
Abstract
Paratuberculosis is a worldwide chronic enteric disease of ruminants, caused by Mycobacterium avium subsp. paratuberculosis (MAP). While MAP has been widely investigated all around the world, little is known about the different strains that circulate in each country. This study describes the genetic diversity of MAP isolates from different bovine and deer herds from Argentina, analyzed by Multiple-Locus Variable number tandem repeat Analysis (MLVA), as well as the phylogenetic relatedness between geographically distant isolates through Whole Genome Sequencing (WGS) and core-genome analysis. A total of 90 MAP isolates were analyzed. The results showed seven different MLVA genotypes, with almost 75% of them belonging to pattern INMV 1, described in all the herds studied. WGS results suggested the presence of a common INMV 1 strain circulating throughout the country. Our results allow confirming the coexistence of different strains in time and space and the mixed infections identified in some animals. These observations suggest the absence of animal monitoring prior to introduction to the herds and the need for a control program in the country. This study represents the first to report WGS of MAP strains in Argentina.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Paratuberculosis (PTB) or Johne’s disease is a widespread intestinal disorder that causes chronic enteritis. The disease is mostly associated to domestic ruminants such as cattle (Gopi et al. 2022; Kumar et al. 2020), sheep (Traveria et al. 2013), deer (Palmer et al. 2019; Paolicchi et al. 2001), goats (Fiorentino et al. 2012) and camelids such as alpaca (Salgado et al. 2016), but it is also associated with other wild and domestic species such as rabbits (Beard et al. 2001; Fox et al. 2018; Shaughnessy et al. 2013), foxes (Matos et al. 2014) and cats (Kukanich et al. 2013). Argentina has the fifth-largest cattle herd worldwide, with great production of meat and dairy products (Espeschit et al. 2017). PTB causes high economic losses for farmers and is thus an important issue for the country (Moreira and Tosi 1995).
The climate and soil conditions of Argentina determine that most of the cows are located in the Pampas region, in the central-east region of the country, where the seroprevalence of PTB ranges from 7.2% to 19.6% (Paolicchii et al. 2003). In contrast, the seroprevalence of PTB in Tierra del Fuego, the southernmost province of Argentina, is unknown and has been considered a bovine tuberculosis-free region since 2011 (SENASA 100/2011 resolution), an uncommon characteristic in the continent. The potential reason for this is that for the past 12 years, no cattle have been introduced and animals are born and raised inside this province.
The etiological agent of PTB is Mycobacterium avium subsp. paratuberculosis (MAP). This pathogen has a strong association to human Crohn’s disease (Singh et al. 2016; Timms et al. 2016). MAP is one of the most common non-tuberculous mycobacteria and a member of the Mycobacterium avium complex. The most frequent route of transmission is the fecal–oral (Gopi et al. 2020), but vertical transmission has also been reported (Vasini Rosell et al. 2020; Whittington and Windsor 2009). A molecular typing technique based on Mycobacterial Interspersed Repetitive Unit (MIRU) and Variable Number Tandem Repeat (VNTR) loci has been developed to analyze the genetic polymorphisms among MAP strains (Thibault et al. 2007). This technique, also called Multiple-Locus Variable number tandem repeat Analysis (MLVA), is a simple and rapid procedure consisting of eight amplifications of different loci and their respective runs in agarose gels to show variations in length (number of repeats) in each locus. This technique could also be applied directly to clinical samples, something that is particularly desirable for MAP because of the very slow-growing nature of this organism. Although more accurate methods to type microorganisms are available, this MIRU-VNTR protocol has been the most used worldwide to type MAP strains to date (Biet et al. 2012; Fernandez-Silva et al. 2011; Gioffre et al. 2015; Imperiale et al. 2017; Inagaki et al. 2009; Radomski et al. 2010; Stevenson et al. 2009; Thibault et al. 2007). On the other hand, the Whole Genome Sequencing (WGS) technology can generate whole genome sequences within a reasonable time frame and provide an extreme resolution of the diversity. However, although the costs associated with WGS have decreased over time, they are still unaffordable for large-scale studies in developing countries as Argentina.
Based on the above, the aim of this study was to analyze the genetic diversity of MAP isolates obtained from bovine and deer herds in Argentina by MLVA and to describe the phylogenetic relatedness between geographically distant isolates through WGS and core-genome analysis.
