
Abstract
Orf virus (ORFV), a typical member of the Parapoxvirus genus within the family Poxviridae, which is the causative agent of Orf, a common epitheliotropic viral disease of sheep, goats, wild ruminants, and humans. In the present study, we sequenced the complete genomic sequences of two ORFV strains (ORFV-SY17, isolated from sheep, and ORFV-NA17, isolated from goat) and conducted the comparative analysis of multiple ORFVs. The complete genomic sequence of ORFV-SY17 was at length of 140,413 bp, including 131 potential open reading frames (ORFs) flanked by inverted terminal repeats (ITRs) of 4267 bp at both ends. The ORFV-NA17 strain displayed the similar genome structure with ORFV-SY17. The whole genomic sequence of ORFV-NA17 strain was 139,287 bp in length and contained 132 ORFs flanked by ITRs of 3974 bp. The overall G+C contents of ORFV-SY17 and ORFV-NA17 genome sequences were about 63.8% and 63.7%, respectively. The ITR sequences analysis showed that ORFV-SY17 and ORFV-NA17 contained the terminal BamHI sites and conserved telomere resolution sequences at both ends of their genome. In addition, comparative analysis of ORFs among ORFV-SY17, ORFV-NA17, and other ORFV strains revealed several sequence variations caused by insertions or deletions, especially in ORFs 005 and 116, which were very likely associated with host species. Phylogenetic analysis based on the complete genome sequences revealed that ORFV-SY17 was genetically closely related to NA1/11 and HN3/12 strains derived from sheep, while ORFV-NA17 was closely related to YX strain derived from goat. The multiple alignment of deduced amino acid sequences further revealed the genetic relationship between host species and genetic variations of ORFV strains. Taken together, the availability of genomic sequences of ORFV-SY17 and ORFV-NA17 strains from Jilin Province will aid in our understanding of the genetic diversity and evolution of ORFV strains in this region and can assist in distinguishing between ORFV strains that originate in sheep and goats.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Orf, also known as contagious ecthyma, contagious pustular dermatitis, scabby mouth or sore mouth, is a non-systemic, highly contagious, and eruptive skin disease in sheep, goats, and various other wild and domestic ruminants [1, 2]. Orf virus (ORFV), the causative agent of Orf, belongs to the prototype member of the genus Parapoxvirus of the family Poxviridae. Clinically, it mainly causes extensive and proliferative lesions on the skin of the lips, tongues, nostrils, breasts, and oral mucosa [2, 3]. Humans can be affected by closely contacting with infected animals or contaminated fomites [4, 5]. Although Orf is considered as a mild disease, high mortality rates are generally due to secondary infections or lesions around mouths and lips of kids and lambs making them reluctant to suck and graze, resulting in rapid emaciation [6].
The genome of the ORFV is linear double-stranded DNA of about 139 kb with 64% G+C content, which contains 132 putative genes including 89 highly conserved genes and some variable genes [7, 8]. Thus far, only several complete or nearly complete genomic sequences of ORFV strains have been publicly reported in the Genbank database. In China, the available whole genomic sequences include four strains originated in goats (GO, YX, NP, and SJ1 strains) from the Fujian province in Southern China [2], a NA1/11 strain originated in sheep from the Jilin province in Northeast China [9] and a HN3/12 strain originated in sheep from the Henan province in the central region of China [10]. However, the entire genomic sequence of ORFV from goats in Jilin province is not currently available.
In the present study, two ORFV strains (named ORFV-SY17 and ORFV-NA17) were successfully isolated from the sheep and goats suspected of Orf infections from Jilin province of China, respectively. Then, we sequenced and obtained the complete genomic sequences of ORFV-SY17 and ORFV-NA17 strains. In addition, the data related to genome structure and phylogenetic relation were discussed to further examine genomic variations between the two ORFVs and other PPVs, which would enable us to understand the diversity of ORFV isolates epidemic in this region or even in the world.
Materials and methods
Tissue samples
In September and October 2016, the two natural outbreaks of ORFV infections occurred in a sheep herd with 145 small-Tailed Han sheep in Songyuan and a goat herd with 106 cashmere goats in Nongan of Jilin Province, northeast of China. The morbidity rates of the outbreaks were 14.5% (21 out of 145) and 8.5% (9 out of 106), respectively. These infected lambs, aged 1 to 4 months, presented with typical clinical features of Orf, including papules, pustules, and scabs on their lips. A total of two clinical lip scabs samples were, respectively, collected from a sheep and a goat suspected to have ORFV infections, and stored in − 80 °C.
