virion formation and a reduction in the formation of mature virions. The consequences of gene silencing in iridovirids have also been studied on several other genes [16,17,18, 32,33,34].
Antibodies produced against specific viral envelope proteins have been used successfully in neutralization assays to identify envelope proteins involved in viral infections [7, 8, 35, 36]. A mouse anti-118L polyclonal antibody exhibited IIV6-neutralizing activity in vitro, suggesting that 118L is involved in IIV6 infection. A similar neutralization study, performed on a 118L homolog in Singapore grouper iridovirus (SGIV), the VP088 envelope protein, also resulted in a significant reduction in virus titer [14].
Iridovirids possess icosahedral virions that are infectious either as non-enveloped or enveloped particles [1]. The fact that homologs of the IIV6 118L protein have been shown to be envelope proteins [5, 11] and the observation that virus infection is significantly reduced upon treatment of IIV6 with 118L-specific antibodies indicates that the 118L is an envelope protein and is necessary for virus replication. However, it is not yet known whether 118L is present in the virus structure in non-enveloped virions or, if not, which protein functions instead.
In conclusion, neutralization of IIV6 with 118L antibody and knockdown of 118L expression led to a marked decrease in IIV6 titer, and all these results, together with the non-producible 118L knockout mutant, suggest that 118L plays an essential role in IIV6 infection. Protein sequence analysis revealed the existence of transmembrane domains, myristoylation and glycosylation sites, and supported the conclusion that 118L likely functions as an envelope protein. Neutralization assays further revealed that anti-118L serum could reduce IIV6 infection in Sf9 cells. We believe that these results will aid our understanding of the envelope structure of iridoviruses, as well as the molecular mechanisms of iridovirus infection. In addition, this study paved the way for further studies to understand the role of 118L at each stage of the viral lifecycle.
Data availability
Not applicable.
References
Chinchar VG, Hick P, Ince IA, Jancovich JK, Marschang R, Qin Q, Subramaniam K, Waltzek TB, Whittington R, Williams T, Zhang QY (2017) ICTV virus taxonomy profile: iridoviridae. J Gen Virol 98:890–891. https://doi.org/10.1099/jgv.0.000818
Ince IA, Boeren SA, van Oers MM, Vervoort JJ, Vlak JM (2010) Proteomic analysis of chilo iridescent virus. Virology 405:253–258. https://doi.org/10.1016/j.virol.2010.05.038
DiMaio D (2014) Viral miniproteins. Annu Rev Microbiol 68:21–43. https://doi.org/10.1146/annurev-micro-091313-103727
Whitley DS, Yu K, Sample RC, Sinning A, Henegar J, Norcross E, Chinchar VG (2010) Frog virus 3 ORF 53R, a putative myristoylated membrane protein, is essential for virus replication in vitro. Virology 405:448–456. https://doi.org/10.1016/j.virol.2010.06.034
Zhao Z, Ke F, Huang YH, Zhao JG, Gui JF, Zhang QY (2008) Identification and characterization of a novel envelope protein in Rana grylio virus. J Gen Virol 89:1866–1872. https://doi.org/10.1099/vir.0.2008/000810-0
He LB, Ke F, Wang J, Gao XC, Zhang QY (2014) Rana grylio virus (RGV) envelope protein 2L: subcellular localization and essential roles in virus infectivity revealed by conditional lethal mutant. J Gen Virol 95:679–690. https://doi.org/10.1099/vir.0.058776-0
Zeng XT, Gao XC, Zhang QY (2018) Rana grylio virus 43R encodes an envelope protein involved in virus entry. Virus Genes 54:779–791. https://doi.org/10.1007/s11262-018-1606-8
Zhang H, Zhou S, Xia L, Huang X, Huang Y, Cao J, Qin Q (2015) Characterization of the VP39 envelope protein from Singapore grouper iridovirus. Can J Microbiol 61:924–937. https://doi.org/10.1139/cjm-2015-0118