Materials and methods
MAP isolates
A total of 90 MAP isolates (Supplementary Table 1) obtained between 1990 and 2017 were selected from the collection of the Bacteriology Unit of the National Institute of Agricultural Technology (INTA)-Balcarce, Argentina. Archived samples corresponding to cattle (n = 85) and deer (n = 5) and isolated from different samples (milk, feces, organs, or tissues) were originally obtained by convenience sampling. Isolates were chosen to maximize geographical diversity within the dataset. The isolates chosen were both from the Pampas region (Buenos Aires, Córdoba, Santa Fe, and La Pampa provinces), considered one of the most productive areas of the country, and from the southernmost and northernmost provinces (Salta and Tierra del Fuego respectively), where according to the National Service of Animal Health (SENASA 2017), productivity is lower (Fig. 1).

Geographical origin of the isolates and number of herds sampled by province
DNA extraction and MAP confirmation
Samples were incubated in Herrold’s egg yolk medium supplemented with mycobactin and pyruvate at 37 °C for at least 2 months until growth. Once colonies were grown, a loop was taken and suspended in sterile distilled water. The cells were lysed by heat shock at 99 °C for 1 h (Mixing block, Bioer) and then centrifuged at 10,000 g for 5 min. Next, 2 µL of the supernatant was used as template for the PCR reactions. The identity of the isolates was confirmed by IS900-PCR in 1% agarose gel electrophoresis (Collins et al. 1993). High-quality genomic DNA was obtained using mini spin columns (Qiagen DNeasy® Blood & Tissue kit), following the kit instructions. DNA quality was tested using the Take3 plate in an Epoch Microplate Spectrophotometer (BioTek).
Genotyping by MLVA
MLVA genotyping was used to test eight different MIRU-VNTR loci, as previously described (Thibault et al. 2007). The loci investigated were VNTR292, MIRUX3, VNTR25, VNTR47, VNTR3, VNTR7, VNTR10, and VNTR32. The primers and PCR conditions were as previously suggested (Thibault et al. 2007), with minor modifications. The mixture consisted of 1X buffer (10 mM Tris–HCl pH 9, 50 mM KCl, 0.1% Triton X-100, 2.5 mM MgCl2), 1 µM of each primer, 0.2 mM of each dNTP and 1.25U GoTaq polymerase (Promega). The mixture for MIRUX3 was supplemented with 2 µL of MgCl2 per reaction, and mixtures for VNTR 47, 3, 7, 10 and 32 were supplemented with dimethyl sulfoxide and betaine (Sigma). The annealing temperatures were as previously described (Thibault et al. 2007), with the exception of VNTR 47, in which, according to a touch-down protocol, the annealing temperature was decreased by 1 °C during the first ten cycles from 69 °C to 59 °C and then set at 64 °C for 35 cycles, and VNTR 292, in which the annealing temperature was decreased by 2 °C and set at 56 °C (Gioffre et al. 2015). The PCR products were revealed with 3.5% agarose gel electrophoresis using a 100-bp DNA marker (INBIO Highway) and a 50-bp DNA marker (Promega) depending on the size of the expected product. A database from the National Institute of Agronomic Research in France (INRA) was consulted to search the INMV pattern/type derived from the numerical profile of each isolate (http://mac-inmv.tours.inra.fr/). DNA from MAP strain ATCC 19,698 (INMV 2) was included as a control.
Discriminatory power
The allelic diversity (D) of each locus and the global discriminatory power of the complete MLVA scheme were determined using the Hunter and Gaston discriminatory index (Hunter 1990; Hunter and Gaston 1988):
where
- N:
-
is the number of unrelated strains tested,
- S:
-
is the total number of different types, and
- xj:
-
is the number of isolates belonging to the jth type. The index was calculated using the online software: http://insilico.ehu.es/mini_tools/discriminatory_power/, University of the Basque Country, Spain.
The relationship between the profiles was determined using the goeBURST algorithm (goeburst.phyloviz.net/) (Francisco et al. 2009). For this, clonal complexes were defined as MLVA linked through single-locus variants. The MLVA genotype associated with most single-locus variants is considered the founder pattern.
WGS and phylogenetic analysis
The following four of the 85 cattle isolates were selected: Map 907-k32 (INMV 1), B35-S34 (INMV 1) and I47-S28 (INMV 2), all three from Buenos Aires, which represents the more productive region of the country, and Map L80 (INMV 1) from Tierra del Fuego. High-quality DNA was obtained as described above. Paired-end Nextera XT libraries were constructed and sequenced in a MiSeq sequencer (2 × 250 bp, Illumina). A quality trimming step was applied to raw reads by using Trimmomatic (Bolger et al. 2014). De novo assembly was performed using SPAdes v3.11.1 (Bankevich et al. 2012). Contigs were oriented using Mauve (Darling et al. 2004; Rissman et al. 2009) and the genome of M. avium subsp. paratuberculosis-K10 (GenBank accession number: SAMN02604086) was used as reference.
