
Abstract
Type 2 diabetes mellitus (DM2) is a chronic condition that affects more than 400 million individuals worldwide. In DM2 patients, an appropriate glycemic control slows the onset and delays the progression of all its micro and macrovascular complications. Even though there are several glucose-lowering drugs, only approximately half of patients achieve glycemic control, while undesirable adverse effects (e.g., low serum glucose) normally affect treatment. Therefore, there is a need for new types of treatments. Sodium-glucose cotransporter 2 inhibitors (SGLT2i) have just been developed for treating DM2. Renal hyperfiltration as a marker of increased intraglomerular pressure in diabetic patients, and the role of renin–angiotensin–aldosterone system (RAAS) in this phenomenon have been studied. Nevertheless, RAAS blockade does not completely reduce hyperfiltration or diabetic renal damage. In this sense, the contribution of renal tubular factors to the hyperfiltration state, including sodium–glucose cotransporter (SGLT), has been currently studied. SGLT2i reduce proximal tubular sodium reabsorption, therefore increasing distal sodium delivery to the macula densa, causing tubule-glomerular feedback activation, afferent vasoconstriction, and reduced hyperfiltration in animal models. In humans, SGLT2i was recently shown to reduce hyperfiltration in normotensive, normoalbuminuric patients suffering from type 1 diabetes mellitus. In DM2 clinical trials, SGLT2 is associated with significant hyperfiltration and albuminuria reduction. The aim of this article is to compile the information regarding SGLT2i drugs, emphasizing its mechanism of renal repercussion.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Glucose renal physiology
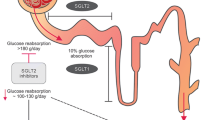
The main role of the kidney is to maintain the Internal Milieu balance through filtration, and the selective secretion and reabsorption of electrolytes, hydrogen, and bicarbonate. Glucose is also filtered and reabsorbed to maintain energy for the body functioning between meals. This occurs in the proximal tubule and it is mediated by two carriers: SGLT1 and SGLT2 [1,2,3,4]. The kidney contributes to glucose homeostasis by three different mechanisms: release of glucose into the circulation via gluconeogenesis; uptake of glucose from the bloodstream to satisfy its energy needs; and glucose reabsorption into the intravascular compartment (IVC) from glomerular filtrate (GF) to preserve it [5]. Under normal circumstances, despite daily fluctuations in the glucose delivery rate into the IVC (e.g., meals, exercise, etc.), glycemia is kept within a relatively narrow range throughout the day [4, 5]. The regulation of glycemia is mainly determined by hormonal and neural factors. In this sense, insulin, glucagon, and catecholamines are the main regulators. Insulin inhibits glucose release in both the liver and kidney through a direct enzyme activation/deactivation mechanism, as well as by decreasing gluconeogenic substrates availability and gluconeogenic inducer activity. Although glucagon has no renal effect, it enhances both hepatic gluconeogenesis and glycogenolysis. Catecholamines normally have a direct effect on renal glucose release only; however, they may indirectly affect both hepatic and renal glucose release by increasing the gluconeogenic substrate availability, glucagon secretion, and suppressing insulin release [5]. On the other hand, a group of counter-regulatory hormones: growth hormone, thyroid hormone and cortisol, as well as catecholamines, have long-term stimulatory influences on hepatic glucose release, by modifying the hepatic, renal, adipose tissue and muscular sensitivity to insulin, glucagon, and catecholamines, and by altering the glycogen stores and gluconeogenesis [5, 6] After prolong overnight fast, glucose, which comes from hepatic glycogenolysis and hepatic–renal gluconeogenesis, is released into the IVC [5]. The release of endogenous glucose decreases by 61% after meal ingestion to inhibit the development of postprandial hyperglycemia. Liver gluconeogenesis reduces by 82% and the synthesized glucose molecules are usually directed into hepatic glycogen. In the postprandial state, despite the endogenous glucose release decreases significantly, renal gluconeogenesis increases twice and accounts for 60% of endogenous glucose release during this period, perhaps to promote an increase in hepatic glycogen stores [5,6,7]. After an overnight fast, most of the released glucose (80%) is of hepatic origin, and the remaining 20% is of renal origin. However, if the duration of fasting increases, hepatic glycogen stores become further depleted, and after 48-h fasting, almost all the glucose released into the IVC derives from gluconeogenesis. It should be taken into account that daily renal glucose production by gluconeogenesis is 15–55 g [5,6,7]. The kidney also contributes to glucose homeostasis by glucose uptake from the IVC to satisfy its energy needs: 25–35 g of glucose are consumed per day by kidneys [4, 6, 8]. Since glucose is not bound to proteins, it is filtered freely. Additionally, since normal glomerular filtration rate (GFR) is about 125 ml/min/1.73 m2 (180 l per day), and the average glycemia in a 24 h period is 100 mg/dl; therefore, the kidneys filtrate 180 g of glucose per day. Practically all of this glucose is reabsorbed into the IVC, which results in zero glucosuria [5,6,7,8,9, 11, 12].. In no DM2 subjects, glycemia rarely increases above the threshold. Thus, glucose renal reabsorption is normally in the range of 150–180 g/day, and even it can be up to 300–450 g/day [8, 10, 12, 13]. Glucose reabsorption happens mainly in the proximal tubule, and is mediated by two different carriers: sodium-glucose cotransporter 1 (SGLT1) and sodium-glucose cotransporter 2 (SGLT2) [13]. The most of the glucose reabsorption (up to 90%) is performed by SGLT2 which is a high-capacity and low-affinity cotransporter. Regarding SGLT1, it handles the remaining glucose renal reabsorption, being a low-capacity and high-affinity glucose transporter [10,11,12,13,14,15]. Once glucose is reabsorbed, it is transported into the IVC through facilitative glucose transporters (GLUTs), which are located at the basolateral membrane of the proximal tubule epithelial cells (GLUT2 in the S1 and S2 segments and GLUT1 in the S3 segment) [5]. Since glucose filters freely in the glomerulus, when glycemia enhances, the filtered glucose increases [13]. This stimulates glucose tubular reabsorption capability by approximately 20% [11]. At a serum glucose value higher than 216 mg/dL (12 mmol/L), tubular glucose load can exceed the maximal reabsorptive capability, resulting in glucosuria [6,7,8,9,10,11].