Huang X, Gong J, Huang Y, Ouyang Z, Wang S, Chen X, Qin Q (2013) Characterization of an envelope gene VP19 from Singapore grouper iridovirus. Virol J 10:354. https://doi.org/10.1186/1743-422X-10-354
Zhou S, Wan Q, Huang Y, Huang X, Cao J, Ye L, Lim TK, Lin Q, Qin Q (2011) Proteomic analysis of Singapore grouper iridovirus envelope proteins and characterization of a novel envelope protein VP088. Proteomics 11:2236–2248. https://doi.org/10.1002/pmic.200900820
Dong CF, Xiong XP, Shuang F, Weng SP, Zhang J, Zhang Y, Luo YW, He JG (2011) Global landscape of structural proteins of infectious spleen and kidney necrosis virus. J Virol 85:2869–2877. https://doi.org/10.1128/JVI.01444-10
Zheng Q, Wang W, Zhao F, Lin S, Chen J (2023) Identification and characterization of an envelope protein 168L in cherax quadricarinatus iridovirus (CQIV). Virus Res 323:198967. https://doi.org/10.1016/j.virusres.2022.198967
Kim YS, Ke F, Lei XY, Zhu R, Zhang QY (2010) Viral envelope protein 53R gene highly specific silencing and iridovirus resistance in fish cells by AmiRNA. PLoS ONE 5(4):10308. https://doi.org/10.1371/journal.pone.0010308
Yuan Y, Wang Y, Liu Q, Zhu F, Hong Y (2016) Singapore grouper iridovirus protein VP088 is essential for viral infectivity. Sci Rep 6:31170. https://doi.org/10.1038/srep31170
Guangchun C, Brian MW, Kwang HY, Chinchar VG, Jacques R (2011) Improved knockout methodology reveals that frog virus 3 mutants lacking either the 18K immediate-early gene or the truncated vIF-2 gene are defective for replication and growth in vivo. J Virol 85:11131–11138. https://doi.org/10.1128/JVI.05589-11
Sample R, Bryan L, Long S, Majji S, Hoskins G, Sinning A, Olivier J, Chinchar VG (2007) Inhibition of iridovirus protein synthesis and virus replication by antisense morpholino oligonucleotides targeted to the major capsid protein, the 18 kDa immediate-early protein, and a viral homolog of RNA polymerase II. Virology 358:311–320. https://doi.org/10.1016/j.virol.2006.07.009
Xie J, Lü L, Deng M, Weng S, Zhu J, Wu Y, Gan L, Chan SM, He J (2005) Inhibition of reporter gene and Iridovirus-tiger frog virus in fish cell by RNA interference. Virology 338:43–52. https://doi.org/10.1016/j.virol.2005.04.040
Dang LT, Kondo H, Hirono I, Aoki T (2008) Inhibition of red seabream iridovirus (RSIV) replication by small interfering RNA (siRNA) in a cell culture system. Antivir Res 77:142–149. https://doi.org/10.1016/j.antiviral.2007.10.007
Ozgen A, Muratoglu H, Demirbag Z, Vlak JM, van Oers MM, Nalcacioglu R (2014) Construction and characterization of a recombinant invertebrate iridovirus. Virus Res 189:286–292. https://doi.org/10.1016/j.virusres.2014.05.012
Darling AJ, Boose JA, Spaltro J (1998) Virus assay methods: accuracy and validation. Biologicals 26:105–110. https://doi.org/10.1006/biol.1998.0134
Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72:248–254. https://doi.org/10.1006/abio.1976.9999
Eaton HE, Metcalf J, Penny E, Tcherepanov V, Upton C, Brunetti CR (2007) Comparative genomic analysis of the family Iridoviridae: re-annotating and defining the core set of iridovirus genes. Virol J 4:11. https://doi.org/10.1186/1743-422X-4-11
Cleverley DZ, Lenard J (1998) The transmembrane domain in viral fusion: essential role for a conserved glycine residue in vesicular stomatitis virus G protein. Proc Natl Acad Sci U S A 95:3425–3430. https://doi.org/10.1073/pnas.95.7.3425
Däubner T, Fink A, Seitz A, Tenzer S, Müller J, Strand D, Seckert CK, Janssen C, Renzaho A, Grzimek NK, Simon CO, Ebert S, Reddehase MJ, Oehrlein-Karpi SA, Lemmermann NA (2010) A novel transmembrane domain mediating retention of a highly motile herpesvirus glycoprotein in the endoplasmic reticulum. J Gen Virol 91:1524–1534. https://doi.org/10.1099/vir.0.018580-0
