
Abstract
We isolated and characterized a novel positive-sense, single-stranded RNA virus from Aedes larvae collected on Okushiri Island, Hokkaido, Japan. This virus, designated Okushiri virus (OKV), replicated in the Aedes albopictus cell line C6/36 with severe cytopathic effects and produced a large number of spherical viral particles that were 50-70 nm in diameter and released into the cell culture medium. The OKV genome consisted of 9,704 nucleotides, excluding the poly(A) tail at the 3′-terminus, and contained three major open reading frames (ORF1, ORF2, and ORF3). ORF1 encoded a putative protein of approximately 268 kDa that included a methyltransferase domain, FtsJ-like methyltransferase domain, helicase domain, and RNA-dependent RNA polymerase domain. The genome organization and results of a phylogenetic analysis based on the amino acid sequence predicted from the nucleotide sequence indicated that OKV is a member of a new insect virus group of negeviruses with a possible evolutionary relationship to some plant viruses. ORF2 and ORF3 were suggested to encode hypothetical membrane-associated proteins of approximately 45 kDa and 22 kDa, respectively. This is the first study on a novel negevirus isolated from mosquito larvae in Japan.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Mosquitoes (Diptera) are one of the most important pest insects that have a serious impact on public health; they are a principal vector of serious infectious diseases including malaria, dengue, chikungunya, Japanese encephalitis, and yellow fever. Approximately 3.2 billion people are estimated to live in malaria-endemic areas, and 584,000 malaria deaths were reported worldwide in 2013 (WHO, Malaria Report 2014).
Vector control is an essential strategy for reducing the transmission of mosquito-borne diseases. Since effective vaccines have yet to be developed for malaria and dengue, the importance of vector control is increasing [35]. Chemicals are a vital component of pesticides controlling mosquitoes; however, the low host specificity of chemical pesticides may lead to non-targeted insects being affected [25, 33]. The appearance of pesticide-resistant insects has also been reported [25, 32]. Mosquito-specific viruses with mosquitocidal activity may be used as an alternative to chemical pesticides. Although some mosquito viruses, such as baculoviruses (family Baculoviridae), densoviruses (family Parvoviridae, subfamily Densovirinae), iridoviruses (family Iridoviridae, genus Chloriridovirus), and cytoplasmic polyhedrosis viruses (family Reoviridae, genus Cypovirus), have been identified [4], molecular biology studies of these viruses are limited because cell lines suitable for investigating the replication of these viruses and effectively producing them in large amounts are not currently available. This has hampered the detailed microbiological analysis and industrial large-scale amplification of these viruses. One exception is mosquito densoviruses (MDV), which are highly specific for mosquitoes and were previously shown to be pathogenic in vitro and in vivo. These features make these viruses potentially attractive candidates as biological control agents for mosquitoes [6]. Since MDVs do not infect all mosquito species and may not be a sufficient measure for resistant insects, the isolation and characterization of novel mosquito viruses are still important steps for the development of environmentally friendly mosquito-management strategies. Novel insect-specific, positive-sense, single-stranded RNA viruses have recently been isolated from naturally infected mosquitoes and sand flies in geographically distant areas, and a new genus, “Negevirus”, has been proposed to include these viruses [2, 5, 12, 23, 34]. Experimental studies are still needed in order to investigate the characteristics of these viruses, such as their pathogenicity in vivo and replication mechanism. Nevertheless, the rapid and high-level replication of negeviruses in some mosquito cells (up to 1010 PFU/ml) [34] implies the possibility of developing some of these viral agents as biological control agents in the future. Here, we report the isolation of a novel negevirus, designated Okushiri virus (OKV), from pools of Aedes larvae collected in the field on Okushiri Island, Hokkaido, Japan.
Materials and methods
Collection of mosquito larvae
Mosquito larvae were captured in small pools in the field during the mosquito season in 2010. The mosquito larvae collected were sub-grouped into three different genera (Aedes, Culex, and Anopheles) based on their external characteristics and stored at -80 °C in 1.5-ml centrifuge tubes with a maximum of six larvae per tube.