Sodium-glucose cotransporters 2 (SGLT2): Mechanism of action and localization
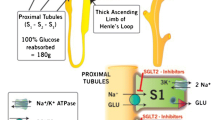
Glucose crosses cell membranes with carriers help, and because of that glucose absorption at the enterocytes, reabsorption at the renal tubules, transport across the blood–brain barrier, and uptake and release by all body cells is accomplished by two groups of glucose transporters (GLUTs) and sodium-glucose cotransporters (SGLTs). GLUTs are passive transporters which work using the glucose gradient, while SGLTs cotransport sodium and glucose into cells using the sodium gradient generated by Na–K-ATPase pump [6, 8,9,10,11,12,13,14]. SGLTs are transmembrane proteins which transport against a glucose concentration gradient, by transporting it concurrently with sodium: when sodium binds at the extracellular surface, a gate opens to trap outside glucose, and then, the protein flips over to the intracellular side, releases sodium and glucose into the cell [15]. This sodium concentration gradient is generated by the Na–K-ATPase pump [16]. SGLT1 is the main glucose transporter in gut epithelium, mainly located in the small intestine, with a secondary role in the kidney (only 10% of glucose reabsorption, mostly in the straight renal proximal tubule: S3 segment). Additionally, SGLT2, selectively expressed in the human kidney, is located predominantly at the apical membrane of renal proximal convoluted tubules (S1 and S2 segments) [4, 12, 15] (Fig. 1). SGLT1 is a high-affinity, low-capacity glucose/galactose transporter with a tenfold greater affinity for galactose. It is expressed mostly in the small intestine, and to a lesser extent in the trachea, lungs, brain, skeletal and heart muscle, liver, and kidneys. As food is digested, SGLT1 absorbs glucose and galactose. On the other hand, SGLT1 found in the late (S3) segment of the proximal renal tubule is responsible for 10% of the renal glucose reabsorption [6, 9, 14]. SGLT2, is a low-affinity, high-capacity sodium-glucose, which is 59% identical to SGLT1 and is expressed almost exclusively in the apical membrane of the early segment (S1) of renal proximal tubule where most of filtered glucose is reabsorbed. SGLT2 plays a major role in the glucose renal reabsorptive mechanism responsible for 90% of renal glucose reabsorption [6,7,8, 13, 14, 17, 18]. Glucose passively leaves the tubular cell through the basolateral membrane into the interstitium, and then enters into the IVC via GLUT2, which participates in the glucose absorption process (Fig. 1) [12]. The glucose transport across the renal tubule is similar to the one known for the small intestine: glucose is first accumulated within the epithelium by an SGLT in the brush-border membrane and then passively diffuses to a concentration gradient out of the cell into the intercellular space, which is in equilibrium with the bloodstream [6,7,8].