Maurer-Stroh S, Eisenhaber F (2004) Myristoylation of viral and bacterial proteins. Trends Microbiol 12:178–185. https://doi.org/10.1016/j.tim.2004.02.006
Maurer-Stroh S, Eisenhaber B, Eisenhaber F (2002) N-terminal N-myristoylation of proteins: refinement of the sequence motif and its taxon-specific differences. J Mol Biol 317:523–540. https://doi.org/10.1006/jmbi.2002.5425
Ojeda S, Senkevich TG, Moss B (2006) Entry of vaccinia virus and cell-cell fusion require a highly conserved cysteine-rich membrane protein encoded by the A16L gene. J Virol 80:51–61. https://doi.org/10.1128/JVI.80.1.51-61.2006
Carbaugh DL, Lazear HM (2020) Flavivirus envelope protein glycosylation: impacts on viral infection and pathogenesis. J Virol 94:e00104-e120. https://doi.org/10.1128/JVI.00104-20
Cao JX, McFadden G (2001) Characterization of the myxoma virus M118L protein: a novel essential poxvirus IMV-associated protein. Virus Genes 23:303–313. https://doi.org/10.1023/a:1012573306916
Armat M, Vaz PK, Browning GF, Noormohammadi AH, Hartley CA, Devlin JM (2022) Construction and characterisation of glycoprotein E and glycoprotein I deficient mutants of Australian strains of infectious laryngotracheitis virus using traditional and CRISPR/Cas9-assisted homologous recombination techniques. Virus Genes 58:540–549. https://doi.org/10.1007/s11262-022-01933-5
He LB, Gao XC, Ke F, Zhang QY (2013) A conditional lethal mutation in Rana grylio virus ORF 53R resulted in a marked reduction in virion formation. Virus Res 177:194–200. https://doi.org/10.1016/j.virusres.2013.07.016
Gencer D, Yesilyurt A, Ozsahin E, Muratoglu H, Acar Yazici Z, Demirbag Z, Nalcacioglu R (2023) Identification of the potential matrix protein of invertebrate iridescent virus 6 (IIV6). J Invertebr Pathol 197:107885. https://doi.org/10.1016/j.jip.2023.107885
Ince IA, Westenberg M, Vlak JM, Demirbaǧ Z, Nalçacioǧlu R, van Oers MM (2008) Open reading frame 193R of chilo iridescent virus encodes a functional inhibitor of apoptosis (IAP). Virology 376:124–131. https://doi.org/10.1016/j.virol.2008.03.009
Whitley DS, Sample RC, Sinning AR, Henegar J, Chinchar VG (2011) Antisense approaches for elucidating ranavirus gene function in an infected fish cell line. Dev Comp Immunol 35:937–948. https://doi.org/10.1016/j.dci.2010.12.002
Schofield DJ, Glamann J, Emerson U, Purchell RH (2000) Identification by phage display and characterization of two neutralizing chimpanzee monoclonal antibodies to the hepatitis E virus capsid protein. J Virol 74:5548–5555. https://doi.org/10.1128/jvi.74.12.5548-5555.2000
Wu WL, Wang L, Zhang XB (2005) Identification of white spot syndrome virus (WSSV) envelope proteins involved in shrimp infection. Virology 332:578–583. https://doi.org/10.1016/j.virol.2004.12.011
Funding
This work was supported by the Scientific and Technological Research Council of Turkey (TUBITAK) (Project no. 119Z209).
Author information
Authors and Affiliations
Contributions
Betul Altun: Methodology, Validation, Data Curation, and Writing—review & editing. Kubra Zengin: Methodology and Investigation. Sevde Yayli Dabag: Methodology and Investigation. Aydin Yesilyurt: Investigation, Visualization, and Writing—original draft. Remziye Nalcacioglu: Methodology, Investigation, and Writing—review & editing. Zihni Demirbag: Project administration, Formal analysis, Writing—review & editing, Supervision, and Funding acquisition.
Corresponding author
Ethics declarations
Conflict of interest
All the authors declared that there are no conflicts.
Additional information
Edited by Sassan Asgari.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Altun, B., Zengin, K., Yayli Dabag, S. et al. Characterization of an envelope protein 118L in invertebrate iridescent virus 6 (IIV6). Virus Genes 60, 549–558 (2024). https://doi.org/10.1007/s11262-024-02082-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11262-024-02082-7