Virus isolation and electron microscopy
The collected scab tissue samples with significant pathological changes were triturated in 0.01 M PBS. The homogenized samples were centrifuged at 3000 r/min for 20 min at 4 °C [11]. Then, 100 μl of the clarified supernatants was passed through 0.45 μm filters and inoculated into a confluent monolayer of primary ovine fetal turbinate cells (OFTu). The inoculated cells were placed in a CO2 incubator supplying 5% CO2. The normal cells for controls were maintained in a similar manner. The cells were observed daily for any cytopathic effects (CPE). Obvious CPE was observed after three blind passages. The CPE-positive cell cultures were negatively stained with 2% phosphor-tungstic acid followed by electron microscopy (EM).
DNA sequencing and assembly
Virions were cultured and purified by sucrose gradient ultra-centrifugation as described previously [12]. Viral DNA was extracted using the innuPREP virus DNA kit (Analytik Jena, Germany) according to the manufacturer’s instructions. The genomes of ORFV-SY17 and ORFV-NA17 strains were sequenced using an Illumina Hiseq2000 (Beijing, China). Raw data were filtered using FastQC v0.10.1 (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/) to remove artificial sequences such as sequencing primers, connectors, and the sequences containing an ambiguous base N. After removing the low-quality regions, contigs were assembled and scaffolds were constructed using clean data with SOAP denovo v2.04 (http://soap.genomics.org.cn/soapdenovo.html). The sequences data were further analyzed using BioEdit software package v7.0.0 (http://www.mbio.ncsu.edu/bioedit/bioedit) to identify gaps between scaffolds. Gaps were closed by primer walking and verified by sequencing of PCR products. The complete genome sequences of ORFV-SY17 and ORFV-NA17 have been submitted to GenBank with accession numbers MG712417 and MG674916, respectively.
Analysis of ORF
The potential open reading frames (ORFs) of ORFV-SY17 and ORFV-NA17 were predicted using the ORF finder program in the website of the National Center for Biotechnology Information (NCBI) (https://www.ncbi.nlm.nih.gov/orffinder/) referring to other known Parapoxvirus (PPV) genomes listed in Table 1 [9]. Using NCBI’s BLAST program (https://blast.ncbi.nlm.nih.gov/Blast.cgi), the predicted ORFs were numbered and named according to ORFV standard strains. In addition, the percent of amino acid sequence identity (%ID) of each ORF was determined by alignment analysis between the two isolated strains and other ORFVs strains using Clustal Omega (https://www.ebi.ac.uk/Tools/msa/clustalo/) [13]. Then, the amino acid sequence of each ORF was further analyzed using software DNAMAN v7.02 in order to determine the mutations and deletions/insertions located in the sequences of ORFV-SY17 and ORFV-NA17 strains. To confirm the reality of the deletions/insertions and mutations, PCR amplifications were performed using primers designed on these regions and PCR products were verified by sequencing.
Analysis of ITR
ORFV genomes contain a large central coding region bounded by two identical inverted terminal repeat (ITR) regions. The ITRs were located at both ends of the ORFV genome, which were considered to have high variable [2, 14]. The ITRs of ORFV-SY17 and ORFV-NA17 were analyzed and compared with other reference strains list in Table 1 using DNAMAN v7.02. The BamHI site (GGATCC) and the telomere resolution sequence (ATTTTTT-N(8)-TAAAT) of ITRs of ORFV-SY17 and ORFV-NA17 were further analyzed to determine the intact of the terminal hairpin loops.
Phylogenetic analysis
The complete genome sequences of ORFV-SY17 and ORFV-NA17 were obtained after sequencing. Individual nucleotide sequences and complete genome sequences (16 PPV strains in total, listed in Table 1) including terminal repetition sequences were aligned by using ClustalW [15]. According to the alignment results, phylogenetic trees based on the complete genome and individual gene were constructed with MEGA v5.05 software, using the neighbor-joining method with 1000 bootstraps [16].
