
Abstract
Chicken anemia virus (CAV) has a ubiquitous and worldwide distribution in the chicken production industry. Our group previously reported a high seroprevalence of CAV in chickens from northern Vietnam. In the present study, tissue samples collected from a total of 330 broiler and breeder commercial chickens in eleven provinces of northern Vietnam were tested for CAV infection. All samples were collected from clinically suspected flocks and diseased birds. The CAV genome was detected in 157 out of 330 (47.58%) chicken samples by real-time PCR. The rate of CAV genome detection in young chickens at 2–3 weeks of age (61.43%), which had not been previously reported in Vietnam, was significantly higher than that in older chickens at 4–11 (44.83%) and 12–28 (35.71%) weeks of age. For nine representative CAV strains from broiler chickens, analysis of the entire protein-coding region of the viral genome was conducted. Phylogenetic analysis of the VP1 gene indicated that the CAVs circulating in northern Vietnam were divided into three distinct genotypes: II, III, and V. Only one of the nine Vietnamese CAV strains clustered with a vaccine strain (Del-Ros), whereas the other eight strains did not cluster with any vaccine strains. Among the three genotypes, genotype III was most widely found in northern Vietnam and this included three sub-genotypes (IIIa, IIIb, and IIIc). The Vietnamese CAV strains were closely related to the Chinese, Taiwanese, and USA strains. One strain was defined to be of genotype V, which is a newly reported CAV genotype. Moreover, recombination analysis suggests that this novel genotype V was generated by recombination between genotype II and sub-genotype IIIc.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Chicken anemia virus (CAV), a non-enveloped, circular, and single-stranded DNA virus, is classified into the Gyrovirus genus of the Anelloviridae family [1]. The viral genome consists of three overlapping open reading frames (ORFs), ORF1, ORF2, and ORF3, that encode the structural protein VP1 (51.6 kDa) and two non-structural proteins VP2 (24 kDa) and VP3 (13.6 kDa), respectively [1, 2]. The major capsid protein VP1 plays a critical role in viral capsid assembly and inducing neutralization antibodies in the host [2, 3]. The VP2 and VP3 proteins serve as scaffolding proteins of VP1 [4, 5] and strong inducers of apoptosis [6], respectively. Previous reports indicated hypervariability of the VP1 gene in the region coding for amino acid (aa) 139–151 and 11 major variable substitutions of aa located at positions 22, 75, 97, 125, 139, 144, 157, 287, 290, 370, and 413. In contrast, VP2 and VP3 genes are highly conserved among isolates [7, 8]. Thus, the VP1 gene has been selected for genetic characterization and evaluation of CAV.
CAV was first isolated from affected chickens in the field from Japan in 1979 [9]; 4 years later, it was successfully propagated in MDCC-MSB1 cells, which is a Marek’s disease virus-transformed chicken lymphoblastoid cell line [10]. Afterward, detection of CAV in chickens was reported in many countries worldwide. CAV has been known to cause economic loss in chicken production [11, 12]. Both horizontal and vertical transmissions have been recorded in CAV infections in chickens [13,14,15]. In the field, vertical transmission in breeders through embryonated eggs results in clinical disease in young chickens at 2 to 3 weeks of age, which leads to increased mortality [16, 17]. CAV-infected young chickens display depression, muscle hemorrhage, pale bone marrow, and thymus atrophy. In addition, horizontal CAV infection occurs in older chickens and results in subclinical disease followed by immunosuppression [18] and reduced immune responses to Newcastle disease virus and Marek’s disease virus vaccines in affected chickens [19, 20].
Phylogenetic analyses of full-length VP1 genes showed that CAV is either divided into three genetically different genotypes (I, II, and III) [21,22,23,24] with two sub-genotypes [21, 22], or four genotypes (I, II, III, and IV) [25]. In addition, Eltahir et al. [26] suggested that CAV consists of four genotypes (A, B, C, and D) and five sub-genotypes (A1, A2, A3, D1, and D2) based on genetic analyses of the full viral genome. Another study demonstrated the presence of intersubtype recombination, which may indicate the appearance of novel genotypes and/or sub-genotypes [27].
