
Abstract
Orf virus (ORFV, species Orf virus) belongs to the typical species of the Parapoxvirus genus of the family Poxviridae, which infects sheep, goats, and humans with worldwide distribution. Although outbreaks of Orf have been reported sequentially in several Chinese provinces, the epidemiology of Orf and genetic diversity of ORFV strains still needs to be further characterized. To further reveal the genomic organization of the ORFV-GZ18 and ORFV-CL18 isolates, the complete genome sequences of two recently obtained ORFV isolates were sequenced using the next-generation sequencing technology and analyzed, which had been deposited in the GenBank database under accession number MN648218 and MN648219, respectively. The complete genomic sequence of ORFV-CL18 was 138,495 bp in length, including 131 potential open reading frames (ORFs) flanked by inverted terminal repeats (ITRs) of 3481 bp at both ends, which has genomic structure typical Parapoxviruses. The overall genomic organization of the fully sequenced genome of ORFV-GZ18 was consistent with ORFV-CL18 genome, with a complete genome size of 138,446 nucleotides, containing 131 ORFs flanked by ITRs of 3469 bp. Additionally, the overall G + C contents of ORFV-GZ18 and ORFV-CL18 genome sequences were about 63.9% and 63.8%, respectively. The phylogenetic analysis showed that both ORFV-GZ18 and ORFV-CL18 were genetically closely related to ORFV-SY17 derived from sheep. In summary, the complete genomic sequences of ORFV-GZ18 and ORFV-CL18 are reported, with the hope it will be useful to investigate the host range, geographic distribution, and genetic evolution of the virus in Southern West and Northern East China.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Orf virus (ORFV), a highly contagious zoonotic pathogen, mainly infects sheep and goats, causing severe proliferative skin lesions around the lips, nostrils, oral mucosa, tongue and muzzle [1,2,3]. Also, it can also infect other wild and domestic ruminants and humans, especially farmers, veterinarians, shepherds, and slaughterhouse workers [4, 5]. ORFV infection is usually self-limiting and the primary lesions resolve spontaneously within 3–4 weeks [6]. However, the morbidity and mortality rates of the disease are higher especially in newly infected kids and lambs due to secondary bacterial and fungal infections [3, 7], resulting in huge economic losses in livestock farming.
ORFV is a highly epitheliotrophic linear double-stranded DNA virus with a genome of approximate 140 kilobase pairs, encoding 132 putative genes [8, 9]. The core region of ORFV genome (ORFs 009-111) is highly conserved and plays critical roles in viral replication, assembly, morphogenesis, and release. Variable genes with potential virulence and immunomodulation functions are mainly found in the terminal region at both ends of each inverted terminal repeat (ITRs) [10,11,12,13]. Although a growing number of representative clinical cases of ORFV infection have been reported around the world [14,15,16,17,18,19], currently available complete genome sequences deposited in the NCBI database are still lacking. Additionally, little is known about their genomic evolution and diversity.
In the present study, we sequenced full genomes of two ORFV isolates, which were, respectively, isolated from infected goat herds in Guizhou Province and sheep flocks in Jilin Province, China, in 2018. Among them, this is the first complete genomic sequence of an ORFV geographic isolate from Guizhou Province, Southern West China. The sequence information obtained herein will provide a valuable resource in genetic research. It is hoped that these data will facilitate future investigations of the molecular characteristics of both ORFV-GZ18 and ORFV-CL18 and will help determine the geographic origin of two ORFV isolates and elucidate their phylogenetic relationship to other ORFV strains.
Materials and methods
Clinical cases and tissue sampling
In this study, the two natural outbreaks of Orf occurred at two farms located in Guizhou Province, Southern West China and Jilin Province, Northern East China, respectively. Tissue samples from the affected animals from the two outbreaks occurred in distinct geographic regions within China were obtained by scraping the affected areas (thick crust skins) showing gross pathological changes consistent with Orf. Samples frozen at − 80 °C were subjected to further virological investigations. Additional tissue sections were preserved in 10% neutral-buffered formalin for histopathologic examinations.
Histopathology
Clinical diagnosis was further confirmed by histopathologic examinations. Briefly, formalin-fixed tissues were processed routinely, and paraffin-embedded sections of scab samples were cut to 4 μm. After staining with hematoxylin and eosin (H&E), the sections were observed and analyzed under a light microscope.
