
Abstract
Selective Oxygen Atom Transfer to α-pinene, β-pinene, (R)-limonene and camphene by dioxo-Mo(VI)Ln complex {Ln = 2-aminothiazole-4-carboxylic acid} anchored on TiO2 nanotubes (TiO2-NT) using O2 as oxidant and UV light was studied at ambient conditions. The Catalytic system was characterized by FTIR, 1H NMR and 13C, and elemental analysis. Molecular oxygen reoxidizes the (Mo(IV)=O) reduced unit in the darkness, forming an oxo-peroxo-Mo intermediate. The catalyst under UV radiation transfers two oxygen atoms to the olefin. The α-pinene is found to be the most reactive monoterpene.
Graphical Abstract

Similar content being viewed by others
Avoid common mistakes on your manuscript.
1 Introduction
Molybdenum enzymes involved in oxygen atom transfer (OAT) reactions are present in nature [1]. The ligand molybdopterin or pyranopterin coordinates to molybdenum via its dithiolene chelate moiety. Remaining coordination sites at molybdenum(VI) are filled by one or two oxido, sulfido or hydroxido ligands as well as coordinating atoms from amino acid side chains (S-Cysteine, Se-Cysteine, O-Serine, O-Asparagine) [2]. Oxo-peroxo and dioxo-Mo(VI) complexes with polydentate ligands containing N, O and S atoms are suitable models to mimic the activity of molybdenum enzymes, where the Mo=O unit is responsible for their catalytic activity in the OAT reactions [3]. Based on these observations there is broad interest in preparing new complexes of molybdenum with sulfur ligands. Isothiazole and thiadiazole complexes have attracted considerable interest, in the synthesis and application as OAT agents [4].
Thiazole-type heterocycles forms 1,3-thiazole or 1,2-thiazole (Fig. 1a, b). Similarly, the thiadiazole ring can assume four different isomers, like Fig. 1c–f.