Our group previously performed the first serological surveillance study of CAV infection in Vietnam and reported that approximately 70% of chicken sera collected in several chicken farms in northern Vietnam were positive for antibody against CAV [28]. The results suggested that CAV infection might be at high prevalence in this country. However, lack of information about vaccination in the studied chickens limited us from fully understanding CAV infection in chickens in Vietnam. From chicken samples mainly collected in Hanoi, a part of northern Vietnam, Dao et al. [29] reported that 63 out of 124 samples were positive for the CAV genome. Based on phylogenetic analyses of the partial VP1 gene (592 bp), they tentatively divided the Vietnamese CAV strains into two groups. In the current study, we investigated CAV infection in broiler and breeder commercial chickens from eleven provinces of northern Vietnam, and furthermore, conducted molecular characterization of identified CAV strains based on the entire protein-coding regions of the viral genome.
Materials and methods
Samples
Tissue samples from a total of 330 chickens obtained in northern Vietnam between 2016 and 2018 were used in this study. For sample collection, chicken farms with slightly increased mortality in young chickens, according to the owners’ personal reports, were selected. From each farm involved in this study, 3 to 6 chickens displaying poor performance and weakness were selected for sample collection. There was no information available about the number of farms tested, but none of the farms involved in this study had previously used CAV vaccines. From each chicken, tissue samples, including liver, spleen, bone marrow and thymus, were collected and pooled into one tube to make individual pooled samples. Among the 330 pooled tissue samples, 328 were obtained from commercial chickens of 2 to 28 weeks of age farmed in eleven provinces in northern Vietnam during 2016–2018: Hanoi (HN), Haiduong (HD), Haiphong (HP), Bacninh (BN), Quangninh (QN), Thaibinh (TB), Namdinh (ND), Hanam (HM), Hoabinh (HB), Phutho (PT), and Vinhphuc (VP). The remaining two samples were obtained from breeder chickens at 12 weeks of age farmed in Hanoi in 2017. Each pooled tissue sample was homogenized in phosphate-buffered saline containing kanamycin (1 mg/mL), gentamycin (100 µg/mL), and amphotericin B (10 µg/mL) as a 20% homogenate.
Real-time PCR
DNA was extracted from the homogenized samples using High Pure PCR Template Preparation Kit (Roche Diagnostics GmbH, Mannheim, Germany). Real-time PCR for the CAV VP1 gene was conducted using primers (CAV-1F, CAV-1R) and a CAV-probe, shown in Table 1, as previously described [23]. In order to prepare a control plasmid for CAV genome quantification, the partial VP1 gene (402 bp) amplified using a pair of primers, CAV-VP1F and CAV-VP1R (Table 1), was inserted into a T-Vector pMD20 (Takara Bio Ind., Shiga, Japan). The constructed plasmid recovered from the transformed E. coli was serially diluted and applied to real-time PCR, using the CAV-1F and CAV-1R primers, to obtain a standard curve. Known amounts of plasmid DNA containing between 5 × 101 and 5 × 106 copies of the control plasmid were used as templates. The standard curve obtained demonstrated high accuracy (R2 > 0.99) and indicated that the assay can detect 50 copies of the gene with the Cq value of 36.8. Samples showing Cq values less than or equal to 36.8 were regarded as positive for the CAV genome.
Nucleotide sequencing and phylogenetic analyses
The full-length protein-coding sequence (1823 bp) of the viral genome was amplified by PCR using two pairs of primers, CAV-CQ1F/CAV-CQ1R and CAV-CQ2F/CAV-CQ2R (Table 1), as described by Zhang et al. [30]. PCR products of 1778 and 831 bp in size, respectively, were separated on agarose gels and purified by GeneClean® II Kit (MP Biomedicals, Santa Ana, CA, USA). Nucleotide sequencing was performed using the BigDye Terminator v3.1 Cycle Sequencing Kit (Life Technologies, Carlsbad, CA, USA) and the Applied Biosystems 3500 Genetic Analyzer (Life Technologies).
The Clustal W multiple alignment tool [31] in BioEdit v.7.2.5 [32] was used to align and analyze the nucleotide sequences and deduced aa sequences derived from CAV. Homology in nucleotide and aa sequences was examined by the GENETYX v.10 software (GENETYX Corp., Tokyo, Japan) and compared with other publicly available sequences using the BLAST program. A maximum likelihood method with Hasegawa-Kishino-Yano model of nucleotide substitutions was used to construct the phylogenetic tree based on nucleotide sequences, and a Jones-Taylor-Thornton model of amino acid substitutions was used to reconstruct the phylogenetic tree based on amino acid sequences of nine currently identified Vietnamese (this study), six previously identified Vietnamese, and 56 foreign CAV strains. The confidence values on phylogenetic trees were assessed by bootstrapping with 1000 replicates using MEGA6 software [33]. Complete protein-coding sequences of the viral genome obtained in this study have been deposited into GenBank under accession numbers MK423866 to MK423874.