Glucose filtration and proximal tubule reabsorption in healthy setting
Diabetes mellitus: mechanism of kidney injury
The main inducing mechanisms of hyperglycemia-induced kidney injury and diabetic nephropathy are oxidative stress, inflammation, fibrosis, and apoptosis [19, 20]. High glucose levels in the proximal tubule can cause excessive glucose, sodium and water reabsorption (Fig. 2), favored by an increase SGTL activity which contributes to the development of diabetic complications [17, 20]. High serum glucose induces synthesis of renal angiotensin II type I receptor (AT1R) in mesangial cells and podocytes, via increasing overall renin level and intraglomerular capillary pressure. High glucose levels and the activation of the renin–angiotensin–aldosterone system (RAAS) lead to the activation of many intracellular transduction systems, which causes hyperplasia and hypertrophy mainly of the proximal tubule, by the intracellular expression of growth factors and cytokines such as transforming growth factor b (TGFb), vascular endothelial growth factor (VEGF), interleukin 6 (IL-6), and monocyte chemotactic protein 1 (MCP1) [9, 13, 19]. It is worth mentioning that TGFβ is the fibrogenic and hypertrophic cytokine involved in the extracellular matrix molecules production (type I and type IV collagen, fibronectin, and laminin) necessary for developing diabetic nephropathy [16]. These cellular mediators have been implicated in renal and vascular proliferative response, increasing local synthesis of SGLT2, glucose transportation to the tubular cell and, consequently, to the IVC [13,14,15,16,17,18,19]. In addition, hyperglycemia leads to non-enzymatic glucose reactions which generate advanced glycation end-products (AGEs) in renal cells, thus contributing to diabetic nephropathy progression [19]. Since DM2 probably increases the expression of SGLT2, there is an increase in glucose reabsorption, which causes the following phenomena [13]:

Glucose filtration and proximal tubule reabsorption in diabetic setting
-
Increased need for oral antidiabetics and insulin.
-
Glucosuria reduction leads to tubulo-glomerular feedback stimulation, perpetuating glomerular vasodilation and glomerular hyperfiltration.
-
Increased glucose proximal tubule reabsorption triggers local production of pro-inflammatory, proliferative, and profibrotic mediators.
It is known that chronic hyperglycemia causes significant morbidity and mortality owing to the resulting macrovascular disease (e.g., atherosclerotic cardiovascular disease) and microvascular disease (e.g., nephropathy, neuropathy, and retinopathy) [21, 22]. The onset of diabetic nephropathy takes many years, and it has several pathophysiologic mechanisms such as: hemodynamic, metabolic, and inflammatory pathways [21]. Hyperglycemia is a crucial factor for inducing renal hyperfiltration, and RAAS activation increases angiotensin II levels, which causes efferent arteriolar vasoconstriction with the subsequent hyperfiltration [17,18,19,20,21]. This phenomenon is associated with renal injury such as glomeruloesclerosis and progressive loss of renal function [17]. There is also increased expression of endothelin-1, another efferent arteriolar vasoconstrictor [21]. Additionally, the increased sodium reabsorption due to SGLT2 activity is sufficient to alter glomerular hemodynamics [23]. Regarding metabolic pathways, hyperglycemia leads to free oxygen radical production, which cause glyceraldehyde-3-phosphate dehydrogenase inhibition, that avoid normal glycolysis, and leads to an accumulation of glycolysis precursors, causing the upregulation of the polyol and hexosamine pathways, cofactors for protein kinase C activation and production of AGEs precursors [21], which induces tubular cells apoptosis, contributing to tubular atrophy and loss in diabetic nephropathy [24]. These metabolic effects are associated to an increase in inflammatory cytokines, renal cell hypertrophy, mesangial matrix proliferation, and glomerular basement membrane damage, all factors which contribute to glomerular hyperfiltration [17,18,19,20,21]. Glucose enters the tubular cells mediated by SGLT2 and potentiates the cells susceptibility to pro-apoptotic effects of AGEs via receptor for AGE (RAGE) expression [24]. Regarding the inflammatory pathways, hyperglycemia causes (a) increased expression of nuclear factor-kB, a transcription factor that regulates gene expression relating to inflammation, immunity, and apoptosis; (b) increased expression of inflammatory cytokines, such as interleukins and tumor necrosis factor-α (TNF-α) [21]. Other pathways which may also promote diabetic nephropathy include defective podocyte autophagic activity and SGLT2 expression upregulation, both associated with hyperglycemia [21]. Glycosuria is toxic for the proximal tubule cells, since excessive glucose rapidly saturates the glycolytic pathways capability drifting towards secondary pathways such as polyols. A considerable amount of NADH is generated through this alternative pathway, and this is also the final product of redox reactions hindered by NADH accumulation. Therefore, triose oxidation (glyceraldehyde 3P) is blocked, and some oxidation intermediates of the Krebs cycle (e.g., succinate, etc.) are blocked too. It has been proposed that succinate accumulation during hyperglycemia could contribute to the afferent arteriole vasodilatation implicated in hyperfiltration [13]. Furthermore, neurohormonal and tubular mechanisms contribute to glomerular hyperfiltration, which is present in 50% of diabetic patients [17]. The blocking of the triose oxidation leads to glycolysis intermediate product accumulation, all of them with glycosylation capability. Intracellular glycosylation of various proteins generates AGEs, which produce oxygen free radicals, pro-inflammatory nuclear transcription factors (NF-k β) activation, cell proliferation, IL-6, PAI-1, and extracellular matrix proteins expression [13]. Since glyceraldehyde 3P drift to dihydroxyacetone phosphate (DHAP), the excess oxidation of glucose enters into the diacylglycerol (DAG) pathway. DAG activates a protein kinase C (CPK) in particular cells (e.g., renal, endothelial, etc.) leading to diabetic tissues damage [13]. Increases in sodium proximal tubule reabsorption also increase the glomerular capillary hydrostatic pressure, and consequently the GFR value. This phenomenon occurs due to the following mechanisms (Fig. 2): [7,8,9,10,11,12,13,14,15,16,17,18,19,20,21,22,23]
-
Sodium flow reduction through the macula densa stimulates tubule-glomerular feedback which dilates the afferent arteriole and increases glomerular hydrostatic pressure.