Cell cultures
The mosquito (Aedes albopictus) cell line C6/36 was used to isolate viruses and analyze their properties. These cells were cultured in minimum essential medium Eagle (MEM, Sigma-Aldrich, St Louis, MO, USA) containing 10% heat-inactivated fetal bovine serum (FBS), 2% non-essential amino acids (NEAA, Sigma-Aldrich), 100 U of penicillin per ml (Gibco BRL, Gaitherburg, MD, USA), and 100 µg of streptomycin per ml (Gibco), and they were then maintained at 26 °C.
Virus isolation from mosquito larvae
Samples of larvae in 500 µl of ice-cold MEM containing 2% NEAA, 100 U of penicillin per ml, and 100 µg of streptomycin per ml were homogenized using a µT-01 bead crusher (Taitec) with stainless steel beads (2 mm, 70 g).
The homogenates were clarified by centrifugation at 1,000 × g at 4 °C for 5 min, and the supernatants were passed through sterile 0.22-µm filters (Ultrafree MC, Millipore, Bedford, MA, USA). The filtrates inoculated onto monolayers of C6/36 cells. The plates were incubated at 26 °C. After inoculation, cell cultures were incubated for approximately 5 days and then observed using phase contrast microscopy. The supernatants of cells with cytopathic effects (CPE) were stored at -80 °C as virus samples. The supernatants from cells without CPE were also stored at -80 °C after a second blind passage.
Purification of viral particles
Viral particles were purified from the culture medium of C6/36 cells inoculated with virus samples. The fluid was clarified from cell debris by low-speed centrifugation as above, and viral particles were precipitated by ultracentrifugation (250,000 × g at 4 °C for 5 hours). Pellets were suspended in TE buffer, layered onto a 10-40% linear sucrose gradient, and centrifuged at 99,600 × g for 2 hours. Viral particles that formed a white band in the gradient were resuspended in TE buffer, collected by centrifugation at 10,000 × g at 4 °C for 12 hours and resuspended in TE buffer.
Electron microscopic observations
Purified viral particles were used for electron microscopic observations. Viral particles were negatively stained and then observed using transmission electron microscopy (Hitachi H-800) as described elsewhere [30].
Characterization of the viral genome
Nucleic acids were extracted from the purified viral particles or supernatants of infected C6/36 cell cultures using TRIzol Reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions. The extracted nucleic acids were treated with DNase I or RNase A at 37 °C for 20 minutes, denatured with formamide and formaldehyde, and electrophoresed on a 1.0 % GTG agarose gel (Lonza). The agarose gels were stained with GelStarTM Nucleic Acid Gel Stain (Lonza) and visualized under ultraviolet illumination.
Complementary DNA strand synthesis, sequencing, and analysis of genome organization
The first complementary DNA (cDNA) strand was synthesized from viral RNA extracted from purified viral particles with random primers or oligo dT primers, using M-MLV reverse transcriptase (Takara), followed by second-strand cDNA synthesis, using DNA polymerase I, after the RNase H treatment. Double-stranded cDNA was trimmed to make blunt ends using T4 DNA polymerase, ligated into the SmaI site in the pGEM 3Zf (+) plasmid (Promega), and sequenced. Some primers designed from the sequences obtained were used in PCR to synthesize additional viral genome sequences (Table 1). The 3´ and 5´ sequences were obtained by rapid amplification of cDNA ends (RACE) using a Gene Racer kit (Invitrogen) according to the manufacturer’s instructions.