Identification and confirmation of recombination
Nine CAV strain sequences from the current study and six other sequences from GenBank were used to identify recombination events using RDP, GENECONV, BootScan, MaxChi, Chimaera, SiScan, Phyl-Pro, LARD, and 3Seq methods implemented in Recombination Detection Program (RDP) version Beta 4.97 [34] with default settings. Recombination events were designated if identified by at least 4 of the 9 methods (p value < 0.05). The recombination events were confirmed by bootscanning in SimPlot v.3.5.1 [35].
Statistical analysis
Fisher’s exact test was used to identify significant differences in the rate of CAV genome detection between age groups. A value of p < 0.05 was considered statistically significant.
Results
In this study, samples were obtained from chickens with poor performance and weakness on farms where slightly increased mortality was observed in young chickens. First, we assessed the CAV infection rates among chicken samples tested using real-time PCR. The CAV genome was detected (Cq value < 36.8) in 157 out of 330 (47.58%) samples tested. The proportion of CAV-positive chickens at 2–3 weeks of age was 61.43% (43/70), which was significantly higher than those at 4–11 weeks (44.83%) (p = 0.02) and 12–28 weeks (35.71%) (p = 0.03) of age. The two samples from breeder chickens at 12 weeks of age were also positive in the CAV genome detection real-time PCR (Table 2).
For genetic characterization, nine CAV-positive samples from broiler chickens obtained at different locations (different farms in HN, BN, HB, HP, PT, and VP) and sampling years (2016, 2017, and 2018) were selected for nucleotide sequencing. Full-length CAV VP1 gene sequences from the nine Vietnamese strains were aligned and compared with other sequences retrieved from GenBank. The nucleotide identity ranged from 94.66 to 99.77% among the Vietnamese CAV strains obtained in the current study. Among these, the highest nucleotide identity was found between Vietnam/HN2/16 and Vietnam/HN1/17 (99.77%) while the lowest was between Vietnam/BN1/17 and Vietnam/BN2/16 (94.66%). Comparing the VP1 gene from the Vietnamese CAV strains in this study and those previously reported, the highest nucleotide identity was 99.70% (Vietnam/HB1/17 vs Vietnam/KP780287.1/BN1/13) while the lowest was 94.37% (Vietnam/BN1/17 vs Vietnam/KP780292.1/VP10/13). Seven out of the nine Vietnamese CAV strains showed nucleotide identity higher than 99.03% with Chinese (KU645516.1, KU645510.1, KU050677.1, and KY486146.1) or Taiwanese (KJ728827.1) isolates. The remaining two strains, Vietnam/VP1/18 and Vietnam/PT1/17, showed nucleotide identity of 98.74% and 97.25% with the American strains 98D02152 (AF311892.2) and 98D06073 (AF311900.3), respectively. Neither mutations nor substitutions were found at either nucleic acid or amino acid levels of VP2 and VP3.
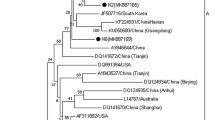
Phylogenetic analysis based on the CAV VP1 gene (1350 bp) indicated that the Vietnamese CAV strains we obtained in northern Vietnam were divided into three genotypes (II, III, and V) and three sub-genotypes (IIIa, IIIb, and IIIc) with high supporting bootstrap values. Genotypes II and V were represented by one strain each. The other seven strains were found to belong to genotype III; one of these CAV strains, Vietnam/VP1/18, was clustered with the US vaccine strain (USA/AF313470/Del-Ros) in sub-genotype IIIb, while the other six genotype III strains belong to sub-genotype IIIa (one strain) or IIIc (five strains). None of the Vietnamese CAV strains were clustered with the vaccine strains, 26P4 and Cuxhaven 1 strains, which have been available in Vietnam (Fig. 1).