Virus isolation, PCR detection, and electron microscopy observation
The scab samples with typical gross pathological changes were triturated in 0.01 M PBS. The homogenized samples of the scab specimens were clarified by centrifugation at 3000 r/min for 20 min at 4 °C. The supernatant fluids were collected and inoculated into a confluent monolayer of primary ovine fetal turbinate cells (OFTu). Subsequently, virus inoculum was removed followed by 2-h incubation at 37 °C to allow virus to adsorb and cells were washed three times with medium and incubated in a CO2 incubator supplying 5% CO2. The normal cells for controls were maintained in a similar manner. The cells were observed daily for any cytopathic effects (CPE). Obvious CPE was observed after five serial passages. The supernatant from CPE-positive cell cultures were submitted for viral identification with negative staining electron microscopy (nsEM) and DNA extraction using the innuPREP virus DNA kit (Analytik Jena, Germany) according to the manufacturer’s instructions. The PCR assay was performed by amplifying ORFV ORF011 and ORF120 genes. The sequences of the primers used in the study are as follows: ORF011-Fw: 5′-ATGTGGCCGTTCTCCTCCATC-3′, ORF011-Rv: 5′-TTAATTTATTGGCTTGCAGAAC-3′ and ORF020-Fw: 5′-ATGGCTTGCGAGTGCGCGTCTC-3′, ORF020-Rv: 5′-TTAGAAGCTGATGCCGCAGTTG-3′. The PCR products were visualized on 1% agarose electrophoretic gels and DNA sequencing (Comate Bioscience Co, China).
Genome characterization
Five micrograms of viral genomic DNA extracted from CPE-positive cell culture supernatant were used to generate next-generation sequencing libraries using Hiseq 2000 cBot flowcell cluster generation. The full-length genome sequences of ORFV-GZ18 and ORFV-CL18 isolates were determined through next-generation sequencing (NGS) technology using Illumina Hiseq 2000 platform (Beijing, China). The raw data were checked using FastQC v0.10.1 (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/) as described previously [20] to remove the low-quality regions. Sequences were assembled and analyzed using BioEdit software package (http://www.mbio.ncsu.edu/BioEdit/bioedit.html). Sequence gaps of each ORFV were identified by linking information from forward and reverse reads and were closed by primer walking, PCR sequencing, and insert subcloning. The complete genome sequences of ORFV-GZ18 and ORFV-CL18 isolates obtained via NGS technique have been deposited in GenBank (the accession number MN648218 and MN648219) and subjected to sequence analyses, respectively. Putative open reading frames (ORFs) of both ORFV-GZ18 and ORFV-CL18 were identified using ORF finder (https://www.ncbi.nlm.nih.gov/orffinder/) and bioinformatics analysis. The predicted ORFs were numbered and named according to ORFV standard strain (OV-IA82). Furthermore, the percent of amino acid identity (% ID) of each ORF was determined by alignment analysis between the two isolates and other ORFV strains using Clustal Omega (https://www.ebi.ac.uk/Tools/msa/clustalo/) [21].
Phylogenetic analyses based on the individual genes and complete genome sequences of ORFV isolates
To determine the potential genetic diversity of ORFV-GZ18 and ORFV-CL18 isolates, multiple sequence alignments were performed using the MAFFT version 7 [22]. In addition, phylogenetic trees based on the individual genes including ORF011 and ORF020 were constructed using maximum likelihood employing GTR + I + G model and 1000 ultrafast bootstrap replicates, implemented by IQ-TREE2 [23].
Additionally, the comparative analysis of the full-length genomes of ORFV-GZ18 isolate, ORFV-CL18 isolate, two previously isolated ORFVs including ORFV-SY17 and ORFV-NA17 strains [20], and one non-Chinese reference strain (OV-IA82 standard strain) in the GenBank database was performed using MAFFT version 7 and IQ-TREE2 [11]. Phylogenetic trees were constructed based on the complete genomic sequences of ORFV-GZ18 and ORFV-CL18 and other 18 ORFV isolates available in GenBank database [24,25,26,27,28].