Viral genome sequences were assembled and analyzed using BLAST with the DDBJ, GenBank, and EMBL databases. Alignments of the sequences obtained with those of other RNA viruses were performed using the Clustal W program [16] included in MEGA5 [31] with the PAM matrix. A gap-opening penalty of 10 and a gap-extension penalty of 0.2 were used. A phylogenetic analysis was performed by the neighbor-joining method using MEGA5 [29], and evolutionary distances were computed using the Poisson correction method. Bootstrap values were calculated from 1,000 replicates [9]. The accession numbers used in the phylogenetic analysis are shown in Table 2b. The nucleotide and amino acid identities for the ORFs were analyzed using ApE software (Utah, United States). The protein sequences encoded in each ORF were characterized by conducting a CD Search using PSI-BLAST (http://blast.ncbi.nlm.nih.gov/Blast.cgi) [3]. A homology search was also carried out using PSI-BLAST. Glycosylation sites were predicted using the NetNGlyc server 1.0 (http://www.cbs.dtu.dk/services/NetNGlyc/), with a threshold value of 0.5. The programs TMHMM (http://www.cbs.dtu.dk/services/TMHMM) and SOSUI (http://harrier.nagahama-i-bio.ac.jp/sosui/sosui_submit.html) [11] were used to analyze the transmembrane domains of putative proteins.
Strand-specific RT-PCR
Total RNAs were extracted from purified OKV particles or OKV-infected C6/36 cells at 6 days postinfection (dpi) using TRIzol Reagent as described above and tested by strand-specific RT-PCR as described by Craggs et al. [8]. Total RNAs from uninfected cells were also extracted and used in strand-specific PCR as a control. Cellular RNAs and RNA extracted from purified viral particles were treated with DNase I at 37 °C for 20 minutes and then reverse transcribed using PrimeScript RTase (Takara) using positive- or negative-strand-specific primers (Table 1) at 50 °C for 15 minutes. The remaining primers and nucleotides in the RT-PCR reaction tubes were digested with ExoSSAP-IT (Usb) at 37 °C for 15 minutes and then used for PCR. RNA 1 strand-specific PCR was carried out using a positive-strand-specific primer set (OKV-F1, Tag) or a negative-strand-specific primer set (OKV-R1, Tag). PCR products were visualized by agarose gel electrophoresis.
Results
Isolation of a virus from mosquito larvae
Extensive CPE with cell globularization and destruction was observed in C6/36 cells inoculated with the homogenates of three (approximately 20 %) field-collected mosquito larval pools 72 hours after inoculation (Fig. 1). Virus-like particles were observed by electron microscopy in the supernatants of C6/36 cell cultures with extensive CPE, but not in the medium of control (uninoculated) cell cultures (data not shown). These virus-like particles purified by sucrose gradient centrifugation were elliptical, with a diameter of approximately 50 to 70 nm and had projection-like structures similar to those of Tanay virus [23] (Fig. 2). The purified particles were infectious to C6/36 cells [10] and caused extensive CPE, similar to those inoculated with the original homogenate (data not shown). The virus isolated here was named OKV after Okushiri Island, where the virus was isolated.

Phase contrast micrographs of control cells, uninfected C6/36 cells (A), and OKV-infected cells at 2 days postinfection (B)

Electron micrographs of viral particles purified by sucrose gradient ultracentrifugation and visualized by negative staining (scale bar, 100 nm)
Characterization of viral nucleic acids
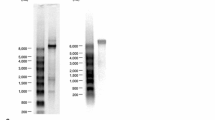
A major band, corresponding to a size of approximately 10 kb, and some smaller bands were detected in the supernatant of inoculated C6/36 cell cultures with CPE by agarose gel electrophoresis under denaturing conditions (Fig. 3A). These bands were also detected in the analysis of nucleic acids extracted from purified viral particles (Fig. 3B). These nucleic acids were subjected to DNase I or RNase A treatment and were analyzed by agarose gel electrophoresis. The bands disappeared when treated with RNase A, but not when treated with DNase I, indicating that they were RNA bands (Fig. 3B). Purified viral particles were subsequently subjected to acridine orange staining, and they emitted a brilliant flame-red fluorescence (data not shown). These results suggested that the viral particles contained single-stranded RNA.