Results
Typical clinical features
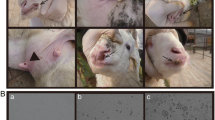
The two natural outbreaks of ORFV infections investigated in this study occurred in September and October 2016, respectively. The first outbreak occurred in a sheep herd with 145 small-Tailed Han sheep in Songyuan where the infected lambs presented with pox lesions with a morbidity rate of 14.5% (21/145). The typical multifocal to coalescing, ulcerated, multiple nodular, or proliferative lesions on the lips or around the mouth were observed when examined (Fig. 1a). The second outbreak of pox disease occurred in a cashmere goat herd in Nongan with a morbidity rate of about 8.5% (9/106). The infected goats presented with the characteristic lesions of Orf on the skin of lips, muzzle, and nostrils (Fig. 1b). In general, all infected animals recovered about 28 days after the first clinical signs appeared. In addition, no human infections were reported in the two outbreak areas.

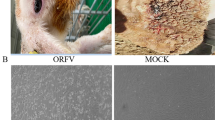
Representative clinical cases of Orf virus infection and electron microscopic examinations of the Orf virus. a Sheep showing multiple nodular lesions on the lips. b Photograph of a cashmere goat with severe, proliferative lesions in the skin of lips and muzzle. c Electron microphotograph of showing the characteristic morphology of an Orf virion from OFTu cell cultures inoculated with the skin lesion of lips collected from a small-Tailed Han sheep (bar = 100 nm). d Electron microphotograph showing the characteristic morphology of an Orf virion from OFTu cell cultures inoculated with the lip scabs collected from a cashmere goat (bar = 100 nm)
Virus identification by electron microscope
Isolation of Orf virus from scab tissue samples was attempted by inoculating tissue suspension into the culture of OFTu cells. After three blind passages, CPE was observed in the OFTu cells (data not shown). Virus particles from the supernatant of the CPE-positive cell cultures were subjected for electron microscopy (EM). Typical parapoxvirus virions with diameters of about 250 nm were observed, which were clearly distinguished from orthopoxviruses (OPVs) because of their characteristic spiral crisscross patterns (Fig. 1c, d). No other viruses were detected. The virus isolates originated from sheep and goat were designated ORFV/Songyuan/2017 (ORFV-SY17) and ORFV/Nongan/2017 (ORFV-NA17), respectively.
Genomic features of ORFV-SY17 and ORFV-NA17
After high-throughput sequencing of ORFV genomes using the Illumina Hiseq2000, the genome sequence of SY17 was assembled into contiguous sequences of 140,413 bp with G+C content of 63.8%, while NA17 was 139,287 bp in length, with 63.7% G+C content. The genomes of SY17 and NA17 were predicted to contain 131 and 132 potential ORFs. As is common to other poxviruses, ORFV-SY17 and ORFV-NA17 genomes contained the relatively conserved regions (ORFs 009-111) and non-conserved terminal regions (ORFs 001-008 and 112-134) (See Supplemental Table 1, Table 2) [9, 17]. The terminal regions of ORFV-SY17 and ORFV-NA17 genomes contained ITRs at both ends. The lengths of ITRs of SY17 and NA17 were 4267 bp and 3974 bp, respectively (Table 1). And the ITR of SY17 is the longest when compared with the corresponding sequences of other published ORFV strains in NCBI.
Comparisons of predicted ORFs of ORFV-SY17 and ORFV-NA17 with other ORFVs
The ORFs of ORFV-SY17 and ORFV-NA17 were predicted by using ORF Finder and BLASTP comparisons of the predicted amino acid sequence with the GenBank database. The amino acid identities of each ORF among SY17, NA17, and other fully sequenced ORFV strains were compared and listed in Fig. 2 and Supplemental Table 1 and Table 2. Compared to other four isolates (NA17, NA1/11, YX, and NZ2), ORFV-SY17 shared < 85% and 85–95% amino acid identities with 3–8 genes and 18–28 genes, respectively (Table 2). Among them, the amino acid identities of all ORFs between SY17 and NA1/11 were above 85%. Meanwhile, ORFV-NA17 shared with < 85% and > 95% amino acid identities with 5–12 genes and 97–123 genes, respectively. The NA17 and YX shared > 95% amino acid identity with 123 genes, however, NA17 only shared > 95% amino acid identity with 97–102 genes of other three ORFV isolates (Table 3). The results suggested that there was a close relationship between SY17 and NA1/11 and between NA17 and YX. In our alignment of ORFV genomes, ORFs 003, 004, 006, and 133 were not used in SY17 and NA17, while ORF 002 was found absent in SY17. Similarities to well-defined proteins in the other ORFVs, the SY17 and NA17 genomes also included two newly recognized genes (12.5 and 107.5) suggested by Mercer et al. [8]. The coding potentials of the ORFV-SY17 and ORFV-NA17 were predicted to contain 131 and 132 genes, respectively (Fig. 2, Supplemental Table 1, Table 2).

