
Abstract
Background
As chronic kidney disease (CKD) progresses, metabolites undergo diverse transformations. Nevertheless, the impact of these metabolic changes on the etiology, progression, and prognosis of CKD remains uncertain. Our objective is to conduct a metabolomics analysis to scrutinize metabolites and identify significant metabolic pathways implicated in CKD progression, thereby pinpointing potential therapeutic targets for CKD management.
Methods
We recruited 145 patients with CKD and determined their mGFR by measuring the plasma iohexol clearance, whereupon we partitioned them into four groups based on their mGFR values. Non-targeted metabolomics analysis was conducted using UPLC-MS/MS assays. Differential metabolites were identified via one-way ANOVA, PCA, PLS-DA, and OPLS-DA analyses employing the MetaboAnalyst 5.0 platform. Ultimately, we performed differential metabolite pathway enrichment analysis, using both the MetaboAnalyst 5.0 platform and the MBRole2.0 database.
Results
According to the findings of the MBRole2.0 and MetaboAnalyst 5.0 enrichment analysis, six amino acid metabolism pathways were discovered to have significant roles in the progression of CKD, with the glycine, serine, and threonine metabolism pathway being the most prominent. The latter enriched 14 differential metabolites, of which six decreased while two increased concomitantly with renal function deterioration.
Conclusions
The metabolic analysis unveiled that glycine, serine, and threonine metabolism plays a pivotal role in the progression of CKD. Specifically, glycine was found to increase while serine decreased with the deterioration of CKD.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Chronic kidney disease (CKD) is a clinical disorder arising from diverse pathogenic factors, including but not limited to glomerulonephritis, lupus nephritis, chronic interstitial nephritis, and diabetic nephropathy. As a major global public health issue, CKD warrants significant attention [1]. For patients diagnosed with CKD, it is essential to have early and accurate detection, followed by prompt intervention. This is crucial to halt the progression of CKD, which if left unattended, could ultimately lead to end-stage renal failure [2].
In line with the research notions of genomics and proteomics, metabolomics involves the quantitative evaluation of low-molecular-weight substances (typically < 1500 Dalton) present in organisms [3]. The identified metabolites pertain to diverse chemical categories, including but not limited to lipids, nucleotides, and amino acids. It is noteworthy that their concentration results from an intricate interplay among genes, the microbiome, and environmental factors [3]. The aberrant characteristics and metabolic pathways identified through metabolomics can offer valuable insight into the underlying disease mechanisms and furnish information regarding pathophysiological alterations in patients with CKD [4]. Therefore, metabolomics is regarded as a valuable tool for nephrology research, particularly in the quest for innovative biomarkers for diagnosing and prognosticating CKD [5].
Besides serving as integral constituents of proteins and polypeptides, amino acids also function as cell signaling molecules and gene expression regulators [6]. A recent investigation has demonstrated that amino acids, alongside their related upstream and downstream metabolites, participate in the pathophysiological processes underlying kidney injury by regulating oxidative stress, inflammation, and immune response [7]. Patients with CKD typically exhibit abnormal serum amino acid metabolic profiles. A study has ascertained that serum leucine levels are markedly diminished in patients with CKD [8], and this observation has been replicated in animal models, including dogs suffering from CKD [9]. According to a case–control study, arginine, methionine, and threonine exhibit potential as metabolic markers of residual renal function and biomarkers of prognosis among patients with nephropathy [10]. 5-methoxy tryptophan (5-MTP) is a potential metabolite for early CKD screening, as its serum level exhibits a robust correlation with the estimated glomerular filtration rate (eGFR) and decreases in tandem with CKD progression. Notably, tryptophan hydroxylase-1 (TPH-1) is an enzyme pivotal in 5-MTP biosynthesis and can attenuate kidney injury by mitigating renal inflammation and interstitial fibrosis. Hence, both 5-MTP and TPH-1 may emerge as viable targets for CKD treatment [11].
Several investigations have identified biomarkers linked with CKD progression, yet limited attention has been paid to plasma amino acids and their corresponding upstream and downstream metabolites regarding their involvement in CKD progression. To address this gap, our study centered on identifying amino acid metabolites and metabolic pathways associated with renal function deterioration.
Methods
Metabolomics samples and mGFR measurement
The study comprised 145 patients fulfilling the inclusion criteria exhibiting diverse levels of chronic renal insufficiency. The diagnosis of CKD was based on the NKFKDOI guidelines. Prior to enrollment, all patients provided informed consent after the Kiang Wu Hospital ethics committee's approval.
We collected 4 mL of peripheral venous blood from each patient to obtain plasma samples for metabolomics analysis. To estimate each patient's glomerular filtration rate (GFR), we employed plasma clearance of iohexol [12]. To conduct this study, we administered 5 mL of iohexol (300 mg/mL, GE Healthcare, Shanghai, China) via intravenous injection into one of the patient's upper limb veins. This injection lasted no more than two minutes, and patients were not in a fasting state at this time. We then collected plasma (6 mL) from the opposite upper limb vein and measured blood concentrations of iohexol at 120 and 240 min after administration using high-performance liquid chromatography (HPLC). When patients exhibited eGFR levels lower than 30 mL/min/1.73 m2 [13], we postponed the second blood collection time to 300 min. All blood samples underwent centrifugation at room temperature for 10 min at 2000g to extract plasma and were stored at − 80 °C until analysis.