Fifty-four MAP whole-genome sequences were downloaded from GenBank (Supplementary Table 2) to provide a global phylogenetic analysis. Roary (http://sanger-pathongens.github.io/Roary) (Page et al. 2015) was used to build a pangenome including the four Argentinian strains, with a threshold of sequence identity ≥ 90%. Core-genome multiple sequence alignment was performed using PRANK and a maximum likelihood phylogenetic tree was generated using RAxML v8.2.11 (Stamatakis 2014). Node support was evaluated with 1,000 bootstraps. The phylogenetic tree, geographical location source, and MLVA type were visualized using iTOL v5 (Letunic and Bork 2016).
MLVA of foreign isolates was performed in silico. MAP whole-genome sequences from the NCBI database were analyzed with the following three different tools to obtain a more accurate result: Unipro UGENE (Okonechnikov et al. 2012), Primer Map (http://www.bioinformatics.org/sms2/primer_map) and in silico PCR amplification tool (http://insilico.ehu.es/mini_tools/PCR/). The hybridization of the eight MIRU-VNTR primer pairs and the putative product size of each locus were evaluated.
Results
MLVA genotyping
IS900-PCR confirmed all the selected isolates as MAP. MLVA of our study sample yielded seven MAP genotypes. The dominant subtype, INMV 1 (n = 68), was present in all the herds sampled. Other genotypes present included INMV 2 (n = 6), INMV 33 (n = 5), INMV 3 (n = 4), INMV 16 (n = 2) and INMV 13 (n = 1) (Table 1). For three isolates from Pergamino and one from Chivilcoy (both localities from Buenos Aires province), the analysis of locus 292 showed two bands corresponding to alleles 3 and 4. The overall loci analysis suggested that these animals could be infected with two different strains, supported by the presence of both INMV 1 and INMV 2 genotypes in the herd, as observed in Table 2.
One out of four of the MAP isolates from Pehuajó (Buenos Aires province) showed the pattern INMV 13, which occurs exclusively in that locality. In the same way, MAP isolates from Las Colonias (Santa Fe province) represented only INMV 16 type (Table 1 and Fig. 2).

GoeBURST clustering of INMV patterns. Different colors represent different locations
Finally, seven of the twenty-five herds analyzed presented more than one pattern (28%). All these herds showed different patterns in the same year, indicating the coexistence of strains with different genotypes.
Allelic diversity and discriminatory power
The discriminatory power (D) was calculated with 37 non-epidemiologically related isolates and reached 0.536. With regards to the discriminatory power of each locus, loci X3, 3, and 32 showed no allelic diversity, whereas locus 292 showed the highest D value with 3 different alleles. These results, shown in Table 2, are in concordance with other studies (Gioffre et al. 2015; Imperiale et al. 2017).
A cluster analysis was performed to study the relationship among MLVA genotypes. The GoeBURST analysis determined that INVM 2 is the primary founder and all other five genotypes are derived from this genotype.
WGS and phylogenetic analysis
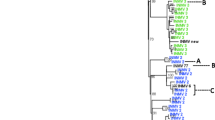
The core-genome phylogenetic tree clearly showed the presence of two lineages: one clustering the classical cattle type (C-type) strains and the other clustering the sheep type (S-type) strains (Fig. 3). All the strains sequenced in this study belong to the C-type lineage, where two branches could be differentiated. One of them grouped 8 out of 45 C-type strains from India, South Korea, Egypt, and the USA, whereas the other grouped most of the C-type strains (37/45). Strains of ovine origin (n = 6) were clustered under the same group (S-type) along with two strains from camelid hosts. This could be explained by horizontal transmission from sheep to other ruminants. The four Argentinian strains from our study were clustered into the broad branch of C-type strains. However, the INMV 1 strains were grouped together and separated from the other Argentinian INMV 2 strain. The INMV 2 strain studied was related to strains from the USA, Germany and Portugal. The in silico MLVA could be achieved for 19 out of 54 strains. Despite the incomplete data, overall results suggest that MLVA does not appear to be in accordance with the clustering obtained with the phylogenetic tree.

The statistics of the four Argentinian strains are shown in Supplementary Table 3.