Phylogenetic tree of VP1 gene sequences (1350 bp) of the Vietnamese CAV strains compared with those available in GenBank. GenBank sequences are indicated by the country name/accession number. The maximum likelihood method in MEGA6 software was used to establish the phylogenetic tree (1000 bootstrap replicates). Number at each branch point indicates bootstrap values ≥ 50% in the bootstrap interior branch test. The current Vietnamese, previous Vietnamese, and vaccine strains are indicated by squares, triangles, and circles, respectively. Five major genotypes and three sub-genotypes were identified and designated as I, II, IIIa, IIIb, IIIc, IV, and V
There was a marked difference in the grouping and subgrouping of sequences between nucleotide and aa levels (Figs. 1 and 2). However, the aa phylogenetic tree also clearly indicated that the Vietnamese CAV strains were divided into several clusters. In addition, in the aa phylogenetic tree, the Vietnamese CAV strains were not clustered with the vaccine strains, 26P4 and Cuxhaven 1 strains (Fig. 2).

Phylogenetic tree of VP1 protein sequences of Vietnamese CAV strains compared with those available in GenBank. GenBank sequences are shown as the country name/accession number. The maximum likelihood method with the Jones-Taylor-Thornton model was used in MEGA6 software to establish the phylogenetic tree (1000 bootstrap replicates). Number at each branch point indicates bootstrap values ≥ 50% in the bootstrap interior branch test. The current Vietnamese, previous Vietnamese, and vaccine strains are indicated by squares, triangles, and circles, respectively
Deduced aa sequences of VP1 of the Vietnamese CAV strains were compared with other CAV strains in various lineages. Nine major variable aa substitutions were detected in VP1 of the Vietnamese CAV strains in the current study, namely 22H/N/Q, 75 V/I, 97 M/L, 125I/L, 139 K/Q, 144E/Q, 287S/A, 370G/S, and 376 L/I. All of the Vietnamese CAV strains had T and Q at position 89 and 394 of VP1, respectively (Table 3).
Recombination analyses suggested that Vietnam/PT1/17, which belongs to the genotype V, resulted from a recombination event. The major and minor parents were Vietnam/VP10/13 of genotype II and Vietnam/VP9/13 of genotype IIIc. This putative recombination event was detected by six out of nine methods using RDP 4 (Table 4 and Fig. 3). Analysis using the SimPlot program also detected recombination. The two breakpoints were detected and located at residues 1040 and 1699 of the protein-coding region of the viral genome, respectively (equivalent to nucleotide positions 534 and 1193 of the VP1 gene) (Fig. 4).

Detection of recombination events using BootScan analysis of the entire protein-coding region of Vietnamese CAV strains. The pairwise distance model with window size 200, step size 20, and 1000 bootstrap replicates were generated by the RDP4 program

The use of distance plot for recombination identification. The Vietnam/PT1/17 strain was used as a query. The pairwise distance of Simplot program with a window size of 200 bp and step size of 20 bp was used. The predicted recombination breakpoints at nucleotide position 1040 and 1699 are indicated by the vertical red lines
Discussion
Understanding CAV infections is important as they cause immunosuppression in infected hosts. The seroprevalence of CAV was first reported in Vietnam by our previous study [28]. Recently, Vietnamese CAV strains were characterized based on partial sequences of the VP1 gene [29]. Other recent studies reported that CAV strains may be divided into more than three genotypes and sub-genotypes [25, 26]. The molecular evolution among Taiwanese CAV strains was also reported based on analyses of coding regions of VP1 and VP2 [25]. The Vietnamese CAV gene sequences are still limited in GenBank, and additional sequence data of protein-coding regions are necessary to further characterize and understand the evolution of CAV strains. This is the first study to characterize nucleotide sequences of the full-length protein-coding region of the viral genome (1823 bp) of Vietnamese CAV strains.