Ultra performance liquid chromatograph tandem mass spectrometry (UPLC-MS/MS) assays
At the Dian Calibra-Metabolon Joint Metabolomics Laboratory (Hangzhou, China), we employed four distinct UPLC-MS/MS methods to test each sample's small molecule metabolites [14]. Sample preparation automated liquid transfer was executed using Hamilton's MicroLab STAR® system (Hamilton, Switzerland). We combined a methanol-based metabolite extraction solution with each sample, followed by shaking using a GenoGrinder 2010 (Spex SamplePrep, USA) and centrifugation for two minutes to precipitate proteins and other debris. The metabolite-containing supernatant was evenly split into four portions corresponding to four UPLC-MS/MS analyses. Two components underwent reversed-phase (RP) UPLC-MS/MS analyses in positive ion electrospray ionization (ESI) mode utilizing the same column (BEH C18 2.1 × 100 mm, 1.7 μm column, Waters) and mobile solutions (water and methanol supplemented with 0.05% perfluoropentanoic acid (PFPA) and 0.1% formic acid (FA)). The third component was analyzed via RP UPLC-MS/MS in negative ion ESI mode, while the last component was evaluated through hydrophilic interaction chromatography (HILIC) UPLC-MS/MS in negative ion ESI mode. After drying each fraction under nitrogen, we dissolved it in an appropriate solution for each UPLC-MS/MS method and then injected it into four UPLC-MS/MS systems. We processed the raw mass spectrometry data using our in-house developed software, extracted and identified peaks and matched the experimental ion characteristics with entries in an internal library constructed using standard compounds to identify metabolites. Metabolite identification matching criteria encompassed retention time index (RI), the molecular ion mass-to-charge ratio (m/z), and MS/MS spectral data. To heighten experiment reliability and minimize error, we applied a stringent matching window to RI and m/z criteria, considering both forward and reverse MS/MS matching scores between standard compound entries and experimental data.
Pretreatment of metabolomics data
The study participants were divided into four groups based on their measured glomerular filtration rate (mGFR) values: group A (mGFR < 30 mL/min/1.73 m2), group B (30 mL/min/1.73 m2 ≤ mGFR < 60 mL/min/1.73 m2), group C (60 mL/min/1.73 m2 ≤ mGFR < 90 mL/min/1.73 m2), and group D (mGFR ≥ 90 mL/min/1.73 m2). We conducted statistical analyses of patients' clinical characteristics and variables using SPSS 26.0 software, with a p-value < 0.05 deemed statistically significant. MetaboAnalyst 5.0 (https://www.metaboanalyst.ca/) is an integrated platform for metabolomics data analysis, capable of conducting comprehensive normalization, statistical analysis, function enrichment analysis, meta-analysis, and other multi-omics analyses for both targeted and non-targeted metabolomic data [15]. We uploaded the comma-separated values (.csv) mass spectrometry data file onto the MetaboAnalyst 5.0 website, comprising a 145 (samples) × 1094 (compounds) data matrix. Before conducting data analysis, we performed data integrity checks, including class label verification, the existence of non-numeric values, missing values, or features with constant values. To mitigate biased results stemming from truncated data omissions, we replaced missing values with 1/5 of the minimum positive value of its corresponding variable.
Data filtering was then implemented to identify and eliminate variables unlikely to be utilized when modeling the data. Specifically, we filtered out variables whose relative standard deviation (RSD) exceeded 25% across the entire sample, removing 40% of the total. Finally, we performed median normalization, logarithmic transformation based on 10, and Pareto scaling to achieve general systematic differences adjustment among samples.
Univariate and multivariate statistical analysis
We conducted a one-way analysis of variance (ANOVA) to preliminarily screen for significant features, setting the ANOVA p-value cutoff at 0.05. Following this, we carried out paired principal component analysis (PCA) among Group A, B, C, and D, and selected the primary comparison group based on the PCA results. We then performed partial least squares-discriminant analysis (PLS-DA), and orthogonal partial least squares-discriminant analysis (OPLS-DA) on this main comparison group.
PCA is an unsupervised dimension reduction method enabling the study of training samples without labeled sample data. It linearly recombines all initially identified metabolites to construct a new set of comprehensive variables, selecting 2–3 comprehensive variables according to analyzed characteristics to reflect original variable information as much as possible, achieving dimension reduction.
PLS-DA is a supervised PCA method that considers grouping information to realize data dimension reduction. It uses both the X matrix and Y matrix by searching for multidimensional directions in the X space that can explain maximum variance in the Y space, better screening differential metabolites among different groups. OPLS-DA is a multivariate statistical analysis method employing supervised pattern recognition. Combining PLS-DA analysis with orthogonal signal correction (OSC) technology eliminates irrelevant influences and effectively screens differential metabolites.