The comparative map of ORFV-SY17 and ORFV-NA17. The percent of amino acid sequence identity (%ID) of predicated ORFs between SY17 and NA17 were indicated by colored (black, blue, and yellow) arrows. Different colors of arrows indicate amino acid identity between SY17 and NA17. The %ID of identical ORFs (> 95%) were indicated in black arrows, 85–95% amino acid identities were present in blue, and < 85% amino acid identities were present in yellow
ITR analysis of ORFV-SY17 and ORFV-NA17
The characteristics of ITRs of ORFV-SY17 and ORFV-NA17 were further analyzed by comparing the corresponding regions of other published ORFV strains in NCBI. The ITRs of SY17 and NA17 contained terminal BamHI sites at both ends, which were similar with SJ1, GO, YX strains. However, the NA1/11 and NP strains only contained the BamHI site at the right end (Fig. 3a). In addition, the ITRs of SY17 and NA17 contained conserved telomere resolution sequences at both ends, however, the NA1/11 and NP strains only contained the telomere-related sequence at the right end (Fig. 3a). The ITRs of SY17 and NA17 both contained conservative BamHI sites and telomere resolution sequences at both ends of genomes, which suggested that the genomic sequences of SY17 and NA17 were relatively intact [2, 14].

A Alignment of ITRs of eleven ORFV genomes. a Regions corresponding to the left terminal sequences (5′-ITRs) of eleven ORFV isolates were aligned using DNAMAN v7.02. The regions of the terminal BamHI site (GGATCC) and telomere resolution sequences (ATTTTTT-N(8)-TAAAT) were indicated by black boxes and black virtual frames, respectively. b Regions corresponding to the right terminal sequences (3′-ITRs) of eleven ORFV isolates were aligned using DNAMAN v7.02. The regions of the terminal BamHI site (GGATCC) and telomere resolution sequences (ATTTTTT-N(8)-TAAAT) were indicated by black boxes and black virtual frames, respectively. B Alignment of ITRs of eight ORFV genomes. a′ Regions corresponding to the left terminal sequences (5′-ITRs) starting at the position of 1730 bp of eight ORFV isolates were aligned using DNAMAN v7.02. The deletion regions in the 5′-ITRs of SY17 and NA17 were indicated by black boxes. b′ Regions corresponding to the left terminal sequences (5′-ITRs) starting at the position of 3170 bp. The deletion regions in the 5′-ITRs of SY17 and NA17 were indicated by black boxes
Considering the difference caused by geographical distance factors, we performed comparison analysis for ITRs of ORFVs originated from different regions. There are several long deletion regions in the ITRs of ORFV-SY17 and ORFV-NA17, which are not present in other six ORFV strains (GO, NA1/11, NP, SJ1, YX, and HN3/12). Although the differences were observed in length of the ITRs of SY17 and NA17, the most of their ITR sequences were consistent with other six ORFV strains (Fig. 3b). By comparing the left-end ITR sequence with the inverted sequence of the right-end ITR of SY17 strain, we found that its inverted right-end ITR existed a deletion (CCTCCGCGGAGTCGGAGTCCT) at the position of 1418 bp, while the left-end ITR deleted a sequence (AGGACTCCGACTCCGCGGAGG) at the position of 1975 bp. The deleted regions are, respectively, located within the ITRs at both ends and are complementary (data not shown).
Phylogenetic analysis
Phylogenetic analysis based on the complete genomic sequences of 16 PPVs revealed that six ORFV strains originated in goats and six ORFV strains originated in sheep, which formed two separate branches with 100% bootstrap support. ORFV-SY17 isolated from sheep showed a close relationship with NA1/11 and HN3/12, while ORFV-NA17 isolated from goat was closer to YX than to other four strains (SJ1, SA00, NP, GO). In addition, the analysis result also showed that 12 ORFVs were more closely related to PCPV than to BPSV (Fig. 4).