Results
Typical gross pathological changes
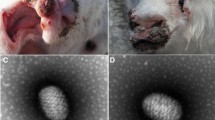
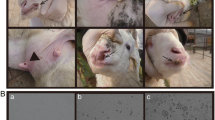
In one of the outbreaks that occurred in Guizhou Province, all the 28 breed of lambs in the flock purchased from a local market developed masses covered by thick crusts after about 20 days (Fig. 1A). One of the affected lambs died due to secondary infection. In the other outbreak in Jilin Province, the affected animals appeared typical nodular lesions in the mucocutaneous interface, including the lips, nostrils, and eyelids, which varied in size from approximately 5–12 mm (Fig. 1B).

Representative clinical cases of Orf virus infection. A Sheep showing multiple nodular lesions on the lips, oral commissures, and nostrils. B Photograph of affected animals with severe, proliferative lesions in the skin of lips and muzzle
Microscopic examination
Microscopic examination of skin lesions showed that thickening spinous cell layer (Fig. 2A), swelling, vacuolation, ballooning degeneration, karyorrhexis and characteristic eosinophilic cytoplasmic inclusion bodies in spinous cells of the stratum spinosum (Fig. 2B), hemorrhage around the hair follicle (Fig. 2C), and follicular necrosis (Fig. 2D). A large amount of hemosiderin particles appeared in the red pulp of spleen (Fig. 2E). In addition, large number of lymphocyte aggregation was observed around the sheathed artery (Fig. 2F). No obvious lesions were observed in other organs.

Microscopic examination of skin lesions. Skin biopsy showing spinous layer thickening (A), swelling, vacuolation, ballooning degeneration and karyorrhexis in spinous cells of the stratum spinosum (B), hemorrhage around the hair follicle (C), and follicular necrosis (D). A large amount of hemosiderin particles appeared in the red pulp of spleen (E), and large number of lymphocyte aggregation was observed around the sheathed artery (F)
Virus isolation and identification
Two viruses were isolated on OFTu cells from the crust skins showing gross pathological changes consistent with Orf. Obvious CPE was observed at the second-day post-inoculation after five blind passages, which was characterized by the diffused degeneration of a monolayer with rounded cells fluctuating in the culture medium (Fig. 3B, E). Furthermore, nsEM of CPE-positive cell culture supernatant revealed the presence of characteristic ovoid-shaped viral particles (Fig. 3C, F), which are related to the Poxviridae family. Finally, the two isolates were identified by PCR and then confirmed by sequencing of the generated amplicons. ORF011 (B2L) is a major envelope protein gene, which is highly conserved in different PPVs including ORFV, BPSV, PCPV, and parapoxvirus of red deer in New Zealand (PVNZ) and usually serves as a common and precise marker for the genome stability of PPVs. Thus, the ORF011 (B2L) gene from ORFV-GZ18 and ORFV-CL18 was successfully amplified by PCR which produced an amplicons of approximately 1137 bp in size which were confirmed by automated DNA sequencing. To further understand the evolutionary relationship among ORFV isolates and with other members of poxviruses, ORF020, an ORFV-encoded early interferon resistance protein (OVIFNR) has important role for viral replication, which is located in the left terminal variable region of PPVs genome and chosen for amplified and produced an amplicons of approximately 552 bp in size which were confirmed by automated DNA sequencing (Fig. 4A, B).

Infected OFTu cells and electron microscopic examinations of the Orf virus. A Mock-infected cells; B obvious cytopathic effects (CPE) was observed at OFTu cells under microscope when inoculated with the supernatant fluid prepared from the skin lesion of infected animals in Jilin Province; C electron microphotograph of showing the characteristic morphology of an Orf virion from OFTu cell cultures inoculated with the skin lesion of lips (bar = 100 nm); D mock-infected cells; E obvious CPE was observed at OFTu cells under microscope when inoculated with the supernatant fluid prepared from the scabs of infected animals in Guizhou Province; F electron micrograph of the orf virus particle showing typical oval-shaped morphology (bar = 100 nm)

PCR amplification of ORF 011 and ORF020 of ORFV-CL18 and ORFV-GZ18 isolates. A Lane M: DL 2000 Marker; Lane 1 and 2: ORF011 gene (1137 bp in size) was amplified from genomic DNA isolated from ORFV-CL18; Lane 3 and 4: ORF020 gene (552 bp in size) was amplified from genomic DNA isolated from ORFV-CL18; B Lane M: DL 2000 Marker; Lane 5 and 6: ORF011 gene (1137 bp in size) was amplified from genomic DNA isolated from ORFV-GZ18; Lane 7 and 8: ORF020 gene (552 bp in size) was amplified from genomic DNA isolated from ORFV-GZ18
Genome assembly
After demultiplexing and quality trimming, sequence fragments were assembled and the obtained contigs annotated using BLASTn and BLASTx against non-redundant protein database (GenBank). The complete genomic sequences of ORFV-GZ18 and ORFV-CL18 were determined to be 138,446 bp (GenBank accession number MN648218) and 138,495 bp (GenBank accession number MN648219) in length, respectively. Both ORFV-GZ18 and ORFV-CL18 have typical genomic organization and structural characteristic of Poxviruses, which contain 131 ORFs, respectively, flanked by ITRs of 3469 bp and 3481 bp at both ends (Fig. 5; Table 1). Additionally, the overall G + C contents of ORFV-GZ18 and ORFV-CL18 genome sequences were about 63.9% and 63.8%, respectively (Table 1).