Discussion
This study describes the analysis by MLVA and WGS of 90 MAP strains from Argentina, introducing MAP isolates from deer for the first time. The MLVA revealed the presence of nine different genotypes in Argentina, with a higher prevalence of INMV 1 over others (Gioffre et al. 2015; Imperiale et al. 2017 and this study). The prevalence of INMV 1 in the region was also reported on a systematic review from Latin America and the Caribbean (Correa-Valencia et al. 2021), together with INMV 2 and INMV 11. In this study, INMV 3 was described in the country for the first time, while INMV 5, INMV 8 and INMV 11, previously reported by Imperiale et al. (2017) and Gioffre et al. (2015), were not present in any of the 90 isolates. Differences could be due to the fact that these four genotypes were found in a low percentage and in few herds, making it more difficult to isolate and possibly not as widespread as others. Genotype INMV 1 seems to be distributed all over the country, and has even reached the southernmost province, Tierra del Fuego. This was unexpected and of particular interest because Tierra del Fuego has been an isolated region considered free of tuberculosis since 2011 (SENASA resolution 100/2011) and because this is the first case of PTB reported in that region. Core-genome phylogenetic analysis demonstrated a close phylogenetic relationship between this southern isolate and others. This strain from Tierra del Fuego was isolated from a dairy cow with clinical PTB, also confirmed by strong positive results by ELISA serology. After diagnosis, this animal was culled and samples were taken to the laboratory where MAP was isolated and genotyped. This is likely an instance of recent introduction of a carrier animal to the province. However, the management practices in the area do not support this hypothesis. Thus, an intermediate host such as wildlife is possible (Corti et al. 2021), although underreporting of the disease in this region cannot be ruled out.
The main criticism around MLVA typing is the limited resolution between isolates and that the polymorphisms detected do not necessarily reflect the phylogenetic relationships between strains (Ahlstrom et al. 2015; Bryant et al. 2016). Despite this, the MLVA approach allowed us to describe some features of the productive system of Argentina. Seven of the twenty-five herds analyzed presented more than one strain, which is evidence of the genetic diversity of strains within herds. These herds showed different patterns in the same year, confirming that the simultaneous presence of multiple MAP genotypes is frequent, as reported previously (Gioffre et al. 2015; Perets et al. 2022). Moreover, a co-infection with two strains within the same animal was also observed in four isolates from two different herds. The coexistence of different strains in the herds strongly suggests the absence of animal monitoring prior to the introduction (Ahlstrom et al. 2016). This represents a major risk factor for infection in herds and could be easily explained by the absence of a control program over the time in the country. The deer herds studied shared a common feeding area with beef cows, a farming practice that could have led to interspecies infection. Previous reports of similar MAP genotypes from deer and cattle in co-grazing conditions provides evidence for interspecies transmission (Fritsch et al. 2012). The results obtained in the present study support the idea that there is no relation between the host and genotype and that MAP can infect a wide variety of species, making its eradication from a herd even more difficult (Shaughnessy et al. 2013).
A frequent concern about MLVA genotyping is the stability of the markers and whether this technique can be trusted for epidemiological studies. A previous study showed different genotypes from the same vaccine strain coming from different laboratories or batches, not only with MLVA, but also with IS900-RFLP (Thibault et al. 2007). Further studies tested genetic stability under controlled conditions, both in vitro and in vivo, and proved that MIRU-VNTR alleles remain stable after several passages (Kasnitz et al. 2013), not only for MAP isolates, but also other mycobacteria like Mycobacterium tuberculosis (Savine et al. 2002). In this regard, the use of these MIRU-VNTR loci is plausible, at least for short-term analysis. A bibliographical search suggests different dynamics of the strains circulating between Argentina and Europe. In Argentina, the frequency of patterns is clearly biased to INMV 1 (Barandiaran et al. 2015; Imperiale et al. 2017), whereas in Europe the INMV 2 is prevalent (Biet et al. 2012; Stevenson et al. 2009; Thibault et al. 2007). This observation could be explained by the different genetic structures of the dominant MAP genotypes, the breed of the host, or the combination of both factors.
An alternative to prevent expansion of the disease is the adoption of test-and-cull-based control strategies. However, it must be considered that the presence of wild animals infected with MAP in the environment could hinder the success of control programs, since animals eliminate the bacteria in feces, representing a persistent and widespread source of infection (Fox et al. 2018). This emphasizes the importance of considering wild animals as reservoirs of the infection in the different environments of Argentina, as diverse as the Pampas and Patagonia regions. Molecular typing tools could help to support these programs and thus contribute to maintaining the herd health status and strengthening the regional economies of developing countries as Argentina. This study represents the first report of whole-genome sequences of MAP in Argentina.
Data availability
The dataset used to run the phylogenetic analysis is available at the NCBI website and is also included in this article as supplementary table 3. Accession numbers for Map L80, Map 907-K32, B35_S34 and I47_S28 strains are SAMN12798883, SAMN12798393, SAMN28119949 and SAMN28119950, respectively.