In the present study, the CAV genome was detected in 47.58% of chicken samples collected in northern Vietnam which is similar to the previous study [29]. However, samples used in this study were collected from chickens with poor performance rather than being randomly collected from a chicken flock. In addition, we do not have enough information about the number of farms tested in this study. Therefore, our CAV detection rate is not relevant for estimating the CAV prevalence in Vietnam. Further studies are needed to reveal the prevalence of CAV infection in Vietnam. Additional studies are needed to clarify the relationship between CAV infection and the clinical signs observed in chickens. It has been reported that the rate of CAV infection varies among countries, such as China (10.2%) [26], India (73.3%) [36], and Brazil (90%) [37]. This variation could be attributed to differences in sampling size, time, location, and sensitivity of detection methods used. Nevertheless, most studies suggest that CAV is circulating at a high prevalence and may be affecting chicken production worldwide. In the current study, we first found a high CAV genome rate in Vietnamese chickens at 2–3 weeks of age (61.43%). The rate of CAV genome detection decreased in older chickens at 4–11 (44.83%) and 12–28 weeks (35.71%) of age. A previous study reported that the presence of maternal antibodies derived from vaccination completely suppressed CAV infection in young chickens [38]. In our study, the high detection rate of the CAV genome in 2–3-week-old chickens may suggest the necessity for a CAV vaccination program in breeder chickens in Vietnam to protect young chickens from CAV infection. It should be noted that CAV live vaccine strains reportedly could revert back to become virulent strains after multiple passages in chickens [39]. Therefore, a precise investigation is required to assess the efficacy of vaccine strains against wild strains as well as the risk of vaccine reversion upon application of the CAV vaccination program. It has been well known that CAV is characterized as a single serotype. On the other hand, CAV strains have been classified into several distinct genotypes and sub-genotypes. Three genotypes of CAV were reported, namely genotypes I, II, and III, based on analyses of the VP1 gene [21,22,23,24], as well as two sub-genotypes of genotype III, in Korean and Nigerian chickens [21, 22]. However, phylogenetic analyses using the complete genome revealed that four genotypes and five sub-genotypes of CAV were found to be circulating in Chinese chickens, namely A, B, C, and D [26, 27]. A novel genotype IV and two sub-genotypes of genotype III were also reported in Taiwanese commercial and native chickens [25]. The phylogenetic analysis in the present study revealed that three distinct genotypes II, III, and V were circulating in Vietnamese chickens from 2016 to 2018. Among those, genotype III was the most dominant, including three sub-genotypes, IIIa, IIIb, and IIIc. Our study is the first to report the only strain in genotype V, which might have resulted from a recombination event between genotype II and sub-genotype IIIc. Currently, genotypes V and I consist of only one Vietnamese strain and two Australian strains, respectively (Fig. 1). Therefore, additional CAV sequence data is required for better understanding the molecular diversity of CAV strains worldwide. One Vietnamese CAV strain of genotype III was closely related to the vaccine strain Del-Ros. Overall, results from the phylogenetic analysis and nucleotide comparison of the CAV VP1 gene suggested that Vietnamese strains identified in this study are closely related to the Chinese, Taiwanese, and USA strains. Breeder chickens have been imported into Vietnam from many countries like USA, China, and Thailand since the 1990s [40, 41]. Approximately 722 thousand (41.7%) from a total of 1.7 million breeder poultry birds were imported into Vietnam from USA in 2014 [42]. Moreover, daily trading of live birds along the border between Vietnam and China has occurred [43], which could trigger outbreaks of the highly pathogenic avian influenza virus in chickens [44]. Such circumstances could have affected the diversity of CAV genotypes in Vietnam.
Recombination is one of the evolutionary processes which has been reported in CAV from several countries such as China [27], Taiwan [25], and Egypt [45] based on sequence data of complete genomes or the protein-coding region of the viral genome. Inter- or intra-genotypic recombination might generate new CAV strains. In addition, the recombination regions could span across both protein-coding or non-protein-coding regions [25, 27, 45]. In this study, we reveal the first evidence supporting a recombination event in Vietnamese CAV strains in the coding region of the VP1 gene. It suggests that recombination between genotype II and sub-genotype IIIc generated a novel genotype V. It has been reported that recombination seemed to occur between strains in geographically related regions [27, 45] which is consistent with the findings in our present study, since the detected recombination could have occurred in VP or PT, which are closely located to each other in northern Vietnam.
Kim et al. [22] previously reported the classification of Korean CAVs using eleven variable amino acids of VP1 and identified two distinct groups of CAVs circulating in breeder chickens within Korea. In the present study, Vietnamese CAVs could not be differentiated into such groups based on the variable amino acids of VP1, which is consistent with previous reports [25, 26]. These results may indicate that there is a substantial difference between the classification of CAVs by phylogenetic analysis of nucleotide sequences and that by molecular analysis of aa sequences.
Regarding the pathogenicity of CAV, Yamaguchi et al. [46] reported that viral pathogenicity could change from highly to less pathogenic if the aa at position 394 in VP1 was substituted from glutamine to histidine. In contrast, later reports demonstrated that significant reduction of pathogenicity in chickens was observed in CAV strains with glutamine at position 394 in VP1 like the attenuated A2, Cloned isolate 10, and P310 strains [46,47,48]. In addition, CAV pathogenicity might be reduced by aa substitution at residue 89T [49]; this substitution was absent in the Vietnamese CAV strains analyzed. However, as the virulence-affecting amino acid substitutions in CAV have not been fully understood, additional animal studies should be conducted to properly evaluate the pathogenicity of Vietnamese CAV strains.