-
Increased proximal tubule sodium reabsorption reduces fluid flow through the proximal tubule and consequently decreases Bowman’s space pressure that opposes filtration.
-
SGLT2 excessive activation increases glucose and sodium proximal tubule reabsorption, which maintains hyperglycemia as a mechanism against glucose osmotic diuresis, and to decrease sodium delivery to distal tubules, which promotes renal hemodynamic dysfunction. Regarding histological changes in the diabetic kidney, particularly tubule-interstitial fibrosis, present a significant correlation with renal function reduction. The most important tubule-interstitial changes observed in DM2 are the thickening of the proximal tubular cell basement membrane, hyperplasia, and hypertrophy in early stages, followed by its subsequent atrophy and interstitial fibrosis [16].
SGLT2i: mechanism of action
Both SGLT2 and renal GLUT2 are upregulated in DM2 patients compared to healthy subjects. This phenomenon of SGLT2 and GLUT2 upregulation could be important adaptations to maintain renal tubular glucose reabsorption. Selective SGLT2 inhibitors (SGLT2is) are oral hypoglycemic agents used to treat patients suffering from DM2. They improve glycemic control in an insulin-independent manner by blocking glucose reabsorption in the renal proximal tubule, inducing glucosuria, osmotic diuresis, and reduced glucose-sodium proximal tubule reabsorption [4, 7, 11, 16, 21, 22]. SGLT2is hinder the glucose cotransporters activity in proximal convoluted tubules, reducing glucose reabsorption, and consequently causing a net loss of 70–80 g of glucose a day [10]. Since SGLT2is act through an insulin-independent mechanism, their therapeutic efficacy does not decline with progressive beta cell dysfunction and/or insulin resistance [11]. Due to that, SGLT2i improves glycemic control in all stages of DM2. Another advantage of these drugs is that they act synergistically with other antihyperglycemic drugs [11]. In addition, hyperglycemia correction reverses glucotoxicity, thus improving β-cell function [4].
SGLT2i: systemic effects
Basic antihyperglycemic SGLT2i efficacy consists of the reduction of serum glucose levels which does not depend on insulin or the β cell action, attenuating glucose toxicity [12,13,14,15,16,17]. As a result of its mechanism of action, this drug increases glycosuria and natriuresis, being its renal and cardiovascular protective effects in DM2 due to glycosuria, which leads to an improved glycemic control, weight loss, and positive effects on insulin activity. Additionally, natriuresis leads to blood pressure (BP), intraglomerular hypertension, and proteinuria reduction [17,18,19,20,21,22,23,24,25]. These protective renal and cardiovascular effects have been observed in DM2 patients treated with empagliflozin (EMPA-REG OUTCOME Trial), canagliflozin (CANVAS Trial), and dapagliflozin (DECLARE–TIMI Trail) [14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30] (Table 1) SGLT2i can help control hypertension, because it reduces systolic and diastolic blood pressure by 3.77 and 1.75 mmHg, respectively [7]. Decreases in systolic blood pressure of up to 5 mmHg have been described in clinical trials of dapagliflozin, empagliflozin, and canagliflozin. Due to the fact that SGLT2i has osmotic diuretic-like effects (volume contraction), they can lower systolic blood pressure by 3–5 mmHg [1, 4, 17]. In addition, antihypertensive effects result in a reduction of arterial stiffness [22]. These drugs induce a daily glucose urinary loss of 70–80 g, consequently leading to a 2–3 kg body weight decline over 3–6 months [11, 22]. Initially, the weight loss is mainly due to fluid depletion, but subsequently there is loss of fat tissue. Nevertheless, this weight loss appears to plateau after about 6 months of treatment [4, 7, 12, 22]. It is worth mentioning that hypoglycemia does not occur with the use of SGLT2is neither in DM2 patients nor in non-diabetic individuals. Therefore, it has been hypothesized that the liver could react to the glycosuria by increasing glucose release [12]. SGLTis might also induce favorable cardiovascular and renal effects by decreasing plasma uric acid levels [11]. Since insulin resistance and hyperinsulinemia reduce uric acid renal excretion, hyperuricemia is commonly observed in patients with DM2. By increasing glucose concentrations in the filtrate, SGLT2 inhibition is suggested to cause glucose transporter to excrete uric acid in exchange for glucose reuptake, causing a reduction in uric acid reabsorption [7]. It has been documented that SGLT2is improve renal and cardiovascular outcome [7]. The EMPA-REG OUTCOME Trial documented that empagliflozin greatly improved the occurrence of major adverse cardiovascular events (cardiovascular death, nonfatal myocardial infarction, and nonfatal stroke) in DB2 patients, compared to the control group [27,28,29,30,31]. Since these benefits occurred too early (within months), they were attributed not to antiatherosclerotic effects but to empagliflozin-induced hemodynamic changes [7, 27] In CANVAS and CANVAS-R trials involving DM2 patients with an elevated risk of cardiovascular disease, patients treated with canagliflozin had a lower risk of cardiovascular events than those who received placebo. However, they had a greater amputation risk, primarily at the level of the toe or metatarsal. An increase in bone fractures has been previously described with canagliflozin, however, only documented in CANVAS but not in CANVAS-R [29]. The DECLARE–TIMI 58 trial documented that dapagliflozin was non-inferior to placebo regarding the composite safety outcome of cardiovascular death, myocardial infarction, or ischemic stroke. However, dapagliflozin did not result in a significantly lower rate of major adverse cardiovascular events; still, it did significantly reduce cardiovascular death and heart failure hospitalization rate in a broad population of DM2 patients. The genital infections rate was higher with dapagliflozin than with placebo, but not Fournier’s gangrene rate [30].