References
Ahlstrom C, Barkema HW, Stevenson K, Zadoks RN, Biek R, Kao R, Trewby H, Haupstein D, Kelton DF, Fecteau G, Labrecque O, Keefe GP, McKenna SL, De Buck J (2015) Limitations of variable number of tandem repeat typing identified through whole genome sequencing of Mycobacterium avium subsp. paratuberculosis on a national and herd level. BMC Genomics 16:161. https://doi.org/10.1186/s12864-015-1387-6
Ahlstrom C, Barkema HW, Stevenson K, Zadoks RN, Biek R, Kao R, Trewby H, Haupstein D, Kelton DF, Fecteau G, Labrecque O, Keefe GP, McKenna SL, Tahlan K, De Buck J (2016) Genome-wide diversity and phylogeography of mycobacterium avium subsp. paratuberculosis in Canadian Dairy Cattle. PLoS One 11(2):e0149017. https://doi.org/10.1371/journal.pone.0149017
Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, Lesin VM, Nikolenko SI, Pham S, Prjibelski AD, Pyshkin AV, Sirotkin AV, Vyahhi N, Tesler G, Alekseyev MA, Pevzner PA (2012) SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol 19(5):455–77. https://doi.org/10.1089/cmb.2012.0021
Barandiaran S, Perez AM, Gioffre AK, Martinez Vivot M, Cataldi AA, Zumarraga MJ (2015) Tuberculosis in swine co-infected with Mycobacterium avium subsp. hominissuis and Mycobacterium bovis in a cluster from Argentina. Epidemiol Infect 143(5):966–74. https://doi.org/10.1017/S095026881400332X
Beard PM, Daniels MJ, Henderson D, Pirie A, Rudge K, Buxton D, Rhind S, Greig A, Hutchings MR, McKendrick I, Stevenson K, Sharp JM (2001) Paratuberculosis infection of nonruminant wildlife in Scotland. J Clin Microbiol 39(4):1517–1521. https://doi.org/10.1128/JCM.39.4.1517-1521.2001
Biet F, Sevilla IA, Cochard T, Lefrancois LH, Garrido JM, Heron I, Juste RA, McLuckie J, Thibault VC, Supply P, Collins DM, Behr MA, Stevenson K (2012) Inter- and intra-subtype genotypic differences that differentiate Mycobacterium avium subspecies paratuberculosis strains. BMC Microbiol 12:264. https://doi.org/10.1186/1471-2180-12-264
Bolger AM, Lohse M, Usadel B (2014) Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30(15):2114–2120. https://doi.org/10.1093/bioinformatics/btu170
Bryant JM, Thibault VC, Smith DG, McLuckie J, Heron I, Sevilla IA, Biet F, Harris SR, Maskell DJ, Bentley SD, Parkhill J, Stevenson K (2016) Phylogenomic exploration of the relationships between strains of Mycobacterium avium subspecies paratuberculosis. BMC Genomics 17:79. https://doi.org/10.1186/s12864-015-2234-5
Collins DM, Hilbink F, West DM, Hosie BD, Cooke MM, de Lisle GW (1993) Investigation of Mycobacterium paratuberculosis in sheep by faecal culture, DNA characterisation and the polymerase chain reaction. Vet Rec 133(24):599–600
Correa-Valencia NM, Moyano RD, Hernandez-Agudelo M, Fernandez-Silva JA (2021) Mycobacterium avium subsp. paratuberculosis (MAP) molecular diversity in cattle, sheep, and goats from Latin America and the Caribbean: a systematic review. Trop Anim Health Prod 53(5):468. https://doi.org/10.1007/s11250-021-02923-9
Corti P, Collado B, Salgado M, Moraga CA, Radic-Schilling S, Tejeda C, Ruiz-Aravena M (2021) Dynamic of Mycobacterium avium subspecies paratuberculosis infection in a domestic-wildlife interface: domestic sheep and guanaco as reservoir community. Transbound Emerg Dis. https://doi.org/10.1111/tbed.14277
Darling AC, Mau B, Blattner FR, Perna NT (2004) Mauve: multiple alignment of conserved genomic sequence with rearrangements. Genome Res 14(7):1394–1403. https://doi.org/10.1101/gr.2289704
Espeschit IF, Schwarz DGG, Faria ACS, Souza MCC, Paolicchi FA, Juste RA, Carvalho IA, Moreira MAS (2017) Paratuberculosis in Latin America: a systematic review. Trop Anim Health Prod 49(8):1557–1576. https://doi.org/10.1007/s11250-017-1385-6