Agarose gel electrophoresis of OKV-specific RNA. Nucleic acids from the supernatant of OKV-inoculated C6/36 cells were separated by agarose gel electrophoresis (A). M, size marker; 1, control (uninfected) C6/36 cells; 2, OKV-infected C6/36 cells. RNAs from purified viral particles treated with RNase A and DNase I (B). M, size marker; 3, control (no treatment); 4, RNase-A-treated; 5, DNase-I-treated. White arrows indicate OKV-specific RNA (vRNA)
Strand-specific RT-PCR with primer sets designed from the sequence of the 10-kb RNA (designated vRNA) was performed in order to determine whether OKV had positive or negative-stranded RNA. The positive- strand- and negative-strand-specific RT-PCRs using RNA extracted from OKV-infected C6/36 cells as the template amplified the sequences, producing amplicons that each had the expected size of approximately 2.4 kb (Fig. 4). On the other hand, only PCR designed for detecting positive-stranded RNA amplified a sequence with the expected size when RNA from purified OKV particles was used as the template. However, both positive- and negative-stranded RNA were detected in OKV-infected C6/36 cells (Fig. 4).

Strand-specific RT-PCR using RNA extracted from purified viral particles (lanes 1 and 2) or OKV-infected C6/36 cells one day postinfection (lanes 3 and 4). Lanes 1 and 3, negative-strand-specific RT-PCR; lanes 2 and 4, positive-strand-specific RT-PCR
Organization of the viral genome RNA (vRNA)
Sequence analysis suggested that the vRNA was composed of a 9,704-kb nucleic acid (DDBJ accession number AB972669) and had three major ORFs (ORF1 at nt 262 to 7,347, ORF2 at nt 7,377 to 8,585, and ORF3 at nt 8,724 to 9,350) (Fig. 5A). A schematic diagram of the strategy for vRNA cloning is shown in Fig. 5. cDNA fragments obtained by 3′ RACE had a poly(A) tail at the 3´end of the vRNA in the range of 28 to 44 nucleotides. The 5′-terminal sequence of the vRNA was determined using 5′ RACE PCR products that were only obtained when vRNA was used as the template after the decapping treatment.

(A) Schematic drawing of the organization of the OKV genome. Boxes with arrowheads indicate the positions of open reading frames (ORFs), with putative conserved domains indicated. Numbers in parentheses indicate nucleotide positions in the ORFs and domains. (B) Predicted transmembrane regions in proteins encoded by ORF3 of OKV and negeviruses (NEGEV, NEGEV174, PIUV, BRJV, and NWTV) and P23/P24 of negevirus-related plant viruses (CILV-C, CILV-C2, HGSV, and BNRBV). The amino acid sequence of OKV P22 was aligned with the deduced amino acid sequences obtained from NCBI GenBank using the Clustal W program [16] included in MEGA5 [31]. Hypothetical transmembrane regions (A-D) predicted by SOSUI [11] are shown in grey. GenBank accession numbers for the sequences used for alignment are given in Table 2a
A BLAST search using the predicted amino acid sequence of ORF1 indicated that it contained four conserved domains: a methyltransferase domain (vMet, at nt 709 to 1,364), an FtsJ-like methyltransferase (ribosomal RNA methyltransferase) domain (FtsJ, at nt 2,722 to 3,177), a viral RNA helicase domain (Hel, at nt 4,414 to 5,196), and an RNA-dependent RNA polymerase domain (RdRp, at 6,046 to 7,212). A BLAST search showed that the RdRp amino acid sequence had high similarity to those of the negeviruses Piura virus (PIUV), Brejeira virus (BRJV), Negev virus (NEGEV), NEGEV-like virus #174 (NEGEV174), Ngewotan virus (NWTV), Loreto virus (LORV), Santana virus (SANV), Dezidougou virus (DEZV), Wallerfield virus (WALV), Tanay virus (TANAV), and Goutanap virus (GANP) (Table 3), and also to those of positive- sense, single-stranded RNA plant viruses, including citrus leprois virus C (CiLV-C), citrus leprosis virus C type 2 (CiLV-C2), and ligustrum ringspot virus (LigRSV), and segmented double-stranded RNA plant viruses, including hibiscus green spot virus (HGSV) and blueberry necrotic ring blotch virus (BNRBV). CiLV-C has not yet been assigned to any family but has been assigned to the genus Cilevirus, and it has been proposed to include LigRSV in this genus [7, 13, 17, 18, 20, 26, 27]. HGSV was very recently classified as a member of the genus Higrevirus [1], and BNRBV is an unclassified virus that has been proposed for inclusion in the proposed genus “Blunervirus” [27].