SGLT2i: renal repercussions
SGLT2is reduce glomerular hyperfiltration in DM2 patients, since this cotransporter inhibition increases delivery of fluid and electrolytes to the macula densa, thereby activating tubule-glomerular feedback [11]. In addition, an increment in urine output is commonly detected with acute SGLT2 inhibition, phenomenon which has been attributed to the osmotic diuresis induced by glucosuria. With chronic SGLT2i administration, excess urine volume appears to settle at 200–600 ml per day. As a consequence, an increased hematocrit is usually observed in patients on SGLT2i, while clinical volume depletion is infrequent [12]. Proteinuria is a crucial prognostic factor for the progression of chronic kidney disease (CKD) [14]. Even though this progression has decreased with advances in hypertension treatment and RAAS inhibition, there are still many DM2 patients suffering from CKD who will progress to end-stage renal disease and renal-replacement treatment (RRT) [21].
The proteinuria reduction mechanism may occur on the basis of direct proximal tubule effects, which causes urinary sodium loss [14]. In the context of diabetes mellitus, there is an increase in SGLT2-mediated transport, which leads to augmented glucose and sodium proximal tubule reabsorption, and reduced distal delivery to the macula densa [14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30]. A decrease in sodium supply at the macula densa is sensed by the juxtaglomerular apparatus as an effective hypovolemia, leading to afferent arteriolar vasodilation via the tubule-glomerular feedback (TGF), which leads to an increase in intraglomerular pressure. By blocking proximal tubular uptake of sodium by SGLT2 inhibition, the resulting increase of distal sodium delivery to the macula densa stimulates TGF-mediated afferent vasoconstriction, contributing to a decrease in intraglomerular hypertension [14,15,16,17,18,19,20,21,22,23,24,25,26]. As a consequence of reducing glomerular hyperfiltration, it is expected to reduce the transport work and renal oxygen consumption, as well as the glomerular capillary hypertension and kidney injury rate [11, 23]. Apparently as a consequence of this decrease in glomerular hypertension, SGLT2i induce albuminuria reduction in patients suffering from DM2 by 30–50% [22].
Inhibition of this cotransporter lowers primary proximal tubule hyper-reabsorption, typical of diabetic patients, thus reducing glomerular hyperfiltration [11]. An acute reduction in GFR (over the first few weeks) was observed with SGLT2is, explained by the hemodynamic response to these drugs [17,18,19,20,21,22,23,24,25,26,27]. SGLT2 blockade can not only improve hyperglycemia by improving urinary glucose excretion, but also reduce glucose overload to the proximal tubule cells, generating beneficial effects on tubular apoptosis and atrophy [17,18,19,20,21,22,23,24]. In addition to direct effects on proximal tubule sodium reabsorption, the glucosuria caused by SGLT2i induces an osmotic diuresis effect, contributing to excess sodium and water excretion [23].