Variable Importance in Projection (VIP) measures the variable weight value of (O) PLS-DA model variables, assessing each metabolite's expression pattern influence intensity and explanatory ability on sample classification and discrimination in each group. VIP describes each differential metabolite's overall contribution to the model, with a VIP value > 1 deemed statistically significant. We set the threshold for VIP value as > 1.
Metabolic pathway analysis
MBRole2.0 (http://csbg.cnb.csic.es/mbrole2/) is an open database for functional enrichment analyses of significant compounds in metabolomics research [16]. We converted differential metabolite names screened by ANOVA analysis into "KEGG compound IDs" in MBROLE 2.0, setting annotations and background as "KEGG pathways" and "Homo sapiens (human)," respectively. Both MetaboAnalyst 5.0 platform and the MBRole2.0 database were utilized to conduct metabolite pathway enrichment analyses.
Results
Clinical characteristics of the study populations
A total of 145 patients aged between 20 and 96 years with CKD were recruited, 86 of whom were women. Based on their mGFR values, 22 patients were assigned to Group A, 47 to Group B, 39 to Group C, and 37 to Group D. We conducted summary statistics on some clinical variables of the patients (Table 1), and the original clinical data of the patients were presented in the form of supplementary documents (Supplementary Table 1). For mGFR, age and body mass index (BMI) of the patients, we calculated the corresponding arithmetic means, standard deviations, and p-values of one-way ANOVA tests. For the dichotomous variables of the use of antiplatelet agents, antilipemic agents, antihypertensive medications, hypoglycemic drugs, and uric acid reduction medicines we showed their proportions and numbers in each group in Table 1, alongside their P-values obtained from chi-square tests.
Univariate analysis
A comprehensive total of 1094 metabolites were successfully identified utilizing non-targeted metabolomics analysis (Fig. 1). Lipids accounted for the highest proportion (40%) among the diverse categories, followed by amino acids (21%) and xenobiotics (21%). From this pool, we screened 673 distinct metabolites via one-way ANOVA analysis and the results of ANOVA analysis were presented in Supplementary Table 2.

The overview of annotated metabolites
PCA, PLS-DA and OPLS-DA
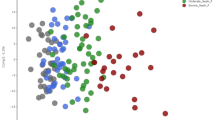
The results of PCA are depicted in Fig. 2A–F. The group A and group D were separated in the principal component 1. Meanwhile, compared with other subgroups, the metabolic spectrum difference between group A and group D was the most significant, signifying that patients' metabolic profiles underwent alterations with the progression of CKD, with the most significant changes noted among those with end-stage renal disease (ESRD).

Scores plot between the selected PCs. The explained variances are shown in brackets. A–F 2D score plots derived from the pairwise PCA analysis of Group A, B, C and D; G 2D score plots derived from the PLS-DA analysis between Group A and D; H 2D score plots derived from the OPLS-DA analysis between Group A and D
Based on the results of PCA, we identified group A and group D as the main comparison group. To further enhance the performance of the PCA model, we carried out PLS-DA (Fig. 2G), which conducts supervised analysis based on sample grouping to illuminate differences in metabolic profiles between groups and pinpoint more valuable differential metabolites. Among the metabolites that increased with mGFR decline, 2-methylcitrate and N-acetyl-1-methylhistidine had the highest VIP scores (Fig. 3A). Conversely, among the metabolites that decreased with mGFR decline, S-allylcysteine and caffeine had the highest VIP scores. We evaluated the quality of our model using the goodness of fit parameter (R2) and predictive power parameter (Q2) values. R2 indicates the interpretation rate of the built model to the X and Y matrix, while Q2 reflects the proportion of variance in the data predicted by the model, which translates into the model's prediction ability. In our study, all comparison groups exhibited Q2 values exceeding 0.8 (Fig. 4A), indicative of the model's satisfactory predictability.

Significant differential metabolites were identified by the ranking of the variable importance in the projection (VIP) scores. A Important features identified by PLS-DA. The colored boxes on the right indicate the relative concentrations of the corresponding metabolite in each group under study; B important features identified by OPLS-DA. The colored boxes on the right indicate the relative concentrations of the corresponding metabolite in each group under study

The validation of the PLS-DA and OPLS-DA model. A The bar plot showing the three performance measures (prediction accuracy, multiple correlation coefficient R2, and explained variance in prediction Q2) obtained after the tenfold cross-validation analysis of multivariate data. The red star indicates the best classifier; B the predictive power evaluation of the OPLS-DA model by 2000 permutation tests
To further highlight the differences between the two groups, we employed OPLS-DA for analysis, and generated score plots (Fig. 2H). OPLS-DA integrates an orthogonal signal correction (OSC) filter with PLS-DA to decompose X matrix information into data related and unrelated to Y and subsequently filter out difference information not associated with the classification. Finally, we used 2000 permutation tests to evaluate model predictability and statistical significance. The Q2 value obtained via cross-validation of the OPLS-DA model exceeded 0.9 (Fig. 4B), indicating its suitability. The OPLS-DA score plot displayed a marked separation between the two comparison groups. Erythronate, N-acetylneuraminate, C-glycosyl-tryptophan, hydroxy-asparagine and 3-(3-amino-3-carboxypropyl)-uridine exhibited the highest VIP scores, all of which increased with the deterioration of CKD (Fig. 3B).