Fernandez-Silva JA, Abdulmawjood A, Bulte M (2011) Diagnosis and molecular characterization of mycobacterium avium subsp. paratuberculosis from dairy cows in Colombia. Vet Med Int 2011:352561. https://doi.org/10.4061/2011/352561
Fiorentino MA, Gioffre A, Cirone K, Morsella C, Alonso B, Delgado F, Paolicchi F (2012) First isolation of Mycobacterium avium subsp. paratuberculosis in a dairy goat in Argentina: pathology and molecular characterization. Small Rumiant Res 108(1–3):133–136. https://doi.org/10.1016/j.smallrumres.2012.06.010
Fox NJ, Caldow GL, Liebeschuetz H, Stevenson K, Hutchings MR (2018) Counterintuitive increase in observed Mycobacterium avium subspecies paratuberculosis prevalence in sympatric rabbits following the introduction of paratuberculosis control measures in cattle. Vet Rec 182(22):634. https://doi.org/10.1136/vr.104638
Francisco AP, Bugalho M, Ramirez M, Carrico JA (2009) Global optimal eBURST analysis of multilocus typing data using a graphic matroid approach. BMC Bioinformatics 10:152. https://doi.org/10.1186/1471-2105-10-152
Fritsch I, Luyven G, Kohler H, Lutz W, Mobius P (2012) Suspicion of Mycobacterium avium subsp. paratuberculosis transmission between cattle and wild-living red deer (Cervus elaphus) by multitarget genotyping. Appl Environ Microbiol 78(4):1132–1139. https://doi.org/10.1128/AEM.06812-11
Gioffre A, Correa Munoz M, Alvarado Pinedo MF, Vaca R, Morsella C, Fiorentino MA, Paolicchi F, Ruybal P, Zumarraga M, Traveria GE, Romano MI (2015) Molecular typing of Argentinian Mycobacterium avium subsp. paratuberculosis isolates by multiple-locus variable number-tandem repeat analysis. Braz J Microbiol 46(2):557–64. https://doi.org/10.1590/S1517-838246220140283
Gopi B, Vir Singh R, Kumar S, Kumar S, Chauhan A, Sonwane A, Kumar A, Bharati J, Vir Singh S (2022) Effect of selected single nucleotide polymorphisms in SLC11A1, ANKRA2, IFNG and PGLYRP1 genes on host susceptibility to Mycobacterium avium subspecies paratuberculosis infection in Indian cattle. Vet Res Commun 46(1):209–221. https://doi.org/10.1007/s11259-021-09849-5
Gopi B, Singh RV, Kumar S, Kumar S, Chauhan A, Kumar A, Singh SV (2020) Single-nucleotide polymorphisms in CLEC7A, CD209 and TLR4 gene and their association with susceptibility to paratuberculosis in Indian cattle. J Genet 99. https://doi.org/10.1007/s12041-019-1172-4
Hunter PR (1990) Reproducibility and indices of discriminatory power of microbial typing methods. J Clin Microbiol 28(9):1903–1905. https://doi.org/10.1128/jcm.28.9.1903-1905.1990
Hunter PR, Gaston MA (1988) Numerical index of the discriminatory ability of typing systems: an application of Simpson’s index of diversity. J Clin Microbiol 26(11):2465–2466. https://doi.org/10.1128/jcm.26.11.2465-2466.1988
Imperiale BR, Moyano RD, Giulio ABDI, Romero MA, AlvaradoPinedo MF, Santangelo MP, Traveria GE, Morcillo NS, Romano MI (2017) Genetic diversity of Mycobacterium avium complex strains isolated in Argentina by MIRU-VNTR. Epidemiol Infect 145(7):1382–1391. https://doi.org/10.1017/S0950268817000139
Inagaki T, Nishimori K, Yagi T, Ichikawa K, Moriyama M, Nakagawa T, Shibayama T, Uchiya K, Nikai T, Ogawa K (2009) Comparison of a variable-number tandem-repeat (VNTR) method for typing Mycobacterium avium with mycobacterial interspersed repetitive-unit-VNTR and IS1245 restriction fragment length polymorphism typing. J Clin Microbiol 47(7):2156–2164. https://doi.org/10.1128/JCM.02373-08
Kasnitz N, Kohler H, Weigoldt M, Gerlach GF, Mobius P (2013) Stability of genotyping target sequences of Mycobacterium avium subsp. paratuberculosis upon cultivation on different media, in vitro- and in vivo passage, and natural infection. Vet Microbiol 167(3–4):573–83. https://doi.org/10.1016/j.vetmic.2013.09.008
Kukanich KS, Vinasco J, Scott HM (2013) Detection of mycobacterium avium subspecies paratuberculosis from intestinal and nodal tissue of dogs and cats. ISRN Vet Sci 2013:323671. https://doi.org/10.1155/2013/323671