There is increasing evidence, suggesting that SGLT2is may have renal protective effects, as reported by diverse clinical trials (EMPA-REG OUTCOME, CANVAS, DECLARE-TIMI 58, and CREDENCE) whose findings are summarized as follows [27,28,29,30,31,32,33,34,35,36,37]. In the EMPA-REG OUTCOME Trial, it was proved that the use of empagliflozin reduces the incidence of CKD, progression to macroalbuminuria, and frequency of RRT initiation, compared to placebo [22,23,24,25,26,27]. The potential main mechanism responsible for these renoprotective effects is probably the empagliflozin renovascular and hemodynamic effects [19, 21]. In the CANVAS trails, albuminuria progression occurred less frequently in the canagliflozin group than in the placebo group:hazard ratio of 0.73 (95% CI 0.67–0.79). However, these effects were greater in CANVAS-R than in CANVAS. In addition, albuminuria regression also occurred more frequently in the canagliflozin group than in the placebo group (hazard ratio, 1.70; 95% CI 1.51–1.91). Regarding the renal outcome, (sustained 40% GFR reduction, renal-replacement therapy need, or death from renal cause) occurred less frequently in the canagliflozin group than in the placebo group (hazard ratio of 0.60; 95% CI 0.47–0.77). In this outcome, no significant difference was documented between both CANVAS trials [29]. Regarding the DECLARE-TIMI 58 trial, its findings supported a possibly lower rate of adverse renal composite outcome (≥ 40% decrease in eGFR to < 60 ml/min/1.73 m2, new end-stage renal disease (ESRD), or death from renal cause) in the dapagliflozin group than in the placebo group (hazard ratio, 0.53; 95% CI 0.43–0.66) [30] Finally, in the CREDENCE trial, the event rate of its primary composite outcome (ESRD, doubling of the creatininemia, or renal or cardiovascular death) was significantly lower in the canagliflozin group than in the placebo group (hazard ratio, 0.70; 95% CI 0.59–0.82. The effects were also consistent across renal components, including the doubling of the serum creatinine level, and the exploratory outcome of dialysis, kidney transplantation, or renal death [36]. Studies in animal models of diabetes mellitus have shown that SGLT2 inhibition can reduce renal growth, inflammation, and injury, which means that SGLT2i could reduce renal and extrarenal glucotoxicity [11]. In this sense, many inflammatory inducing mechanisms, which are not mutually exclusive, that could be inhibited by SGLT2 inhibition, have been proposed: body overweight, postprandial hyperglycemia, high plasma insulin and uric acid levels, low serum ketone and beta-hydroxybutyrate levels, and endothelial pro-inflammatory chemikine/cytokine secretion [32, 33] (Table 2). Another SGLT2i effect consists of reducing glucose accumulation in the renal cells. This phenomenon leads to a reduction in reactive oxygen species (ROS), and glucose-induced inflammatory and fibrotic markers in proximal tubule cells, expressed by the reduced expressions of Toll-like receptor-4 (TLR4), type IV collagen, interleukin-6 secretion, and nuclear factor kappa B (NF-kB). These results indicated that SGLT2i can prevent proximal tubular damage linked to glucotoxicity. Finally, since AGEs resulting from chronic hyperglycemia can induce protein modifications and activation of inflammatory signaling pathways, SGLT2i could reduce their production (Table 2) [4,5,6,7,8,9,10,11,12,13,14,15,16,17,18,19,20]. Renal impairment reduces SGLT2i efficacy, since its activity depends on the number of nephrons; therefore, the main renal drug handbooks do not recommend SGLT2i use in patients with a glomerular filtration rate (eGFR) < 40 ml/min/1.73 m2 [1, 34]. However, it is worth emphasizing that empagliflozin has been accepted for treating patients with eGFR ≥ 60, although it can be continued until eGFR of 45 ml/min. Even after the CREDENCE study publication, canagliflozin has been accepted to be initiated in patients with eGFR of 30 ml/min, despite the fact that this is not universally accepted [27, 32,33,34]. Mild and transitory GFR changes (about 5 ml/min), albumin-to-creatinine ratio, and serum urea were observed at the beginning of a study in stage-3 CKD patients, but after a 26-week treatment with SGLT2is, a return of these parameters to baseline levels was registered, along with a rise in serum potassium and magnesium in such patients [1]. In DM2 patients, SGLT2is can reduce hyperfiltration and could have a long-term renal protection effect [1, 17]. A number of studies reported that there is no evidence of empagliflozin-induced nephrotoxicity, and conversely it was even found that this drug reduces this risk [7]. However, regardless of the renal safety outcomes reported in the EMPA-REG study, a few reports suggest that there could be a risk for renal damage associated to this drug which has been attributed to the trans-glomerular pressure reduction, and mild decline in GFR, which is paradoxically the basis of its long-term renal protection effect [27, 28, 31,32,33,34,35,36]. Conversely, the CREDENCE Trial proved that DB2 patients who received canagliflozin had a lower risk of renal failure than the patients who received placebo [36].
Conclusion
SGLT2 inhibitors are now widely used in patients with type 2 diabetes mellitus to improve glycated hemoglobin levels and to reduce cardiovascular risk. They are also the most promising drugs to confer renoprotection. Evidence has shown that these glucose-lowering agents attenuate the glomerular hyperfiltration associated with diabetes mellitus and reduce the glucose tubular toxicity, ameliorate renal growth and, subsequently, reduce albuminuria, probably explaining its renoprotective effects.