Phylogenetic analyses based on whole genome sequences of PPVs. a The phylogenetic tree was constructed by the neighbor-joining method using MEGA v5.05. The numbers above or below the branch points indicate the bootstrap support calculated for 1000 replicates. Filled triangle SY17 isolated in this study; filled circle NA17 isolated in this study. b The circular phylogenetic tree was constructed by the neighbor-joining method using MEGA v5.05. The numbers above or below the branch points indicate the bootstrap support calculated for 1000 replicates. Filled triangle SY17 isolated in this study; filled circle NA17 isolated in this study
To investigate the genetic characteristics and phylogenetic relationships, we further analyzed the phylogenetic trees of each gene sequences of ORFVs. Phylogenetic analysis based on individual gene sequences of 16 PPVs revealed that 19 ORFs (008, 009, 012, 022, 025, 028, 049, 061, 062, 064, 065, 081, 107, 111, 113, 114, 115, 123, and 129) could be easily used for separating goat from sheep origins (Fig. 5, Supplementary Figs. S1, S2). The analysis results indicated that the genetic relationship of ORFV strains derived from the same host species was closer.

Phylogenetic analysis based on individual gene of ORFVs. The phylogenetic trees were respectively constructed by the neighbor-joining method using MEGA v5.05. The numbers above or below the branch points indicate the bootstrap support calculated for 1000 replicates. Filled triangle SY17 isolated in this study; filled circle NA17 isolated in this study. a ORFV008. b ORFV009. c ORFV012. d ORFV022. e ORFV025. f ORFV028. g ORFV049. h ORFV061. i ORFV062
Multiple alignment of ORFV-SY17 and ORFV-NA17
Alignment of ORFs among ORFV-SY17, ORFV-NA17, and other ORFVs reference strains revealed several sequence variations caused by insertions or deletions in ORFs 001, 005, 115, 116, 120, 132, and 134, especially in ORFs 005 and 116. ORFs 001, 005, 115, 116, 120, 132, and 134 all located at both ends of ORFV genome, which were confirmed to have a low %ID (Tables 2, 3). Multiple alignment of the amino acid sequences of individual genes 005, 115, and 116 further revealed the unique amino acid residues in the coding regions of ORFVs derived from sheep or goats (Fig. 6). Only fewer unique amino acid residues were found in genes 001, 120, and 132, and no unique amino acid residue was found in gene 134. As expected, amino acid mutations were found in multiple alignments of these genes.

Multiple alignments of deduced amino acid sequences. The unique amino acid residues were indicated by black virtual frames. a ORF 005. b ORF 115. c ORF 116
Discussion
Orf is a common epitheliotropic viral disease of sheep, goats, and wild ruminants, which has a worldwide distribution and is characterized by the formation of papules, nodules, or vesicles [18]. In the present study, we successfully isolated and identified two ORFV strains from infected sheep and goats in Jilin province of China, which were named ORFV-SY17 and ORFV-NA17, respectively. The complete genome sequences of SY17 and NA17 were sequenced and analyzed. The results will help to better understand the heterogeneity of ORFVs circulating in China.
Genomic sequences of ORFV-SY17 (140,413 bp, GenBank accession number MG712417) and ORFV-NA17 (139,287 bp, GenBank accession number MG674916) were obtained and analyzed by the comparative genomic analysis. Compared with other reported ORFV reference strains, the genome length of SY17 was the longest. As is common to other previously reported ORFV strains, SY17 and NA17 genomes both contained the large central coding region which were bounded by two identical inverted terminal repeat (ITR) regions on either end of the ORFV genome [14, 19]. Additionally, there are differences in the length of ITR between different ORFVs. The lengths of ITRs of ORFV-SY17 and ORFV-NA17 were 4267 bp and 3974 bp, respectively. Among them, the ITR of ORFV-SY17 is the longest when compared with the corresponding sequences of other published ORFV strains in NCBI, which indicated that natural variations occurred in this genomic region. Although the ITR region of ORFV genome was considered to be highly variable [9, 19], the relatively conserved telomere resolution sequences (ATTTTTT-N(8)-TAAAT) and the BamHI sites (GGATCC) were found at the both ends of ORFV-SY17 and ORFV-NA17 genomes, which were consistent with that of the vaccinia virus [20] and might be served as a characteristic sequence of the ITRs of ORFV.