Pathway analysis
To identify the metabolic pathways underlying CKD progression, 673 compounds screened via ANOVA underwent ID conversion in MBRole2.0, with annotations set to "KEGG pathways" and background as "Homo sapiens (human)." To mitigate errors and omissions stemming from enrichment in any single database, we imported metabolites after ID conversion into the MetaboAnalyst 5.0 platform for enrichment analysis. Table 2 delineates the results of enrichment analysis on the MetaboAnalyst 5.0 platform, revealing twelve significantly enriched (raw p-value < 0.05) metabolic pathways, six of which pertain to amino acid metabolism, including glycine, serine, and threonine metabolism, phenylalanine, tyrosine, and tryptophan biosynthesis, arginine, and proline metabolism, phenylalanine metabolism, arginine biosynthesis, and tyrosine metabolism. The bubble map of metabolic pathway enrichment highlights that the most substantial bubbles corresponding to glycine, serine, and threonine metabolism and caffeine metabolism are situated at the top right of the diagram. This suggests that these two pathways are the most crucial in CKD progression (Fig. 5A). Meanwhile, MBRole2.0's enrichment outcomes are presented in Fig. 5B. Among the ten most important pathways, five belong to amino acid metabolism. In contrast to the other three pathways, arginine and proline metabolism, and glycine, serine, and threonine metabolism are considered the most two critical amino acid metabolic pathways. Fourteen metabolites are enriched in the glycine, serine, and threonine metabolism pathway (Fig. 5C), where the levels of dimethylglycine and glycine increase with renal function deterioration, while the levels of 3-phosphoglycerate, glycerate, pyruvate, sarcosine, serine, and tryptophan decline with worsening renal function.

The results of significant metabolites pathway enrichment. A Bubble plot for pathway analysis depicted in MetaboAnalyst 5.0 platform. The vertical axis represents the log (p) value, while the horizontal axis represents the pathway impact. The pathway impact is calculated by adding up the important measures of each of the matched metabolites and then dividing by the sum of the important measures of all metabolites in each pathway. The larger the pathway impact, the more meaningful the metabolic pathway is. The pathway in the upper right corner of the bubble chart is the most reliable; B The results of metabolite set enrichment in MBRole2.0. Set: total number of selected metabolites; In set: the number of differential metabolites contained in this pathway; C KEGG’s metabolic pathway for the glycine, serine, and threonine metabolism. The red circle represents the enriched metabolites; the green boxes are metabolites that rise as mGFR decreases; the red boxes are metabolites that decrease as mGFR decreases. KEGG Kyoto Encyclopedia of Genes and Genomes
Discussion
Numerous studies have demonstrated several plasma metabolites' involvements during CKD progression towards ESRD [17, 18]. Some delay CKD progression, but most accumulate as CKD progresses and exacerbate renal function deterioration examples include indoxyl sulphate (IS), p-cresyl sulphate (PCS), among others [19, 20]. Some significant uremic toxins are created and incorporated into the gut microbiota of CKD patients through amino acids in their diet, such as IS and indole-3-acetic acid (IAA) from tryptophan, while PCS is derived from tyrosine or phenylalanine [21]. The kidneys play a pivotal role in homeostasis, metabolism, and regulation of plasma amino acid concentrations. Plasma amino acid concentrations in CKD patients significantly differ from those of healthy subjects; for instance, CKD patients exhibit lower essential amino acid concentrations and higher non-essential amino acid levels than normal individuals [22]. Patients with CKD experience numerous changes in their amino acid metabolic profiles; however, the significance of these changes in CKD progression remains unclear. Thus, this study aims to screen plasma amino acids and related metabolites that affect CKD progression using metabolomic methods.
The results of PCA analysis showed that the metabolic profiles of patients in group A and group D were most significantly different. However, this result may be influenced by the patient's age and BMI value, especially age. One study showed a gradual decrease in serum L-histidine concentration with increasing age (participants aged 32–81 years) [23]. For children and adolescents from 3 months to 18 years of age, the mean concentrations of aspartate, glycine, and valine all increased significantly with age [24]. Meanwhile, this study also found a significant positive correlation between BMI and the levels of branched-chain and aromatic amino acids [24]. This suggests that factors such as age and BMI can influence the metabolic profile and should be considered when interpreting the results.
At the same time, the impact of medications on metabolic pathways in the human body should not be underestimated. For example, atorvastatin can alter amino acid metabolism by affecting self-regulatory mechanisms in hyperlipidemia rats [25]. In animals treated with high doses of fenofibrate, plasma lysine, methionine and branched-chain amino acids were reduced [26]. Three months of dual insulin sensitizer therapy (metformin plus pioglitazone) has been found to reduce plasma concentrations of phenylalanine, tyrosine, and arginine in patients with fasting hyperglycemia [27].