References
Halimi S, Vergès B (2014) Adverse effects and safety of SGLT-2 inhibitors. Diabetes Metab. 40(6 Suppl 1):S28–S34. https://doi.org/10.1016/S1262-3636(14)72693-X
World Health Organization [Internet] (2018) https://www.who.int/news-room/fact-sheets/detail/diabetes. Accessed 28 Aug 2018
Škrtić M, Cherney D (2015) Sodium–glucose cotransporter-2 inhibition and the potential for renal protection in diabetic nephropathy. Curr Opin Nephrol Hypertens 24(1):96–103
Poudel R (2013) Renal glucose handling in diabetes and sodium glucose cotransporter 2 inhibition. Indian J Endocrinol Metab 17(4):588–593
Gerich JE (2010) Role of the kidney in normal glucose homeostasis and in the hyperglycaemia of diabetes mellitus: therapeutic implications. Diabet Med 27(2):136–142
Salvatore T, Carbonara O, Cozzolino D, Torella R, Nasti R, Lascar N, Sasso FC (2011) Kidney in diabetes: from organ damage target to therapeutic target. Curr Drug Metab 12(7):658–666. https://doi.org/10.2174/138920011796504509
Van Bommel E, Muskiet M, Tonneijck L, Kramer M, Nieuwdorp M, van Raalte D (2017) SGLT2 Inhibition in the diabetic kidney—from mechanisms to clinical outcome. Clin J Am Soc Nephrol. 12(4):700–710
Komala MG, Panchapakesan U, Pollock C, Mather A (2013) Sodium glucose cotransporter 2 and the diabetic kidney. Curr Opin Nephrol Hypertens 22(1):113–119
De Nicola L, Gabbai F, Liberti ME, Sagliocca A, Conte G, Minutolo R (2014) Sodium/glucose cotransporter 2 inhibitors and prevention of diabetic nephropathy: targeting the renal tubule in diabetes. Am J Kidney Dis 64(1):16–24
Gómez-Fernández P, Fernández-García D (2016) Renal safety profile of sodium-glucose type 2 cotransporter inhibitors and other safety data. Med Clin (Barc). 147(Suppl 1):44–48. https://doi.org/10.1016/S0025-7753(17)30625-5
Vallon V, Thomson S (2016) Targeting renal glucose reabsorption to treat hyperglycaemia: the pleiotropic effects of SGLT2 inhibition. Diabetologia 60(2):215–225
Ferrannini E, Solini A (2012) SGLT2 inhibition in diabetes mellitus: rationale and clinical prospects. Nat Rev Endocrinol 8(8):495–502
Tejedor-Jorge A (2016) Implicaciones hemodinámicas y renales de los inhibidores del cotransportador sodio-glucosa tipo 2 en el contexto de la diabetes mellitus tipo 2. Med Clín 147(S1):35–43. https://doi.org/10.1016/S0025-7753(17)30624-3
Rajasekeran H, Cherney D, Lovshin JA (2017) Do effects of sodium–glucose cotransporter-2 inhibitors in patients with diabetes give insight into potential use in non-diabetic kidney disease? Curr Opin Nephrol Hypertens 26(5):358–367
Ferrannini E (2017) Sodium-glucose co-transporters and their inhibition: clinical physiology. Cell Metab 26(1):27–38
Panchapakesan U, Pegg K, Gross S, Komala M, Mudaliar H, Forbes J, Mather A (2013) Effects of SGLT2 inhibition in human kidney proximal tubular cells-renoprotection in diabetic nephropathy? PLoS One 8(2):e54442
Cherney D, Perkins B (2014) Sodium-glucose cotransporter 2 inhibition in type 1 diabetes: simultaneous glucose lowering and renal protection? Can J Diabetes. 38(5):356–363
Ferrannini E, Veltkamp S, Smulders R, Kadokura T (2013) Renal glucose handling: impact of chronic kidney disease and sodium-glucose cotransporter 2 inhibition in patients with type 2 diabetes. Diabetes Care 36(5):1260–1265
Jaikumkao K, Pongchaidecha A, Chatsudthipong V, Chattipakorn S, Chattipakorn N, Lungkaphin A (2017) The roles of sodium-glucose cotransporter 2 inhibitors in preventing kidney injury in diabetes. Biomed Pharmacother 94:176–187
Osorio H, Coronel I, Arellano A, Pacheco U, Bautista R, Franco M, Escalante B (2012) Sodium-glucose cotransporter inhibition prevents oxidative stress in the kidney of diabetic rats. Oxid Med Cell Longev 2012:542042. https://doi.org/10.1155/2012/542042
Wanner C (2017).EMPA-REG OUTCOME: the nephrologist’s point of view. 120(1S):S59–S67. https://doi.org/10.1016/j.amjcard.2017.05.012
Heerspink H, Kosiborod M, Inzucchi S, Cherney D (2018) Renoprotective effects of sodium-glucose cotransporter-2 inhibitors. Kidney Int 94(1):26–39