ORF2 was suggested to code for a putative glycoprotein with an estimated size of approximately 45 kDa (designated P45) with two potential N-linked glycosylation sites and three transmembrane domains, which is a common feature of hypothetical proteins encoded by ORF2 of negeviruses [12, 15].
ORF3 encoded a hypothetical protein of approximately 22 kDa (designated P22) that showed a high level of similarity in its primary sequence (from 38 to 71 %) to hypothetical proteins encoded by ORF 3 of NEGEV, NEGEV174, PIUV, BRJV, LORV, and NWTV. It also showed moderate sequence similarity (from 19 to 32%) to P23/P24 of the plant viruses HGSV [21], CiLV-C2 [20], CILV-C [17], and BNRBV [27]. Furthermore, transmembrane domains (two or four domains) were predicted for those proteins (Fig. 5B). This structural feature was also commonly observed in hypothetical proteins encoded in ORF3 of all other negeviruses (data not shown).
Phylogenetic analysis
Phylogenetic analysis based on the conserved RdRp domain demonstrated that OKV formed a monophyletic clade together with LORV as well as NEGV, NWTV, BRJV, and PIUV, while the other members of the proposed genus Negevirus, SANV, TANAV, GANP, DEZV, and WALV, formed a different phylogenetic clade (Fig. 6). Phylogeny also revealed a sister group relationship between negeviruses and the plant-infecting viruses HGSV (Higrevirus), CiLV-C, CiLV-C2, LigRSV (Cilevirus), and BNRBV (Blunervirus) on a solitary branch. The phylogenetic relationships of these viruses did not contradict the results of the analysis based on whole ORF1 sequences (data not shown) or those reported previously [2, 5, 12, 23, 34]. The topology of these viruses was also well maintained in a phylogenetic tree including members of the family Virgaviridae as an outgroup, which was confirmed by an analysis based on a combination of the Bayesian approach and the maximum-likelihood procedure in MEGA5, except that BNRBV showed a closer relationship to negeviruses in group II (data not shown).

Phylogenetic analysis based on the conserved amino acid sequence of the RdRp region. The phylogenetic tree was constructed by the neighbor-joining method based on the alignment of the RdRp region (amino acid positions 1900-2342) of OKV ORF1 (DDBJ accession number AB972669) with the deduced amino acid sequence obtained from NCBI GenBank. The bootstrap test (1,000 replicates) results are shown next to the branches. The tree was drawn to scale, with branch lengths in the same units as those of the evolutionary distances used to infer the phylogenetic tree. The scale indicates the number of nucleotide substitutions per site. The sequence of OKV from this study is in boldface. GenBank accession numbers for the sequences used to construct the phylogenetic tree are given in Table 2b
Discussion
OKV, which was newly isolated from Aedes larvae collected on Okushiri Island, Japan, is a spherical virus of approximately 50 to 70 nm in diameter that contains a positive-sense single-stranded RNA of 9,704 nt composed of three ORFs separated by intergenic regions (Fig. 5A). Some lower-molecular-weight RNAs were detected by electrophoresis, even in the experiment using RNA extracted from purified OKV particles (Fig. 3B). Similar smaller RNAs have also been detected in NEGV [34]; however, more detailed analysis is needed. Strand-specific PCR confirmed that OKV replicated in the Aedes albopictus cell line C6/36, causing severe CPE (Figs. 1 and 4). The genome organization and amino acid sequences encoded in the ORFs showed a high degree of similarity to those of viruses belonging to a proposed new genus of insect-specific, positive-single-stranded RNA viruses, “Negevirus” (Fig. 6). Positive-single-stranded RNA viruses have been divided into three groups [14], and OKV contained sequences that are conserved in the Met, Hel, and RdRp domains of viruses in the groups including plant viruses (such as members of the families Virgaviridae, Bromoviridae, and Closteroviridae), mosquito-borne viruses (family Togaviridae, genus Alphavirus), mammal-infecting viruses (family Hepeviridae), and family Togaviridae, genus Rubivirus), and newly isolated insect viruses belonging to the proposed genus Negevirus [12, 23]. Phylogenetic analysis clearly grouped the negeviruses into two major clades (Fig. 6) corresponding to the negevirus phylogenetic topologies presented in previous studies [2, 5, 12, 23, 34]. OKV formed one of the two major negevirus clades (group I) together with NEGV, NWTV, BRJV, PIUV, and LORV (Fig. 6). Furthermore, the genome organization of OKV (non-segmented, positive-sense ssRNA genome with three major ORFs, four domains: vMet, FtsJ, Hel, and RdRP predicted for the first ORF) was similar to those of the other negeviruses reported to date. Potential glycosylation sites and/or transmembrane domains were also commonly predicted in the proteins P45 (ORF 2) and P22 (ORF 3) of OKV and the negeviruses reported previously. These results implied that both ORFs encoded putative viral membrane proteins, confirming the conservation of genome organization among negeviruses. These results suggest that OKV is a negevirus.