The coding potentials of the ORFV-SY17 and ORFV-NA17 were predicted to contain 131 and 132 genes, respectively. After the comparison of ORFs between ORFVs, the results of amino acid identity showed that there was a close relationship between SY17 and NA1/11 and between NA17 and YX, which might be associated with host species. Multiple alignments of amino acid sequences of ORFs among different ORFVs revealed several amino acid residue variations caused by insertions or deletions, which indicated close relationship of the virus evolution and its self-protection mechanism. The phylogenetic tree was constructed based on the complete genomic sequences to analyze the evolutionary patterns of our two isolates ORFVs and other PPVs. The results showed that the six goat ORFVs and six sheep ORFVs formed distinctly separate branches. SY17 had close relations to NA1/11 and HN3/12 than to the others, while NA17 was more similar with YX. These results may imply the close relations between SY17, NA17, and other ORFVs in evolution, and also further confirmed the genetic differences between ORFV strains might be closely related to host species. The observed genetic differences including genomic deletions or insertions mainly located in the terminal regions of the genomes, which have been confirmed to encode factors that determine host range, pathogenesis, and virulence [7, 21]. Thus, we speculated that the distinct genetic differences in virus isolates from sheep compared to goats might be associated with ORFV genome structural features.
In conclusion, the new genomic information of the two ORFV strain originated from sheep and goat in Northeast China’s Jilin province were obtained. The availability of ORFV-SY17 and ORFV-NA17 nucleotide sequences will promote the development of future comparative genomic studies. We expect that the usability of the two full-length virus genome sequences will be helpful for more comprehensive understanding of ORFV biology and epidemiology.
References
Haig DM, Mercer AA (2002) Immunity and counter-immunity during infection with the parapoxvirus orf virus. Virus Res 88:3–16
Chi X, Zeng X, Li W, Hao W, Li M, Huang X, Huang Y, Rock DL, Luo S, Wang S (2015) Genome analysis of orf virus isolates from goats in the Fujian Province of southern China. Front Microbiol 6:1135
Spyrou V, Valiakos G (2015) Orf virus infection in sheep or goats. Vet Microbiol 181(1–2):178–182
Hosamani M, Scagliarini A, Bhanuprakash V, McInnes CJ, Singh RK (2009) Orf: an update on current research and future perspectives. Expert Rev Anti Infect Ther 7(7):879–893
Bodilsen J, Leth S (2013) Orf parapoxvirus can infect humans after relevant exposure. Ugeskr Laeger 175(16):1121–1122
Gumbrell RC, McGregor DA (1997) Outbreak of severe fatal orf in lambs. Vet Rec 141(6):150–151
Delhon G, Tulman ER, Afonso CL, Lu Z, de la Concha-Bermejillo A, Lehmkuhl HD, Piccone ME, Kutish GF, Rock DL (2004) Genomes of the parapoxviruses orf virus and bovine papular stomatitis virus. J Virol 78(1):168–177
Mercer AA, Ueda N, Friederichs SM, Hofmann K, Fraser KM, Bateman T, Fleming SB (2006) Comparative analysis of genome sequences of three isolates of Orf virus reveals unexpected sequence variation. Virus Rec 116(1–2):146–158
Li W, Hao W, Peng Y, Duan C, Tong C, Song D, Gao F, Li M, Rock DL, Luo S (2015) Comparative genomic sequence analysis of Chinese orf virus strain NA1/11 with other parapoxviruses. Arch Virol 160(1):253–266
Chen H, Li W, Kuang Z, Chen D, Liao X, Li M, Luo S, Hao W (2017) The whole genomic analysis of orf virus strain HN3/12 isolated from Henan province, central China. BMC Veterinary Research 13:260
Zhao K, Song D, He W, Lu H, Zhang B, Li C, Chen K, Gao F (2010) Identification and phylogenetic analysis of an Orf virus isolated from an outbreak in sheep in the Jilin province of China. Vet Microbiol 142(3–4):408–415
Li W, Ning Z, Hao W, Song D, Gao F, Zhao K, Liao X, Li M, Rock DL, Luo S (2012) Isolation and phylogenetic analysis of orf virus from the sheep herd outbreak in northeast China. BMC Vet Res 8:229