In summary, in this study, CAV infection was detected at a high rate among poorly performing flocks of young chickens in northern Vietnam. It is possible that unvaccinated breeder chickens were infected with CAV and subsequently transmitted the virus to commercial chickens. Phylogenetic and molecular analyses revealed that the Vietnamese CAV strains belong to genotypes II, IIIa, IIIb, IIIc, and V. This is the first report of CAV genotype V in the world. None of the Vietnamese CAV strains were related to the vaccine strains currently used in the country. The novel genotype V was likely generated from a recombination event between genotypes II and III. Further investigation should be conducted to understand the possible evolutionary mechanisms acting on CAV and improve the control of CAV infection in chicken production across Vietnam.
References
Rosario K, Breitbart M, Harrach B, Segales J, Delwart E, Biagini P, Varsani A (2017) Revisiting the taxonomy of the family Circoviridae: establishment of the genus Cyclovirus and removal of the genus Gyrovirus. Arch Virol 162:1447–1463
Noteborn MH, de Boer GF, van Roozelaar DJ, Karreman C, Kranenburg O, Vos JG, Jeurissen SH, Hoeben RC, Zantema A, Koch G (1991) Characterization of cloned chicken anemia virus DNA that contains all elements for the infectious replication cycle. J Virol 65:3131–3139
Todd D, Creelan JL, Mackie DP, Rixon F, McNulty MS (1990) Purification and biochemical characterization of chicken anaemia agent. J Gen Virol 71(Pt 4):819–823
Noteborn MH, Verschueren CA, Koch G, Van der Eb AJ (1998) Simultaneous expression of recombinant baculovirus-encoded chicken anaemia virus (CAV) proteins VP1 and VP2 is required for formation of the CAV-specific neutralizing epitope. J Gen Virol 79(Pt 12):3073–3077
Koch G, van Roozelaar DJ, Verschueren CA, van der Eb AJ, Noteborn MH (1995) Immunogenic and protective properties of chicken anaemia virus proteins expressed by baculovirus. Vaccine 13:63–770
Noteborn MH, Todd D, Verschueren CA, de Gauw HW, Curran WL, Veldkamp S, Douglas AJ, McNulty MS, van der Eb AJ, Koch G (1994) A single chicken anemia virus protein induces apoptosis. J Virol 68:346–351
Farkas T, Tanaka A, Kai K, Kanoe M (1996) Cloning and sequencing of the genome of chicken anaemia virus (CAV) TK-5803 strain and comparison with other CAV strains. J Vet Med Sci 58:681–684
Renshaw RW, Soine C, Weinkle T, O’Connell PH, Ohashi K, Watson S, Lucio B, Harrington S, Schat KA (1996) A hypervariable region in VP1 of chicken infectious anemia virus mediates rate of spread and cell tropism in tissue culture. J Virol 70:8872–8878
Yuasa N, Taniguchi T, Yoshida I (1979) Isolation and some characteristics of an agent inducing anemia in chicks. Avian Dis 23:366–385
Yuasa N (1983) Propagation and infectivity titration of the Gifu-1 strain of chicken anemia agent in a cell line (MDCC-MSB1) derived from Marek’s disease lymphoma. Natl Inst Anim Health Q (Tokyo) 23:13–20
McIlroy SG, McNulty MS, Bruce DW, Smyth JA, Goodall EA, Alcorn MJ (1992) Economic effects of clinical chicken anemia agent infection on profitable broiler production. Avian Dis 36:566–574
Schat KA, van Santen VL (2008) Chicken infectious anemia virus and other Circovirus infections. In: Saif AMFYM, Glisson JR, McDougald LR, Nolan LK, DE Swayne (eds) Diseases of Poultry, 12th edn. Blackwel, Iowa, pp 211–235
Hoop RK (1992) Persistence and vertical transmission of chicken anaemia agent in experimentally infected laying hens. Avian Pathol 21:493–501
Rosenberger JK, Cloud SS (1989) The effects of age, route of exposure, and coinfection with infectious bursal disease virus on the pathogenicity and transmissibility of chicken anemia agent (CAA). Avian Dis 33:753–759
Yuasa N, Yoshida I (1983) Experimental egg transmission of chicken anemia agent. Natl Inst Anim Health Q (Tokyo) 23:99–100