Phylogenetic relationships between the negeviruses and some plant viruses of the genera Higrevirus and Cilevirus and the proposed genus Blunervirus were identified based on the conserved domain sequences (Fig. 6) as reported previously [2, 5, 12, 23, 34]; however, the genome architecture differs between negeviruses and plant viruses (plant viruses have a segmented genome). The relationships between these viruses may also be imagined based on previous findings in which P22 of negeviruses and the putative structural proteins P23/P24 of the plant viruses [2, 26] shared topological and sequence similarities (Fig. 5B). Previous studies investigated plant-virus-like mosquito viruses other than negeviruses. CuTLV, a mosquito virus that was isolated from Culex mosquitoes, has the ability to cause CPE in the mosquito cell line C6/36 and shares sequence similarity with plant viruses of the family Tymoviridae [36]. These findings suggest possible transmission of some plant viruses by mosquitoes through the behavior of feeding on plant nectar and juices [22]; however, a mosquito-borne plant virus or a virus that replicates in both plants and mosquitoes has not yet been identified. This is supported by the findings that economically important viruses of the genera Cilevirus [17] and Higrevirus [1], the proposed genus Blunervirus [27], and the family Tymoviridae may be transmitted by mites, such as Brevipalpus sp. [28] and insects such as leafhoppers [19], respectively. Furthermore, a metagenomic survey of DNA viruses using mosquitoes also showed the presence of plant virus (geminivirus and nanovirus)-like sequences [24]. OKV isolated from Aedes larvae may be transmitted by feeding at the larval stage (horizontal infection) and/or through their parents (vertical infection); however, further studies are needed to examine this in more detail.
In conclusion, we have isolated a novel negevirus, Okushiri virus (OKV), in a subarctic region of Japan and carried out genomic characterization. OKV formed one of the two phylogenetic clades of negeviruses, together with viruses isolated from Israel (NEGV), Portugal (NEGV174), North America (NEGV), South America (BRJV, PIUV, LORV), and Indonesia (NWTV). The worldwide distribution of negeviruses with possible phylogenetic relationships to plant viruses has prompted us to speculate about unknown intimate relationships between mosquitoes and plants. Further studies are needed in order to elucidate the nature of negeviruses for a deeper understanding of the potential risk of mosquitoes not only to public health but also to agriculture.