Genome map of the 2 new isolates (ORFV-CL18 and ORFV-GZ18) and other reference ORFV isolates. The predicated ORFs are indicated by colored (green, black, red, and yellow) arrows. The ORFs located in left terminal variable regions are present in green, and the right terminal variable regions are presented in yellow. The conserved regions are shown in black and red
Sequence alignments of ORFs and ITRs
The amino acid identifies of each ORF among ORFV-GZ18, ORFV-CL18, and other fully sequenced representative ORFV strains were compared and listed in Tables 2 and 3 [11, 25, 26]. Comparative analyses of the complete sequences of ORFV-GZ18 and ORFV-CL18 revealed that the two isolates exhibit remarkably limited genomic variability and share higher amino acid identity in equivalent ORFs especially in the central regions of the complete genomic sequence. ORFs 002, 005, 058, 103, 116, 118, 119, and 132 were found to have a lower % ID (below 60%), which mainly located at both ends of ORFV genome usually termed as terminal variable region. Additionally, sequence alignment showed that ORFV-GZ18 and ORFV-CL18, respectively, shared nucleotide identify 84–97% and 86–96% with other isolates at the ITRs. The ITR of the two ORFV strains share highly 95.31% identity between each other, including the BamH I site (GGATCC) and the relatively conserved telomere resolution sequences (ATTTTTT-N(8)-TAAAT), which indicated that ORFV-GZ18 and ORFV-CL18, respectively, isolated from two different areas of China probably have a more recent last common ancestor than the last common ancestor they share with the other. In addition, this genetic structure might correspond to geographic structure or host specificity.
Phylogenetic analysis
To determine the evolutionary relationship of the two isolates to other known isolates worldwide, phylogenetic analysis based on 20 PPVs entire genome sequences was subsequently performed using IQ-TREE2 [11, 24,25,26,27,28]. The results indicated that all the ORFV strains analyzed were divided into 3 clusters, and the two newly identified isolates (ORFV-GZ18 and ORFV-CL18) shared the closest relationship with ORFV-SY17, which previously was isolated from Jilin Province of China (Fig. 6). Additionally, phylogenetic trees based on the individual genes including the full-length nucleotide sequence of ORF011 and ORF020 genes were also constructed to further explore the genetic relationship among these ORFV strains. As shown in Fig. 7B, phylogenetic analysis based on ORF020 gene also showed that ORFV-GZ18 and ORFV-CL18 also had a close relationship to ORFV-SY17 isolate. Overall, these results suggested that ORFV isolated in Jilin Province and Guizhou Province might have the same origin.

Phylogenetic analyses based on the complete genomic sequences of ORFV-GZ18 and ORFV-CL18 and other 19 ORFV isolates available in GenBank database. A The phylogenetic tree was constructed using maximum likelihood employing GTR + I + G model and 1000 ultrafast bootstrap replicates, implemented by IQ-TREE2 software.  CL18 isolated in this study;
CL18 isolated in this study;  GZ18 isolated in this study. B The circular phylogenetic tree was constructed by the maximum likelihood method using IQ-TREE2 software. The numbers above or below the branch points indicate the bootstrap support calculated for 1000 replicates.