Sievers F, Higgins DG (2014) Clustal omega, accurate alignment of very large numbers of sequences. Methods Mol Biol 1079:105–116
Mercer AA, Fraser K, Barns G, Robinson AJ (1987) The structure and cloning of orf virus DNA. Virology 157(1):1–12
Thompson JD, Higgins DG, Gibson TJ (1994) CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res 22:4673–4680
Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S (2011) MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 28(10):2731–2739
Wang G, He W, Song D, Li J, Bao Y, Lu R, Bi J, Zhao K, Gao F (2014) In vitro RNA interference targeting the DNA polymerase gene inhibits orf virus replication in primary ovine fetal turbinate cells. Arch Virol 159(5):915–920
Robinson AJ, Balassu TC (1981) Contagious pustular dermatitis (orf). Vet Bull 51(10):771–782
Fraser KM, Hill DF, Mercer AA, Robinson AJ (1990) Sequence analysis of the inverted terminal repetition in the genome of the parapoxvirus, orf virus. Virology 176:379–389
Merchinsky M (1990) Mutational analysis of the resolution sequence of vaccinia virus DNA: essential sequence consists of two separate AT-rich regions highly conserved among poxviruses. J Virol 64(10):5029–5035
Fleming SB, Wise LM, Mercer AA (2015) Molecular genetic analysis of Orf virus: a Poxvirus that has adapted to skin. Viruses 7:1505–1539
McGuire MJ, Johnston SA, Sykes KF (2012) Novel immune-modulator identified by a rapid, functional screen of the parapoxvirus ovis (Orf virus) genome. Proteome Sci 10(1):4
Hautaniemi M, Ueda N, Tuimala J, Mercer AA, Lahdenperä J, McInnes CJ (2010) The genome of pseudocowpoxvirus: comparison of a reindeer isolate and a reference strain. J Gen Virol 91(6):1560–1576
Huang T, Tulman ER, Diel DG, Khatiwada S, Sims W, Edwards JF, Wen X, Kutish GF, Rock DL, Delhon G (2015) Coinfection with multiple strains of bovine papular stomatitis virus. Arch Virol 160(6):1527–1532
Acknowledgements
This study was supported by the National Natural Science Foundation of China (Grants 31,672,554).
Author information
Authors and Affiliations
Contributions
JZ and KZ conceived the study and participated in its design and coordination. JZ, SS, YZ, SC, ZW, SZ, MX, XW, and YG performed the research. JZ, JG, SZ, and WH analyzed and interpreted data. JZ and KZ wrote the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
This article does not contain any studies with human participants performed by any of the authors. All animal experiments were in accordance with the Animal Welfare Ethical Committee of the College of Veterinary Medicine, Jilin University.
Additional information
Edited by Joachim Jakob Bugert.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
11262_2019_1666_MOESM1_ESM.tif
Supplementary Fig. S1 Phylogenetic analysis based on single genes of ORFVs. The phylogenetic trees were constructed by the neighbor-joining method using MEGA v5.05. The numbers above or below the branch points indicate the bootstrap support calculated for 1000 replicates. ▲SY17 isolated in this study; ●NA17 isolated in this study. (A) ORFV064. (B) ORFV065. (C) ORFV081. (D) ORFV107. (E) ORFV111. (F) ORFV113. (G) ORFV114. (H) ORFV115. (I) ORFV123. (J) ORFV129. 1 (TIFF 641 kb)
11262_2019_1666_MOESM2_ESM.tif
Supplementary Fig. S2 Phylogenetic analysis based on single genes of ORFVs. The phylogenetic trees were constructed by the neighbor-joining method using MEGA v5.05. The numbers above or below the branch points indicate the bootstrap support calculated for 1000 replicates. ▲SY17 isolated in this study; ●NA17 isolated in this study. (A) ORFV064. (B) ORFV065. (C) ORFV081. (D) ORFV107. (E) ORFV111. (F) ORFV113. (G) ORFV114. (H) ORFV115. (I) ORFV123. (J) ORFV129. 1 (TIFF 249 kb)
Rights and permissions
About this article

Cite this article
Zhong, J., Guan, J., Zhou, Y. et al. Genomic characterization of two Orf virus isolates from Jilin province in China. Virus Genes 55, 490–501 (2019). https://doi.org/10.1007/s11262-019-01666-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11262-019-01666-y