N-acetyl-1-methylhistidine, an amino acid metabolite associated with the gene NAT8, had the second highest VIP score in PLS-DA analysis. High levels of circulating N-acetyl-1-methylhistidine have been associated with lower eGFR and increased CKD incidence rates in African American populations [28]. Notably, there exists a significant correlation between NAT8 gene variations and N-acetylated amino acids [29]. Targeted drug development targeting NAT8 gene mutations and N-acetyl-1-methylhistidine metabolite may present novel strategies for CKD treatment, potentially delaying CKD progression.
Simultaneously, we also discovered that among the metabolites that decreased with the decline of mGFR, S-allylcysteine had the highest VIP score. S-allylcysteine is an organosulfur phytochemical derived from aged garlic extract and can provide renal protection in patients with chronic renal failure. It achieves this by inhibiting matrix remodeling mediated by the TGF-β1/Smad3 signaling pathway [30]. This suggests that bioactive compounds such as S-allylcysteine could potentially be utilized in the management of chronic kidney disease to slow down its progression.
Through OPLS-DA analysis, we identified hydroxy-asparagine and C-glycosyl-tryptophan as two amino acid-related metabolites with high VIP scores. Both increase with renal function deterioration. Hydroxy-asparagine is a rare amino acid formed via post-translational asparagine hydroxylation mainly found in vitamin K-dependent protein S epidermal growth factor-like repeats [31]. C-glycosyl-tryptophan results from tryptophan translation modifications, specifically generated through glucose and tryptophan connection using a carbon–carbon single bond. Studies reveal that low eGFR is associated with high C-glycosyl-tryptophan levels, and elevated C-glycosyl-tryptophan concentrations may heighten eGFR decline risk [32, 33]. However, few studies have explored these two metabolites' implications concerning CKD progression, and their underlying mechanisms require further molecular biological studies coupled with related experiments for clarification.
Our pathway analysis of differential metabolites revealed the significance of glycine, serine, and threonine metabolism, phenylalanine metabolism, phenylalanine, tyrosine, and tryptophan biosynthesis, and arginine and proline metabolism during CKD. Specifically, glycine, serine, and threonine metabolism represent the most crucial pathway in CKD progression. We further observed increased levels of dimethylglycine and glycine alongside renal function deterioration in the glycine, serine, and threonine metabolism pathway, whereas 3-phosphoglycerate, glycerate, pyruvate, sarcosine, serine, and tryptophan concentrations decreased with renal function deterioration.
The kidney plays a crucial role in both glycine anabolism and glycine-to-serine conversion [34]. Elevated glycine levels in CKD patients suggest that renal function deterioration causes glycine accumulation in the circulation. Additionally, elevated glycine cycling concentrations have been observed in rats, cats with CKD, and humans presenting renal insufficiency [35]. However, a previous study found that serum levels of serine, glycine, γ-aminobutyric acid, and tryptophan were all significantly lower in patients with diabetic nephropathy, hypertensive nephropathy, and chronic nephritis compared to healthy controls [36]. This implies that changes in amino acid metabolic pathways in CKD patients may be very complex, and more research is needed to understand the underlying mechanisms. IS metabolite accumulation occurs during CKD progression and promotes glomerulosclerosis progression. It also induces free radical production and nuclear factor-κB (NF-κB) activation, leading to renal tubulointerstitial fibrosis [37]. Glycerate is a monosaccharide present in tomatoes, plantains, grapes, and peanuts and positively correlates with healthy eating patterns. Glycerate can control blood sugar by reducing insulin resistance, thus managing diabetic nephropathy progression. Consuming foods with high glycerate content may reduce CKD-related complications incidence [38]. The remaining five metabolites' accumulation significance in CKD has not been reported, and further studies are necessary to determine their association with CKD.
Nonetheless, our study faces certain limitations. Firstly, our sample size is relatively small; therefore, future research should recruit more subjects to mitigate small sample size bias. Secondly, while our work provides evidence of amino acids as important plasma metabolites in CKD patients, the exact mechanism linking amino acids to CKD progression remains incompletely understood. Lastly, metabolites represent the end products of various biological processes in the human body, and metabolomics can only reflect changes at the end of the reaction pathway [39]. In the future, we need to test the accuracy of our results and uncover the underlying molecular biological mechanisms by combining genomics, transcriptomics, proteomics, and further translational research experiments, such as cell studies and animal studies.
Conclusions
In conclusion, metabolic profiling revealed that glycine, serine, and threonine metabolism constitute the most significant amino acid metabolic pathway during CKD progression. Reduced renal function correlated with diminished 3-phosphoglycerate, glycerate, pyruvate, sarcosine, serine, and tryptophan levels, alongside elevated dimethylglycine and glycine levels. Glycine and serine emerged as the most crucial metabolites during CKD progression.
Data availability
The original contributions presented in the study are included in the article material, further inquiries can be directed to the corresponding authors.
Code availability
Not applicable.