GZ18 isolated in this study. B The circular phylogenetic tree was constructed by the maximum likelihood method using IQ-TREE2 software. The numbers above or below the branch points indicate the bootstrap support calculated for 1000 replicates.  CL18 isolated in this study;
CL18 isolated in this study;  GZ18 isolated in this study
GZ18 isolated in this study

Phylogenetic analysis based on the full length of ORF011 (A) and ORF020 (B). The phylogenetic trees based on the full length of ORF011 and ORF020 gene were constructed via the maximum likelihood method using IQ-TREE2 software, and 1000 bootstrap replicates were subjected to nucleotide sequence distance (cut-off value of 50% from 1000 bootstrap replicates). Only bootstrap values > 50% are shown at each tree node.  : GZ18 isolated in this study;
: GZ18 isolated in this study;  CL18 isolated in this study
CL18 isolated in this study
Discussion
Orf is a highly contagious zoonotic disease of small ruminants and it is usually prevalent in the regions where livestock farming is practiced [29]. Also, it is an occupational hazard for farmers, shepherds, slaughterhouse workers, and veterinarians. The occurrence of Orf disease has led to huge economic losses to goat and sheep livestock industry by reducing the productivity and health-related losses [3, 30]. In recent years, various Orf virus strains of different origins have been reported in many countries, which have become of great concern to some extent because of border crossing and its zoonotic potential [1,2,3,4,5]. In the present study, we successfully isolated and identified two ORFV strains, respectively, from infected goat and sheep from the two outbreaks occurred in distinct geographic regions within China, which were named ORFV-GZ18 and ORFV-CL18, respectively.
To more comprehensively understand the genetic diversity and the evolution of ORFVs from different regions in China, the complete genome sequences of the ORFV-GZ18 and ORFV-CL18 isolates from two naturally infected cases were obtained using next-generation sequencing (NGS) technology. The full-length genomic sequences of ORFV-GZ18 (isolated from Guizhou Province, Southern West China) and ORFV-CL18 (isolated from Jilin Province, Northern East China) were determined to be 138,446 bp (GenBank accession number MN648218) and 138,495 bp (GenBank accession number MN648219), respectively. Comparative analysis of ORFV-GZ18 and ORFV-CL18 entire genomes showed that both of them contained a large central coding region bounded by two identical inverted terminal repeat (ITR) regions of 3469 bp and 3481 bp at both ends [20, 31], which had typical genomic organization and structural characteristic of the member of the genus Parapoxvirus. In addition, sequence analyses also revealed that the two new isolates exhibit remarkably limited genomic variability and share higher amino acid identity in equivalent ORFs especially in the central regions of the complete genomic sequence. Due to its terminal location, the ITR regions usually were considered to be highly variable, which might play an important role in virulence, viral replication, host range, and regulation of gene transcription and expression [4, 31]. Of note, the ITRs of the two newly identified ORFV isolates share highly 95.31% identity between each other, including the BamH I site (GGATCC) and the relatively conserved telomere resolution sequences (ATTTTTT-N(8)-TAAAT), which were also consistent with those of ORFV-SY17 and ORFV-NA17 strains [20] and might be considered as a potential characteristic motif of ORFV ITRs.
To fully determine the evolutionary relationship of the two isolates to other known isolates worldwide, phylogenetic analysis based on 20 PPVs entire genome sequences was subsequently performed using the maximum likelihood method implemented in IQ-TREE2 [11, 24,25,26,27,28]. The results indicated that all the ORFV strains analyzed were divided into 3 clusters, and the two newly identified isolates (ORFV-GZ18 and ORFV-CL18) shared the closest relationship with ORFV-SY17, which previously was isolated from Jilin Province of China. Additionally, phylogenetic trees based on the nucleotide sequences of the full-length nucleotide sequence of ORF011 and ORF020 genes were also constructed to further explore the genetic relationship among these ORFV strains. Compared to other ORFV isolates, ORFV-GZ18 and ORFV-CL18 also had a close relationship to ORFV-SY17 isolate, suggesting they may derive from the same origin. Of note, ORFV-GZ18 (isolated from goat) shares a relatively closer relationship with ORFV-CL18 (isolated from goat) comparing to some other viral strains prevalent in various regions of the world, which indicates ORFVs may be having host-specific adaptations to allowing the viruses to be transmitted more effectively in either sheep or goats. Thus, the results may provide new insight into the origin and evolution of ORFV.