Hoop RK, Guscetti F, Keller B (1992) An outbreak of infectious chicken anemia in fattening chickens in Switzerland. Schweiz Arch Tierheilkd 134:485–489
Yuasa N, Imai K, Watanabe K, Saito F, Abe M, Komi K (1987) Aetiological examination of an outbreak of haemorrhagic syndrome in a broiler flock in Japan. Avian Pathol 16:521–526
Adair BM (2000) Immunopathogenesis of chicken anemia virus infection. Dev Comp Immunol 24:247–255
De Boer GF, Van Roozelaar DJ, Moormann RJ, Jeurissen SH, Wijngaard JC, Hilbink F, Koch G (1994) Interaction between chicken anaemia virus and live Newcastle disease vaccine. Avian Pathol 23:263–275
Zhang Y, Cui N, Han N, Wu J, Cui Z, Su S (2017) Depression of vaccinal immunity to Marek’s disease by infection with chicken infectious anemia virus. Front Microbiol 8:1863
Ducatez MF, Owoade AA, Abiola JO, Muller CP (2006) Molecular epidemiology of chicken anemia virus in Nigeria. Arch Virol 151:97–111
Kim HR, Kwon YK, Bae YC, Oem JK, Lee OS (2010) Molecular characterization of chicken infectious anemia viruses detected from breeder and broiler chickens in South Korea. Poult Sci 89:2426–2431
Olszewska-Tomczyk M, Swieton E, Minta Z, Smietanka K (2016) Occurrence and phylogenetic studies of chicken anemia virus from Polish broiler flocks. Avian Dis 60:70–74
Snoeck CJ, Komoyo GF, Mbee BP, Nakoune E, Le Faou A, Okwen MP, Muller CP (2012) Epidemiology of chicken anemia virus in Central African Republic and Cameroon. Virol J 9:189
Ou SC, Lin HL, Liu PC, Huang HJ, Lee MS, Lien YY, Tsai YL (2018) Epidemiology and molecular characterization of chicken anaemia virus from commercial and native chickens in Taiwan. Transbound Emerg Dis 65(6):1493–1501
Eltahir YM, Qian K, Jin W, Wang P, Qin A (2011) Molecular epidemiology of chicken anemia virus in commercial farms in China. Virol J 8:145
Eltahir YM, Qian K, Jin W, Qin A (2011) Analysis of chicken anemia virus genome: evidence of intersubtype recombination. Virol J 8:512
Trinh DQ, Ogawa H, Bui VN, Nguyen TT, Gronsang D, Baatartsogt T, Kizito MK, AboElkhair M, Yamaguchi S, Nguyen VK, Imai K (2015) Development of a blocking latex agglutination test for the detection of antibodies to chicken anemia virus. J Virol Methods 221:74–80
Dao DT, Cao TBP, Vu TN, Nguyen VG, Huynh TML (2018) Prevalence of chicken infectious anemia virus (CIAV) circulating in Hanoi and surrounding provinces. Vietnam J Agri Sci 16:36–45
Zhang X, Liu Y, Wu B, Sun B, Chen F, Ji J, Ma J, Xie Q (2013) Phylogenetic and molecular characterization of chicken anemia virus in southern China from 2011 to 2012. Sci Rep 3:3519
Thompson JD, Higgins DG, Gibson TJ (1994) CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res 22:4673–4680
Hall TA (1999) BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp Ser 41:95–98
Tamura K, Stecher G, Peterson D, Filipski A, Kumar S (2013) MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol 30:2725–2729
Martin DP, Murrell B, Golden M, Khoosal A, Muhire B (2015) RDP4: detection and analysis of recombination patterns in virus genomes. Virus Evol 1:1–5
Lole KS, Bollinger RC, Paranjape RS, Gadkari D, Kulkarni SS, Novak NG, Ingersoll R, Sheppard HW, Ray SC (1999) Full-length human immunodeficiency virus type 1 genomes from subtype C-infected seroconverters in India, with evidence of intersubtype recombination. J Virol 73:152–160
Wani MY, Dhama K, Barathidasan R, Gowthaman V, Tiwari R, Bhatt P, Mahajan NK, Chawak MM, Singh SD, Kataria JM (2013) Molecular detection and epidemiology of chicken infectious anaemia virus in India. South Asian J Exp Biol 3:145–151
Simionatto S, Lima-Rosa CA, Binneck E, Ravazzolo AP, Canal CW (2006) Characterization and phylogenetic analysis of Brazilian chicken anaemia virus. Virus Genes 33:5–10