Abbreviations
- ANOVA:
-
Analysis of variance
- BMI:
-
Body mass index
- CKD:
-
Chronic kidney disease
- eGFR:
-
Estimated glomerular filtration rate
- ESI:
-
Electrospray ionization
- FA:
-
Formic acid
- GFR:
-
Glomerular filtration rate
- HILIC:
-
Hydrophilic interaction chromatography
- IAA:
-
Indole-3-acetic acid
- IS:
-
Indoxyl sulphate
- mGFR:
-
Measured glomerular filtration rate
- 5-MTP:
-
5-Methoxy tryptophan
- m/z :
-
Mass to charge ratio
- NF-κB:
-
Nuclear factor-κB
- OPLS-DA:
-
Orthogonal partial least squares-discriminant analysis
- OSC:
-
Orthogonal signal correction
- PCA:
-
Principal component analysis
- PCS:
-
p-Cresyl sulphate
- PFPA:
-
Perfluoropentanoic acid
- PLS-DA:
-
Partial least squares-discriminant analysis
- RP:
-
Reversed-phase
- TPH-1:
-
Tryptophan hydroxylase-1
- UPLC-MS/MS:
-
Ultra performance liquid chromatograph tandem mass spectrometry
- VIP:
-
Variable importance in the projection
References
Wolf G (2006) Renal injury due to renin-angiotensin-aldosterone system activation of the transforming growth factor-beta pathway. Kidney Int 70(11):1914–1919. https://doi.org/10.1038/sj.ki.5001846
Chasapi SA, Karagkouni E, Kalavrizioti D, Vamvakas S, Zompra A, Takis PG, Goumenos DS, Spyroulias GA (2022) NMR-based metabolomics in differential diagnosis of chronic kidney disease (CKD) subtypes. Metabolites. https://doi.org/10.3390/metabo12060490
Kalim S, Rhee EP (2017) An overview of renal metabolomics. Kidney Int 91(1):61–69. https://doi.org/10.1016/j.kint.2016.08.021
Johnson CH, Ivanisevic J, Siuzdak G (2016) Metabolomics: beyond biomarkers and towards mechanisms. Nat Rev Mol Cell Biol 17(7):451–459. https://doi.org/10.1038/nrm.2016.25
Hocher B, Adamski J (2017) Metabolomics for clinical use and research in chronic kidney disease. Nat Rev Nephrol 13(5):269–284. https://doi.org/10.1038/nrneph.2017.30
Wu G, Zhong J, Chen L, Gu Y, Hong Y, Ma J, Zheng N, Liu AJ, Sheng L, Zhang W, Li H (2020) Effects of the Suxiao Jiuxin pill on acute myocardial infarction assessed by comprehensive metabolomics. Phytomedicine 77:153291. https://doi.org/10.1016/j.phymed.2020.153291
Song Y, Hu T, Gao H, Zhai J, Gong J, Zhang Y, Tao L, Sun J, Li Z, Qu X (2021) Altered metabolic profiles and biomarkers associated with astragaloside IV-mediated protection against cisplatin-induced acute kidney injury in rats: an HPLC-TOF/MS-based untargeted metabolomics study. Biochem Pharmacol 183:114299. https://doi.org/10.1016/j.bcp.2020.114299
Gupta N, Yadav DK, Gautam S, Kumar A, Kumar D, Prasad N (2023) Nuclear magnetic resonance-based metabolomics approach revealed the intervention effect of using complementary and alternative medicine (CAM) by CKD patients. ACS Omega 8(8):7722–7737. https://doi.org/10.1021/acsomega.2c06469
Parker VJ, Fascetti AJ, Klamer BG (2019) Amino acid status in dogs with protein-losing nephropathy. J Vet Intern Med 33(2):680–685. https://doi.org/10.1111/jvim.15436
Rhee EP, Clish CB, Wenger J, Roy J, Elmariah S, Pierce KA, Bullock K, Anderson AH, Gerszten RE, Feldman HI (2016) Metabolomics of chronic kidney disease progression: a case-control analysis in the chronic renal insufficiency cohort study. Am J Nephrol 43(5):366–374. https://doi.org/10.1159/000446484
Chen DQ, Cao G, Chen H, Argyopoulos CP, Yu H, Su W, Chen L, Samuels DC, Zhuang S, Bayliss GP, Zhao S, Yu XY, Vaziri ND, Wang M, Liu D, Mao JR, Ma SX, Zhao J, Zhang Y, Shang YQ, Kang H, Ye F, Cheng XH, Li XR, Zhang L, Meng MX, Guo Y, Zhao YY (2019) Identification of serum metabolites associating with chronic kidney disease progression and anti-fibrotic effect of 5-methoxytryptophan. Nat Commun 10(1):1476. https://doi.org/10.1038/s41467-019-09329-0
Shah VO, Townsend RR, Feldman HI, Pappan KL, Kensicki E, Vander Jagt DL (2013) Plasma metabolomic profiles in different stages of CKD. Clin J Am Soc Nephrol 8(3):363–370. https://doi.org/10.2215/cjn.05540512
Chen YY, Chen DQ, Chen L, Liu JR, Vaziri ND, Guo Y, Zhao YY (2019) Microbiome-metabolome reveals the contribution of gut-kidney axis on kidney disease. J Transl Med 17(1):5. https://doi.org/10.1186/s12967-018-1756-4