Brady JA, Hallow KM (2018) Model-based evaluation of proximal sodium reabsorption through SGLT2 in health and diabetes and the effect of inhibition with canagliflozin. J Clin Pharmacol. 58(3):377–385. https://doi.org/10.1002/jcph.1030
Maeda S, Matsui T, Takeuchi M, Yamagishi S (2013) Sodium-glucose cotransporter 2-mediated oxidative stress augments advanced glycation end products-induced tubular cell apoptosis. Diabetes/Metab Res Rev 29(5):406–412
Goldenberg R, Berall M, Chan C, Cherney D, Lovshin J, McFarlane P, Weinstein J (2018) Managing the course of kidney disease in adults with type 2 diabetes: from the old to the new. Can J Diabetes 42(3):325–334
Cherney D, Lund S, Perkins B, Groop P, Cooper M, Kaspers S, von Eynatten M (2016) The effect of sodium glucose cotransporter 2 inhibition with empagliflozin on microalbuminuria and macroalbuminuria in patients with type 2 diabetes. Diabetologia 59(9):1860–1870
Zinman B, Wanner C, Lachin JM, Fitchett, Bluhmki E, Hantel S, Mattheus M, Devins T, Johansen OE, Woerle H, Broedl UC, Inzucchi SE (2015) Empagliflozin, cardiovascular outcomes, and mortality in type 2 diabetes. N Engl J Med. 373:2117–2128
Ingelfinger J, Rosen C (2019) Clinical credence—SGLT2 inhibitors, diabetes, and chronic kidney disease. N Engl J Med 380(24):2371–2373. https://doi.org/10.1056/NEJMe1904740
Neal B, Perkovic V, Mahaffey K, de Zeeuw D, Fulcher G, Erondu N, Shaw W, Law G, Desai M, Matthews D, Phil D (2017) Canagliflozin and Cardiovascular and Renal Events in Type 2 Diabetes. N Engl J Med 377:2097–2099. https://doi.org/10.1056/NEJMc1712572
Wiviott S, Raz I, Bonaca M, Mosenzon O, Kato E, Cahn A, Silverman M, Zelniker T, Kuder J, Murphy S, Bhatt D, Leiter L, McGuire D, Wilding J, Ruff C, Gause-Nilsson I, Fredriksson M, Johansson P, Langkilde A, Sabatine M (2019) Dapagliflozin and cardiovascular outcomes in type 2 diabetes. N Engl J Med 380:347–357. https://doi.org/10.1056/nejmoa1812389
Vallon V, Richter K, Blantz RC, Thomson S, Osswald H (1999) Glomerular hyperfiltration in experimental diabetes mellitus: potential role of tubular reabsorption. J Am Soc Nephrol 10:2569–2576
Bonnet F, Scheen AJ (2018) Effects of SGLT2 inhibitors on systemic and tissue low-grade inflammation: the potential contribution to diabetes complications and cardiovascular disease. Diabetes Metab. 44(6):457–464. https://doi.org/10.1016/j.diabet.2018.09.005
Prattichizzo F, De Nigris V, Micheloni S, Sala L, Ceriello A Increases in circulating levels of ketone bodies and cardiovascular protection with SGLT2 inhibitors: Is low-grade inflammation the neglected component. Diabetes Obes Metab 20(11): 2515–2522. https://doi.org/10.1111/dom.13488
Ashley C, Dunleavy A, Cunningham J (2019) The renal drug handbook. Taylor & Francis Group, New York
Szalat A, Perlman A, Muszkat M, Khamaisi M, Abassi Z, Heyman S (2017) Can SGLT2 inhibitors cause acute renal failure? Plausible role for altered glomerular hemodynamics and medullary hypoxia. Drug Saf 41(3):239–252
Perkovic V, Jardine MJ, Neal B, Bompoint S, Heerspink HJL, Charytan DM, Edwards R, Agarwal R, Bakris G, Bull S, Cannon CP, Capuano G, Chu PL, de Zeeuw D, Greene T, Levin A, Pollock C, Wheeler DC, Yavin Y, Zhang H, Zinman B, Meininger G, Brenner BM, Mahaffey KW (2019) Canagliflozin and renal outcomes in type 2 diabetes and nephropathy. N Engl J Med. 380(24):2295–2306. https://doi.org/10.1056/NEJMoa1811744
Prattichizzo F, La Sala L, Rydén L, Marx N, Ferrini M, Valensi P, Ceriello A (2019) Glucose-lowering therapies in patients with type 2 diabetes and cardiovascular diseases. Eur J Prev Cardiol 26(2):73–80. https://doi.org/10.1177/2047487319880040
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
All the authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Castañeda, A.M., Dutra-Rufato, A., Juarez, M.J. et al. Sodium-glucose cotransporter 2 inhibitors (SGLT2i): renal implications. Int Urol Nephrol 53, 291–299 (2021). https://doi.org/10.1007/s11255-020-02585-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11255-020-02585-w