In conclusion, two newly identified ORFV isolates, respectively, originated goat from Guizhou Province, Southern West China and sheep from Jilin Province, North east China were obtained. Meanwhile, this is the first complete genomic sequence of an ORFV geographic isolate from Guizhou Province. The availability of ORFV-GZ18 and ORFV-CL18 complete genomic sequence information will provide a valuable resource in genetic research. It is hoped that these data will facilitate future investigations of the molecular characteristics of both ORFV-GZ18 and ORFV-CL18, which also can be served as the basis from which to determine the evolutionary history of these two lineages.
References
Bergqvist C, Kurban M, Abbas O (2017) Orf virus infection. Rev Med Virol 27(4):e1932
Duan C, Liao M, Wang H, Luo X, Shao J, Xu Y, Li W, Hao W, Luo S (2015) Identification, phylogenetic evolutionary analysis of GDQY orf virus isolated from Qingyuan City, Guangdong Province, southern China. Gene 555:260–268
Bala JA, Balakrishnan KN, Abdullah AA, Mohamed R, Haron AW, Jesse FF, Noordin MM, Mohd-Azmi ML (2018) The re-emerging of orf virus infection: a call for surveillance, vaccination and effective control measures. Microb Pathog 120:55–63
Mercer A, Fleming S, Robinson A, Nettleton P, Reid H (1997) Molecular genetic analyses of parapoxviruses pathogenic for humans. Arch Virol 13:25–34
Long M, Wang Y, Chen D, Wang Y, Wang R, Gong D, He H, Rock DL, Hao W, Luo S (2018) Identification of host cellular proteins LAGE3 and IGFBP6 that interact with orf virus protein ORFV024. Gene 661:60–67
Haig DM, McInnes CJ (2002) Immunity and counter-immunity during infection with the parapoxvirus orf virus. Virus Res 88(4):3–16
Gumbrell RC, McGregor DA (1997) Outbreak of severe fatal orf in lambs. Vet Rec 141(6):150–151
Spyrou V, Valiakos G (2015) Orf virus infection in sheep or goats. Vet Microbiol 181(1–2):178–182
Martins M, Cargnelutti JF, Weiblen R, Flores EF (2014) Pathogenesis in lambs and sequence analysis of putative virulence genes of Brazilian orf virus isolates. Vet Microbiol 174:69–77
Mercer AA, Fraser K, Barns G, Robinson AJ (1987) The structure and cloning of orf virus DNA. Virology 157:1–12
Delhon G, Tulman ER, Afonso CL, Lu Z, de la Concha-Bermejillo A, Lehmkuhl HD, Piccone ME, Kutish GF, Rock DL (2004) Genomes of the parapoxviruses Orf virus and bovine popular stomatitis virus. J Virol 78:168–177
Wang X, Xiao B, Zhang J, Chen D, Li W, Li M, Hao W, Luo S (2016) Identification and characterization of a cleavages site in the proteolysis of Orf virus 086 protein. Front Microbiol 7:538
Muhsen M, Protschka M, Schneider LE, Muller U, Kohler G, Magin TM, Buttner M, Alber G, Siegemund S (2019) Orf virus (ORFV) infection in a three-dimensional human skin model: characteristic cellular alterations and interference with keratinocyte differentiation. PLoS ONE 14:e0210504
Veraldi S, Esposito L, Pontini P, Vaira F, Nazzaro G (2019) Feast of Sacrifice and Orf, Milan, Italy, 2015–2018. Emerg Infect Dis 25(8):1585–1586
Tryland M, Beckmen KB, Burek-Huntington KA, Breines EM, Klein J (2018) Orf virus infection in Alaskan mountain goats, Dall’s sheep, muskoxen, caribou and Sitka black-tailed deer. Acta Vet Scand 60:12
Yogisharadhya R, Bhanuprakash V, Kumar A, Mondal M, Shivachandra SB (2018) Comparative sequence and structural analysis of Indian orf viruses based on major envelope immuno-dominant protein (F1L), an homologue of pox viral p35/H3 protein. Gene 663:72–82
Olivero N, Reolon E, Arbiza J, Berois M (2018) Genetic diversity of Orf virus isolated from sheep in Uruguay. Arch Virol 163:1285–1291
Simulundu E, Mtine N, Kapalamula TF, Kajihara M, Qiu Y, Ngoma J, Zulu V, Kwenda G, Chisanga C, Phiri IK, Takada A, Mweene AS (2017) Genetic characterization of orf virus associated with an outbreak of severe orf in goats at a farm in Lusaka, Zambia (2015). Arch Virol 162:2363–2367