Shen B, Yi X, Sun Y, Bi X, Du J, Zhang C, Quan S, Zhang F, Sun R, Qian L, Ge W, Liu W, Liang S, Chen H, Zhang Y, Li J, Xu J, He Z, Chen B, Wang J, Yan H, Zheng Y, Wang D, Zhu J, Kong Z, Kang Z, Liang X, Ding X, Ruan G, Xiang N, Cai X, Gao H, Li L, Li S, Xiao Q, Lu T, Zhu Y, Liu H, Chen H, Guo T (2020) Proteomic and metabolomic characterization of COVID-19 patient sera. Cell 182(1):59-72.e15. https://doi.org/10.1016/j.cell.2020.05.032
Pang Z, Zhou G, Ewald J, Chang L, Hacariz O, Basu N, Xia J (2022) Using MetaboAnalyst 5.0 for LC-HRMS spectra processing, multi-omics integration and covariate adjustment of global metabolomics data. Nat Protoc 17(8):1735–1761. https://doi.org/10.1038/s41596-022-00710-w
López-Ibáñez J, Pazos F, Chagoyen M (2016) MBROLE 2.0-functional enrichment of chemical compounds. Nucleic Acids Res 44(W1):W201-204. https://doi.org/10.1093/nar/gkw253
Ramezani A, Massy ZA, Meijers B, Evenepoel P, Vanholder R, Raj DS (2016) Role of the gut microbiome in uremia: a potential therapeutic target. Am J Kidney Dis 67(3):483–498. https://doi.org/10.1053/j.ajkd.2015.09.027
Wang X, Yang S, Li S, Zhao L, Hao Y, Qin J, Zhang L, Zhang C, Bian W, Zuo L, Gao X, Zhu B, Lei XG, Gu Z, Cui W, Xu X, Li Z, Zhu B, Li Y, Chen S, Guo H, Zhang H, Sun J, Zhang M, Hui Y, Zhang X, Liu X, Sun B, Wang L, Qiu Q, Zhang Y, Li X, Liu W, Xue R, Wu H, Shao D, Li J, Zhou Y, Li S, Yang R, Pedersen OB, Yu Z, Ehrlich SD, Ren F (2020) Aberrant gut microbiota alters host metabolome and impacts renal failure in humans and rodents. Gut 69(12):2131–2142. https://doi.org/10.1136/gutjnl-2019-319766
Rysz J, Franczyk B, Ławiński J, Olszewski R, Ciałkowska-Rysz A, Gluba-Brzózka A (2021) The impact of CKD on uremic toxins and gut microbiota. Toxins (Basel). https://doi.org/10.3390/toxins13040252
Barreto FC, Barreto DV, Liabeuf S, Meert N, Glorieux G, Temmar M, Choukroun G, Vanholder R, Massy ZA (2009) Serum indoxyl sulfate is associated with vascular disease and mortality in chronic kidney disease patients. Clin J Am Soc Nephrol 4(10):1551–1558. https://doi.org/10.2215/cjn.03980609
Letourneau P, Bataille S, Chauveau P, Fouque D, Koppe L (2020) Source and composition in amino acid of dietary proteins in the primary prevention and treatment of CKD. Nutrients. https://doi.org/10.3390/nu12123892
Kumar MA, Bitla AR, Raju KV, Manohar SM, Kumar VS, Narasimha SR (2012) Branched chain amino acid profile in early chronic kidney disease. Saudi J Kidney Dis Transpl 23(6):1202–1207. https://doi.org/10.4103/1319-2442.103560
Yu Z, Zhai G, Singmann P, He Y, Xu T, Prehn C, Römisch-Margl W, Lattka E, Gieger C, Soranzo N, Heinrich J, Standl M, Thiering E, Mittelstraß K, Wichmann HE, Peters A, Suhre K, Li Y, Adamski J, Spector TD, Illig T, Wang-Sattler R (2012) Human serum metabolic profiles are age dependent. Aging Cell 11(6):960–967. https://doi.org/10.1111/j.1474-9726.2012.00865.x
Hirschel J, Vogel M, Baber R, Garten A, Beuchel C, Dietz Y, Dittrich J, Körner A, Kiess W, Ceglarek U (2020) Relation of whole blood amino acid and acylcarnitine metabolome to age, sex, BMI, puberty, and metabolic markers in children and adolescents. Metabolites. https://doi.org/10.3390/metabo10040149
Wang Y, Zhao M, Wang M, Zhao C (2016) Profiling analysis of amino acids from hyperlipidaemic rats treated with Gynostemma pentaphyllum and atorvastatin. Pharm Biol 54(10):2254–2263. https://doi.org/10.3109/13880209.2016.1152278
Holeček M, Vodeničarovová M (2020) Effects of low and high doses of fenofibrate on protein, amino acid, and energy metabolism in rat. Int J Exp Pathol 101(5):171–182. https://doi.org/10.1111/iep.12368
Irving BA, Carter RE, Soop M, Weymiller A, Syed H, Karakelides H, Bhagra S, Short KR, Tatpati L, Barazzoni R, Nair KS (2015) Effect of insulin sensitizer therapy on amino acids and their metabolites. Metabolism 64(6):720–728. https://doi.org/10.1016/j.metabol.2015.01.008
Yu B, Li AH, Metcalf GA, Muzny DM, Morrison AC, White S, Mosley TH, Gibbs RA, Boerwinkle E (2016) Loss-of-function variants influence the human serum metabolome. Sci Adv 2(8):e1600800. https://doi.org/10.1126/sciadv.1600800