Yuasa N, Noguchi T, Furuta K, Yoshida I (1980) Maternal antibody and its effect on the susceptibility of chicks to chicken anemia agent. Avian Dis 24:197–201
Todd D, Creelan JL, Connor TJ, Ball NW, Scott AN, Meehan BM, McKenna GF, McNulty MS (2003) Investigation of the unstable attenuation exhibited by a chicken anaemia virus isolate. Avian Pathol 32:375–382
Duc NV, and Long T (2008) Poultry production systems in Vietnam. FAO’s website. http://www.fao.org/3/al693e/al693e00.pdf. Accessed 20 Sept 2018
Vang ND, and Ly LV (2000) A review on poultry production in Vietnam. FAO’s website. http://www.fao.org/tempref/AG/Reserved/PPLPF/Docs/Reports%20&%20Papers/PAP_GE_EA_UP_00_Vietnam%20Poultry%20Production_Vang.doc. Accessed 20 Sept 2018
Vietnam ministry of agriculture and rural development (2015) Breeder animals in Vietnam and developing the stategies. Vietnam ministry of agriculture and rural development’s website. https://mard.gov.vn/_CONTROLS/ESPORTAL/PubAnPhamTTChiTiet/Service.svc/download/L0FuUGhhbVRUL0xpc3RzL0FuUGhhbVRU/64. Accessed 20 Sept 2018
Desvaux S, Nguyen CO, Vu DT, Henriquez C, Ky VD, Roger F, Fenwick S, Goutard F (2016) Risk of introduction in northern Vietnam HPAI viruses from China: description, pattern and drivers of illegal poultry trade. Transbound Emerg Dis 63:389–397
Wang J, Vijaykrishna D, Duan L, Bahl J, Zhang JX, Webster RG, Peiris JS, Chen H, Smith GJ, Guan Y (2008) Identification of the progenitors of Indonesian and Vietnamese avian influenza A (H5N1) viruses from Southern China. J Virol 82:3405–3414
Erfan AM, Selim AA, Naguib MM (2018) Characterization of full genome sequences of chicken anemia viruses circulating in Egypt reveals distinct genetic diversity and evidence of recombination. Virus Res 251:78–85
Yamaguchi S, Imada T, Kaji N, Mase M, Tsukamoto K, Tanimura N, Yuasa N (2001) Identification of a genetic determinant of pathogenicity in chicken anaemia virus. J Gen Virol 82:1233–1238
Scott AN, McNulty MS, Todd D (2001) Characterisation of a chicken anaemia virus variant population that resists neutralisation with a group-specific monoclonal antibody. Arch Virol 146:713–728
Meehan BM, Todd D, Creelan JL, Connor TJ, McNulty MS (1997) Investigation of the attenuation exhibited by a molecularly cloned chicken anemia virus isolate by utilizing a chimeric virus approach. J Virol 71:8362–8367
Todd D, Scott AN, Ball NW, Borghmans BJ, Adair BM (2002) Molecular basis of the attenuation exhibited by molecularly cloned highly passaged chicken anemia virus isolates. J Virol 76:8472–8474
Acknowledgements
We would like to thank Ms. Sachiko Matsuda from the Department of Veterinary Medicine, Obihiro University of Agriculture and Veterinary Medicine, for her excellent technical assistance.
Author information
Authors and Affiliations
Contributions
HVD, HO, LTMH, and KI designed the research. HDV, GTHT, and TDD performed the research. GVN, VNB, and LHTM contributed to sample collection. HVD, YT, and HO wrote the manuscript. All the authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interests.
Research involving human participants and/or animals
This article does not contain any studies with human participants. Collection of chicken tissue samples was conducted by Vietnam National University of Agriculture under the institutional approval, and permission from the owners of chickens.
Additional information
Edited by William Dundon.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Van Dong, H., Tran, G.T.H., Van Nguyen, G. et al. Chicken anemia virus in northern Vietnam: molecular characterization reveals multiple genotypes and evidence of recombination. Virus Genes 55, 643–653 (2019). https://doi.org/10.1007/s11262-019-01686-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11262-019-01686-8