Gelaye E, Achenbach JE, Shiferaw J, Gelagay A, Belay A, Yami M, Loitsch A, Grabherr R, Diallo A, Lamien CE (2016) Molecular characterization of orf virus from sheep and goats in Ethiopia, 2008–2013. Virol J 13:34
Zhong J, Guan J, Zhou Y, Cui S, Wang Z, Zhou S, Xu M, Wei X, Gao Y, Zhai S, Song D, He W, Gao F, Zhao K (2019) Genomic characterization of two Orf virus isolates from Jilin province in China. Virus Genes 55(4):490–501
Sievers F, Higgins DG (2014) Clustal omega, accurate alignment of very large numbers of sequences. Methods Mol Biol 1079:105–116
Katoh K, Rozewicki J, Yamada KD (2019) MAFFT online service: multiple sequence alignment, interactive sequence choice and visualization. Brief Bioinform 20:1160–1166
Minh BQ, Schmidt HA, Chernomor O, Schrempf D, Woodhams MD, von Haeseler A, Lanfear R (2020) IQ-TREE 2: new models and efficient methods for phylogenetic inference in the genomic era. Mol Biol Evol 37:1530–1534
Li W, Hao W, Peng Y, Duan C, Tong C, Song D, Gao F, Li M, Rock DL, Luo S (2015) Comparative genomic sequence analysis of Chinese orf virus strain NA1/11 with other Parapoxviruses. Arch Virol 160:253–260
Chen H, Li W, Kuang Z, Chen D, Liao X, Li M, Luo S, Hao W (2017) The whole genomic analysis of orf virus strain HN3/12 isolated from Henan province, central China. BMC Vet Res 13:260
Mercer AA, Ueda N, Friederichs SM, Hofmann K, Fraser KM, Bateman T, Fleming SB (2006) Comparative analysis of genome sequences of three isolates of Orf virus reveals unexpected sequences variation. Virus Res 116:146–158
Chi X, Zeng X, Li W, Hao W, Li M, Huang X, Huang Y, Rock DL, Luo S, Wang S (2015) Genome analysis of orf virus isolates from goats in the Fujian Province of Southern China. Front Microbiol 6:1135. https://doi.org/10.3389/fmicb.2015.01135
McGuire MJ, Johnston SA, Sykes K (2012) Novel immune-modulator identified by a rapid, functional screen of the parapoxvirus ovis (orf virus) genome. Proteome Sci 10:4. https://doi.org/10.1186/1477-5956-10-4
Kumar R, Trivedi R, Bhatt P, Khan S, Khurana S, Tiwari R, Karthik K, Malik Y, Dhama K, Chandra R (2015) Contagious pustular dermatitis (orf disease)—Epidemiology, diagnosis, control and public health concerns. Adv Anim Vet Sci 3(12):649–676
Nadeem M, Curran P, Cooke R, Ryan C, Connolly K (2010) Orf: contagious pustular dermatitis. Ir Med J 103(5):152–153
Fraser KM, Hill DF, Mercer AA, Robinson AJ (1990) Sequence analysis of the inverted terminal repetition in the genome of the parapoxvirus, orf virus. Virology 176:379–389. https://doi.org/10.1016/0042-6822(90)90008-F
Acknowledgements
This study was supported by the National Natural Science Foundation of China (Grant No. 31672554), the Scientific and Technological Project of Jilin Province (Grant No. 20200402052NC), and a program supported by Jilin University for the Excellent Youth Scholars (Grant No. 419080520316).
Author information
Authors and Affiliations
Contributions
YZ and KZ conceived the study and participated in its design and coordination. YZ, LL, HC, MSX, SW, ZY, RZ, SH, ZF, JZ, SC, and SY performed the research. YZ, KZ, JG, DS, WH, and FG analyzed and interpreted the data. YZ and KZ wrote the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
This article does not contain any studies with human participants performed by any of the authors. All animal experiments were in accordance with the Animal Welfare Ethical Committee of the College of Veterinary Medicine, Jilin University.
Additional information
Edited by Joachim Jakob Bugert.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Zhou, Y., Guan, J., Lv, L. et al. Complete genomic sequences and comparative analysis of two Orf virus isolates from Guizhou Province and Jilin Province, China. Virus Genes 58, 403–413 (2022). https://doi.org/10.1007/s11262-022-01918-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11262-022-01918-4