Kumar S, Kumar S, Singh RV, Chauhan A, Kumar A, Bharati J, Singh SV (2020) Association of genetic variability in CD209 gene with bovine paratuberculosis disease: a case-control study in the Indian cattle population. Anim Biotechnol: 1–8. https://doi.org/10.1080/10495398.2020.1823400
Letunic I, Bork P (2016) Interactive tree of life (iTOL) v3: an online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res 44(W1):W242–W245. https://doi.org/10.1093/nar/gkw290
Matos AC, Figueira L, Martins MH, Loureiro F, Pinto ML, Matos M, Coelho AC (2014) Survey of Mycobacterium avium subspecies paratuberculosis in road-killed wild carnivores in Portugal. J Zoo Wildl Med 45(4):775–781. https://doi.org/10.1638/2014-0010.1
Moreira AR, Tosi JC (1995) Paratuberculosis Bovina: Importancia de la enfermedad en la Region. Informe Técnico Nro 1, CERBAS INTA, Argentina, pp 14
Okonechnikov K, Golosova O, Fursov M, Ugene team (2012) Unipro UGENE: a unified bioinformatics toolkit. Bioinformatics 28(8):1166–1167. https://doi.org/10.1093/bioinformatics/bts091
Page AJ, Cummins CA, Hunt M, Wong VK, Reuter S, Holden MT, Fookes M, Falush D, Keane JA, Parkhill J (2015) Roary: rapid large-scale prokaryote pan genome analysis. Bioinformatics 31(22):3691–3693. https://doi.org/10.1093/bioinformatics/btv421
Palmer MV, Kanipe C, Cox R, Robbe-Austerman S, Thacker TC (2019) Characteristics of subclinical Mycobacterium avium ssp. paratuberculosis infection in a captive white-tailed deer herd. J Vet Diagn Invest 31(6):844–851. https://doi.org/10.1177/1040638719873028
Paolicchi F, Vagnozzi A, Morsella C, Verna A, Massone AR, Portiansky EL, Gimeno EJ (2001) Paratuberculosis in red deer (Cervus elaphus): an immunohistochemical study. Zoonoses Public Health 48(4):313–320. https://doi.org/10.1046/j.1439-0450.2001.00458.x
Paolicchii FA, Zumarraga MJ, Gioffre A, Zamorano P, Morsella C, Verna A, Cataldi A, Alito A, Romano M (2003) Application of different methods for the diagnosis of paratuberculosis in a dairy cattle herd in Argentina. J Vet Med B Infect Dis Vet Public Health 50(1):20–26. https://doi.org/10.1046/j.1439-0450.2003.00606.x
Perets V, Allen A, Crispell J, Cassidy S, O’Connor A, Farrell D, Browne JA, O’Mahony J, Skuce R, Kenny K, Gordon SV (2022) Evidence for local and international spread of Mycobacterium avium subspecies paratuberculosis through whole genome sequencing of isolates from the island of Ireland. Vet Microbiol 268:109416. https://doi.org/10.1016/j.vetmic.2022.109416
Radomski N, Thibault VC, Karoui C, de Cruz K, Cochard T, Gutierrez C, Supply P, Biet F, Boschiroli ML (2010) Determination of genotypic diversity of Mycobacterium avium subspecies from human and animal origins by mycobacterial interspersed repetitive-unit-variable-number tandem-repeat and IS1311 restriction fragment length polymorphism typing methods. J Clin Microbiol 48(4):1026–1034. https://doi.org/10.1128/JCM.01869-09
Rissman AI, Mau B, Biehl BS, Darling AE, Glasner JD, Perna NT (2009) Reordering contigs of draft genomes using the Mauve aligner. Bioinformatics 25(16):2071–2073. https://doi.org/10.1093/bioinformatics/btp356
Salgado M, Sevilla I, Rios C, Crossley J, Tejeda C, Manning E (2016) Presence of Mycobacterium Avium Subsp. Paratuberculosis in Alpacas (Lama Pacos) Inhabiting the Chilean Altiplano. J Zoo Wildl Med 47(1):12–6. https://doi.org/10.1638/2012-0255.1
Savine E, Warren RM, van der Spuy GD, Beyers N, van Helden PD, Locht C, Supply P (2002) Stability of variable-number tandem repeats of mycobacterial interspersed repetitive units from 12 loci in serial isolates of Mycobacterium tuberculosis. J Clin Microbiol 40(12):4561–4566. https://doi.org/10.1128/JCM.40.12.4561-4566.2002