Juhanson P, Kepp K, Org E, Veldre G, Kelgo P, Rosenberg M, Viigimaa M, Laan M (2008) N-acetyltransferase 8, a positional candidate for blood pressure and renal regulation: resequencing, association and in silico study. BMC Med Genet 9:25. https://doi.org/10.1186/1471-2350-9-25
Samynathan R, Subramanian U, Venkidasamy B, Shariati MA, Chung IM, Thiruvengadam M (2022) S-allylcysteine (SAC) exerts renoprotective effects via regulation of TGF-β1/Smad3 pathway mediated matrix remodeling in chronic renal failure. Curr Pharm Des 28(8):661–670. https://doi.org/10.2174/1381612828666220401114301
Stenflo J, Lundwall A, Dahlbäck B (1987) beta-Hydroxyasparagine in domains homologous to the epidermal growth factor precursor in vitamin K-dependent protein S. Proc Natl Acad Sci USA 84(2):368–372. https://doi.org/10.1073/pnas.84.2.368
Manabe S, Marui Y, Ito Y (2003) Total synthesis of mannosyl tryptophan and its derivatives. Chemistry 9(6):1435–1447. https://doi.org/10.1002/chem.200390163
Cheng Y, Li Y, Benkowitz P, Lamina C, Köttgen A, Sekula P (2020) The relationship between blood metabolites of the tryptophan pathway and kidney function: a bidirectional Mendelian randomization analysis. Sci Rep 10(1):12675. https://doi.org/10.1038/s41598-020-69559-x
Fürst P (1989) Amino acid metabolism in uremia. J Am Coll Nutr 8(4):310–323. https://doi.org/10.1080/07315724.1989.10720307
Ruberti B, Machado DP, Vendramini THA, Pedrinelli V, Marchi PH, Jeremias JT, Pontieri CFF, Kogika MM, Brunetto MA (2022) Serum metabolites characterization produced by cats CKD affected, at the 1 and 2 stages, before and after renal diet. Metabolites. https://doi.org/10.3390/metabo13010043
Zeng L, Yu Y, Cai X, Xie S, Chen J, Zhong L, Zhang Y (2019) Differences in serum amino acid phenotypes among patients with diabetic nephropathy, hypertensive nephropathy, and chronic nephritis. Med Sci Monit 25:7235–7242. https://doi.org/10.12659/msm.915735
Hasegawa S, Jao TM, Inagi R (2017) Dietary metabolites and chronic kidney disease. Nutrients. https://doi.org/10.3390/nu9040358
Kim H, Yu B, Li X, Wong KE, Boerwinkle E, Seidelmann SB, Levey AS, Rhee EP, Coresh J, Rebholz CM (2022) Serum metabolomic signatures of plant-based diets and incident chronic kidney disease. Am J Clin Nutr 116(1):151–164. https://doi.org/10.1093/ajcn/nqac054
Li T, Zhang W, Hu E, Sun Z, Li P, Yu Z, Zhu X, Zheng F, Xing Z, Xia Z, He F, Luo J, Tang T, Wang Y (2021) Integrated metabolomics and network pharmacology to reveal the mechanisms of hydroxysafflor yellow A against acute traumatic brain injury. Comput Struct Biotechnol J 19:1002–1013. https://doi.org/10.1016/j.csbj.2021.01.033
Funding
This study was supported by grants from the National Natural Science Foundation of China (Grant No. 81873631, 81370866, 81070612) and Science and Technology Plan Project of Guangzhou (Grant No. 202002020047, 202007040003), The Science and Technology Development Fund, Macau SAR (File no. 0032/2018/A1).
Author information
Authors and Affiliations
Contributions
All authors have read and approved the manuscript. JK and XG: Conceptualization, methodology, and writing—original draft preparation. XL and HP: Funding acquisition. YD and CA: Visualization and Supervision. JL and TsT: Investigation. LT and TT: Data curation and Validation. XL and HP: Writing-review, funding, and editing.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Ethical approval
The studies involving human participants were reviewed and approved by the Institutional Review Board of Ethics Commission of Kiang Wu Hospital (KWH 2018-001). The patients/participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Kang, J., Guo, X., Peng, H. et al. Metabolic implications of amino acid metabolites in chronic kidney disease progression: a metabolomics analysis using OPLS-DA and MBRole2.0 database. Int Urol Nephrol 56, 1173–1184 (2024). https://doi.org/10.1007/s11255-023-03779-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11255-023-03779-8