References
Adams MJ, Lefkowitz EJ, King AMQ, Carstens EB (2014) Ratification vote on taxonomic proposals to the International Committee on Taxonomy of Viruses (2014). Archives of Virology 159(10):2831–2841
Auguste AJ, Carrington CV, Forrester NL, Popov VL, Guzman H, Widen SG, Wood TG, Weaver SC, Tesh RB (2014) Characterization of a novel Negevirus and a novel Bunyavirus isolated from Culex (Culex) declarator mosquitoes in Trinidad. J Gen Virol 95:481–485
Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic acids research 25(17):3389–3402
Becnel JJ, White SE (2007) Mosquito pathogenic viruses–the last 20 years. J Am Mosq Control Assoc 23:36–49
Carapeta S, do Bem B, McGuinness J, Esteves A, Abecasis A, Lopes Â, Parreira R (2015) Negeviruses found in multiple species of mosquitoes from southern Portugal: Isolation, genetic diversity, and replication in insect cell culture. Virology 483:318–328
Carlson J, Suchman E, Buchatsky L (2006) Densoviruses for control and genetic manipulation of mosquitoes. Adv Virus Res 68:361–392
Carstens EB (2010) Ratification vote on taxonomic proposals to the International Committee on Taxonomy of Viruses (2009). Archives of virology 155(1):133–146
Craggs JK, Ball JK, Thomson BJ, Irving WL, Grabowska AM (2001) Development of a strand-specific RT-PCR based assay to detect the replicative form of hepatitis C virus RNA. Journal of virological methods 94(1):111–120
Felsenstein J (1985) Confidence limits on phylogenies: An approach using the bootstrap. Evolution 39:783–791
Gorchakov RV, Tesh RB, Weaver SC, Nasar F (2014) Generation of an infectious Negev virus cDNA clone. J Gen Virol 95:2071–2074
Hirokawa T, Boon-Chieng S, Mitaku S (1998) SOSUI: classification and secondary structure prediction system for membrane proteins. Bioinformatics 14(4):378–379
Kallies R, Kopp A, Zirkel F, Estrada A, Gillespie TR, Drosten C, Junglen S (2014) Genetic characterization of goutanap virus, a novel virus related to negeviruses, cileviruses and higreviruses. Viruses 6:4346–4357
King AM, Adams MJ, Lefkowitz EJ (eds) (2012) Virus taxonomy: ninth report of the International Committee on Taxonomy of Viruses, vol 9. Elsevier
Koonin EV (1991) The phylogeny of RNA-dependent RNA polymerases of positive-strand RNA viruses. J Gen Virol 72(Pt 9):2197–2206
Kuchibhatla DB, Sherman WA, Chung BY, Cook S, Schneider G, Eisenhaber B, Karlin DG (2014) Powerful sequence similarity search methods and in-depth manual analyses can identify remote homologs in many apparently “orphan” viral proteins. J Virol 88:10–20
Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ, Higgins DG (2007) Clustal W and Clustal X version 2.0. Bioinformatics 23:2947–2948
Locali-Fabris EC, Freitas-Astúa J, Machado MA (2012) Genus Cilevirus. Ninth Report of the International Committee on Taxonomy of Viruses, Virus taxonomy, pp 1169–1172
Locali-Fabris EC, Freitas-Astúa J, Souza AA, Takita MA, Astúa-Monge G, Antonioli-Luizon R, Rodrigues V, Targon ML, Machado MA (2006) Complete nucleotide sequence, genomic organization and phylogenetic analysis of Citrus leprosis virus cytoplasmic type. J Gen Virol 87:2721–2729
Martelli GP, Sabanadzovic S, Abou-Ghanem Sabanadzovic N, Edwards MC, Dreher T (2002) The family Tymoviridae. Arch Virol 147:1837–1846
Melzer MJ, Simbajon N, Carillo J, Borth WB, Freitas-Astúa J, Kitajima EW et al (2013) A cilevirus infects ornamental hibiscus in Hawaii. Archives of virology 158(11):2421–2424
Melzer MJ, Sether DM, Borth WB, Hu JS (2012) Characterization of a virus infecting Citrus volkameriana with citrus leprosis-like symptoms. Phytopathology 102:122–127
Muller G, Schlein Y (2005) Plant tissues: the frugal diet of mosquitoes in adverse conditions. Med Vet Entomol 19:413–422