Shaughnessy LJ, Smith LA, Evans J, Anderson D, Caldow G, Marion G, Low JC, Hutchings MR (2013) High prevalence of paratuberculosis in rabbits is associated with difficulties in controlling the disease in cattle. Vet J 198(1):267–270. https://doi.org/10.1016/j.tvjl.2013.08.030
Singh SV, Kuenstner JT, Davis WC, Agarwal P, Kumar N, Singh D, Gupta S, Chaubey KK, Kumar A, Misri J, Jayaraman S, Sohal JS, Dhama K (2016) Concurrent resolution of chronic diarrhea likely due to Crohn’s Disease and infection with Mycobacterium avium paratuberculosis. Front Med (lausanne) 3:49. https://doi.org/10.3389/fmed.2016.00049
Stamatakis A (2014) RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30(9):1312–3. https://doi.org/10.1093/bioinformatics/btu033
Stevenson K, Alvarez J, Bakker D, Biet F, de Juan L, Denham S, Dimareli Z, Dohmann K, Gerlach GF, Heron I, Kopecna M, May L, Pavlik I, Sharp JM, Thibault VC, Willemsen P, Zadoks RN, Greig A (2009) Occurrence of Mycobacterium avium subspecies paratuberculosis across host species and European countries with evidence for transmission between wildlife and domestic ruminants. BMC Microbiol 9:212. https://doi.org/10.1186/1471-2180-9-212
Thibault VC, Grayon M, Boschiroli ML, Hubbans C, Overduin P, Stevenson K, Gutierrez MC, Supply P, Biet F (2007) New variable-number tandem-repeat markers for typing Mycobacterium avium subsp. paratuberculosis and M. avium strains: comparison with IS900 and IS1245 restriction fragment length polymorphism typing. J Clin Microbiol 45(8):2404–10. https://doi.org/10.1128/JCM.00476-07
Timms VJ, Daskalopoulos G, Mitchell HM, Neilan BA (2016) The association of Mycobacterium avium subsp. paratuberculosis with inflammatory bowel disease. PLoS One 11(2):e0148731. https://doi.org/10.1371/journal.pone.0148731
Traveria GE, Zumarraga M, Etchechoury I, Romano MI, Cataldi A, Pinedo MF, Pavlik I, Pribylova R, Romero JR (2013) First identification of Mycobacterium avium paratuberculosis sheep strain in Argentina. Braz J Microbiol 44(3):897–899. https://doi.org/10.1590/s1517-83822013005000066
Vasini Rosell B, Lagleyze B, Morsella C, Mendez L, Bresky F, Gioffre A, Paolicchi F (2020) Evidence of in utero infection by Mycobacterium avium subsp. paratuberculosis using Multiple-Locus Variable-number tandem-repeat Analysis - First report in Argentina. Braz J Vet Res Anim Sci 57(1):e161653. https://doi.org/10.11606/issn.1678-4456.bjvras.2020.161653
Whittington RJ, Windsor PA (2009) In utero infection of cattle with Mycobacterium avium subsp. paratuberculosis: a critical review and meta-analysis. Vet J 179(1):60–9. https://doi.org/10.1016/j.tvjl.2007.08.023
Acknowledgements
The authors are grateful to Dr. Karina Caimi for technical advice.
Funding
This study was funded by ANPCyT (PICT grant, project PICT2015-1541), CONICET (PIP grant, project PIP11220150100316CO) and INTA (project 2019-PD-E5-I103-001).
Author information
Authors and Affiliations
Contributions
Molecular analysis, data processing and writing (Brenda Vasini); phylogenetic analysis and writing (Pablo Farace); WGS (Ariel Amadio, Pablo Fresia, Gregorio Iraola); molecular analysis (Karina Cirone); culture and isolation (Laura Mendez, Claudia Morsella); analysis of results and writing (Andrea Gioffre, Fernando Paolicchi).
Corresponding author
Ethics declarations
Ethical approval
Not applicable
Consent to participate
Not applicable
Consent to publish
Not applicable
Conflict of interests
The authors declare no potential conflicts of interest.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Vasini, B., Farace, P., Ariel, A. et al. Phylogenetic and Multiple-Locus Variable number tandem repeat analysis of Mycobacterium avium subsp. paratuberculosis isolates from Argentina. Vet Res Commun 46, 1121–1129 (2022). https://doi.org/10.1007/s11259-022-09983-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11259-022-09983-8