Nabeshima T, Inoue S, Okamoto K, Posadas-Herrera G, Yu F, Uchida L, Ichinose A, Sakaguchi M, Sunahara T, Buerano CC, Tadena FP, Orbita IB, Natividad FF, Morita K (2014) Tanay virus, a new species of virus isolated from mosquitoes in the Philippines. J Gen Virol 95:1390–1395
Ng TF, Willner DL, Lim YW, Schmieder R, Chau B, Nilsson C, Anthony S, Ruan Y, Rohwer F, Breitbart M (2011) Broad surveys of DNA viral diversity obtained through viral metagenomics of mosquitoes. PLoS One 6:e20579
Nicholson GM (2007) Fighting the global pest problem: preface to the special Toxicon issue on insecticidal toxins and their potential for insect pest control. Toxicon 49:413–422
Pascon RC, Kitajima JP, Breton MC, Assumpcao L, Greggio C, Zanca AS, Okura VK, Alegria MC, Camargo ME, Silva GG, Cardozo JC, Vallim MA, Franco SF, Silva VH, Jordao H Jr, Oliveira F, Giachetto PF, Ferrari F, Aguilar-Vildoso CI, Franchiscini FJ, Silva JM, Arruda P, Ferro JA, Reinach F, da Silva AC (2006) The complete nucleotide sequence and genomic organization of Citrus Leprosis associated Virus, Cytoplasmatic type (CiLV-C). Virus Genes 32:289–298
Quito-Avila DF, Brannen PM, Cline WO, Harmon PF, Martin RR (2013) Genetic characterization of Blueberry necrotic ring blotch virus, a novel RNA virus with unique genetic features. J Gen Virol 94:1426–1434
Roy A, Hartung JS, Schneider W, Shao J, Leon G, Melzer MJ et al (2015). Role bending: complex relationships between viruses, hosts and vectors related to citrus leprosis, an emerging disease. Phytopathology (ja)
Saitou N, Nei M (1987) The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol 4:406–425
Sugiharti M, Ono C, Ito T, Asano SI, Sahara K, Pujiastuti Y, Bando H (2011) Isolation of the Thosea asigna virus (TaV) from the epizootic Setothosea asigna larvae collected in South Sumatra and a study on its pathogenicity to Limacodidae larvae in Japan. Journal of insect biotechnology and sericology 79(3):3_117–3_124
Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S (2011) MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 28:2731–2739
Van Bortel W, Trung HD, le Thuan K, Sochantha T, Socheat D, Sumrandee C, Baimai V, Keokenchanh K, Samlane P, Roelants P, Denis L, Verhaeghen K, Obsomer V, Coosemans M (2008) The insecticide resistance status of malaria vectors in the Mekong region. Malar J 7:102
Van Wijngaarden RP, Brock TC, Van den Brink PJ (2005) Threshold levels for effects of insecticides in freshwater ecosystems: a review. Ecotoxicology 14:355–380
Vasilakis N, Forrester NL, Palacios G, Nasar F, Savji N, Rossi SL, Guzman H, Wood TG, Popov V, Gorchakov R, Gonzalez AV, Haddow AD, Watts DM, da Rosa AP, Weaver SC, Lipkin WI, Tesh RB (2013) Negevirus: a proposed new taxon of insect-specific viruses with wide geographic distribution. J Virol 87:2475–2488
Vaughan AM, Kappe SH (2012) Malaria vaccine development: persistent challenges. Curr Opin Immunol 24:324–331
Wang L, Lv X, Zhai Y, Fu S, Wang D, Rayner S, Tang Q, Liang G (2012) Genomic characterization of a novel virus of the family Tymoviridae isolated from mosquitoes. PLoS One 7:e39845
Acknowledgments
This work was supported in part by JSPS and VAST under the Japan-Vietnam Research Cooperation Program.
Author information
Authors and Affiliations
Corresponding author
Additional information
Kota Kawakami and Yudistira Wahyu Kurnia have equally contributed to this work.
Rights and permissions
About this article

Cite this article
Kawakami, K., Kurnia, Y.W., Fujita, R. et al. Characterization of a novel negevirus isolated from Aedes larvae collected in a subarctic region of Japan. Arch Virol 161, 801–809 (2016). https://doi.org/10.1007/s00705-015-2711-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00705-015-2711-9