Chemical structure of a 1,3-thiazole, b 1,2-thiazole (isothiazole), c 1,2,3-thiadiazole, d 1,2,4-thiadiazole, e 1,2,5- thiadiazole and f 1,3,4-thiadiazole
Complexes of molybdenum with thiadiazole ligands have been used as catalysts in olefin epoxidation in solution with TBHP as oxidant with different olefins with high yields (> 90%) and selectivity (> 90%) [5]. Molybdenum(VI) complexes with thiazole-hydrazone ligand encapsulated in zeolite Y have been evaluated in the oxidation of primary alcohols, ethylbenzene and toluene, using TBHP (70% solution in H2O as an oxidant) [6].
In a previous work we have evaluated the OAT activity of some Mo complexes supported on TiO2, showing a photoinduced oxygen transfer, which allows the selective oxidation of monoterpene-type olefins [7,8,9,10,11,12,13]. In this work, in order to evaluate the effect of the N and S heteroatoms of the ligands structure of the dioxo-Mo(VI) complex in OAT reactions, 2-aminothiazole-4-carboxylic acid was used as ligand and anchored on TiO2 nanotubes. The selective photo-epoxidation of α-pinene, β-pinene, (R)-limonene and camphene using O2 as oxidant and UV light at room pressure and temperature was studied. Additionally, the intermediate species formed after reoxidation of the Mo-complex reduced were characterized.
2 Experimental
2.1 Materials and Reagents
TiO2 (anatase nanopowder, Sigma-Aldrich), NaOH (Merck), MoO2Cl2 (Sigma-Aldrich), bis(trimethylsilyl)urea (98%, Alfa Aesar), 2-aminothiazole-4-carboxylic acid (Sigma-Aldrich), (R)-(+)-Limonene (98%, Alfa Aesar), (S)-(−)-limonene (98%, Alfa Aesar) and other reagents were obtained from commercial sources and were used as received. Acetonitrile was bubbled with N2 before use in each reaction. The preparation of the complex and its anchoring procedure were carried out under oxygen- and water-free argon atmosphere using standard Schlenk techniques.
2.2 Instrumentation
TiO2-NT support was characterized by powder X-ray diffraction (XRD) using a Bruker AXS D8 Advance with monochromatized Cu Kα radiation (λ = 1.5418 Å) at 40 kV and 30 mA. The diffraction pattern was recorded at 2θ value range (20°–60°) with a step size of 0.01° and a step time of 0.4 s. The micrographics were done in the Transmission Electron Microscope (TEM) Tecnai F20 Super Twin TMP, field emission source, resolution of 0.1 nm at 200 Kv, maximum magnification at TEM 1.0 MX, GATAN US 1000XP-P camera. 13C CP-MAS NMR spectra were obtained on a Bruker NMR spectrometer Advance 400 MHz, operating at a resonance frequency of 101.6 MHz for 13C. The formation of peroxo-Mo and superoxo-Mo species in the reoxygenation step was followed with FT-IR Spectroscopy (Brucker Tensor 27). As well, Raman spectra were performed using an integrated confocal Raman system (LabRAM HR Evolution HORIBA Scientific) spectrometer using a laser with an excitation wavelength at 532 nm, 10 × objective and a power of 10 mW; ten accumulations of 2s were used in each sample. In addition, C, H, N, S elemental analysis of catalyst was carried out in an Elementar, Vario El Cube equipment. X-Ray Photoelectron Spectroscopy (XPS) analysis were carried out in a SPECS® XPS/ISS/UPS Surface Characterization Platform. The samples were analyzed using a monochromatic Al Kα X-ray source operated at 200 W/12 kV. The pass energy of the hemi- spherical analyzer was set at 60 eV for the high-resolution spectra. Charge compensation was performed using Flood Gun. The reference scale was calibrated by adjusting the carbon adventitious C–H to 284.8 eV. The Relative Sensitivity Factors (RSF) used for quantification procedures were: Mo 3d5/2 (5.73), Ti 2p (7.57) and S 2p (1.73). The pass energy of the hemispherical analyzer was set at 60 eV for high resolution spectra and 100 eV for survey spectra. The XPS data was analyzed using the CasaXPS software. All signals were analyzed using Shirley background. Plus, during nucleophilic and electrophilic model reactions, the catalyst was separated, dried and kept in an N2 atmosphere until the IR and Raman characterization was carried out.
2.3 TiO2 Nanotubes Preparation
TiO2 nanotubes were prepared by hydrothermal treatment. 0.8 g of TiO2 nanopowder (99.5%, Aldrich) was mixed with 40 mL of a solution containing NaOH (10 M). Afterwards, the suspension was constantly stirred during 30 min and left in an autoclave for 24 h at 110 °C. Then, the solid was filtered and washed with water until neutral pH. The catalyst was then dispersed on 500 mL of a nitric acid solution (0.1 M) and stirred for 24 h. Finally, the material was filtered, washed until neutral pH and dried for 6 h at 110 °C. In the second step of the synthesis, the obtained hydrogen titanates phase was calcined at 400 °C for 6 [14].
2.4 Mo(VI)(atca)2Cl2O2/TiO2-NT Catalysts Preparation
2.4.1 Preparation of bis(2-aminothiazole-4-carboxylato)dichlorodioxomolybdenum(VI) Complex Anchored on TiO 2 Nanotubes
The preparation of the complex was carried out following a previously reported methodology [10,11,12,13], in the first stage, the acid group of the 2-aminothiazole-4-carboxylic acid was silylated using Bis(trimethylsilyl)urea (BTSU) as a silylating agent and in the second stage, MoO2Cl2 was used to form the homogeneous complex in solution (Fig. 2).

Preparation route of the bis(2-aminothiazole-4-carboxylato) dichlorodioxo molybdenum(VI) complex anchored on TiO2 nanotubes
1 mmol of BTSU was added to a benzene solution (40 mL) containing the respective aminothiazole ligand (1 mmol) and the mixture was heated under reflux for 4 h. The suspension obtained was filtered and the resulting solution was slowly evaporated until obtaining the respective solids of the silylated ligands. Then, 1 mmol of the trimethylsilylated aminothiazole ligand was added to a benzene solution with 1 mmol of MoO2Cl2 previously dissolved. The reaction mixture was stirred for 4 h at 40 °C. The solvent was evaporated under vacuum until the respective trimethylsilylated dioxo-Mo complex was obtained. Finally, a benzene solution (50 mL) with the bis(trimethylsilyl-2-aminothiazole-4-carboxylato) dichlorodioxo molybdenum(VI) complex was added to the wholly dehydrated TiO2 nanotubes (80 °C, 10–5 mbar and 24 h). The suspension was slowly stirred at 25 °C for 24 h, the support was filtered, washed with benzene (2 × 30 mL) and dried under vacuum to obtain the respective heterogeneous catalytic system.
2.5 Monoterpenes Photo-Oxidation
The monoterpenes oxidation with O2 was performed in a 15 mL glass batch microreactor (ACEGLASS) equipped with a mercury lamp (UV PenRay, λ = 360 nm). Typically, the reactor was loaded with 10 mL 1 × 10−2 M monoterpene solution in CH3CN and 15 mg of catalyst and bubbled with O2 at 19 °C. The oxidation reaction under O2 during 18 h was performed 3 times. Samples were taken every 2 h and the products were quantified using toluene as standard. 400 µL of the standard solution 1 M was added in each reaction. Liquid samples were withdrawn, immediately filtered and analyzed by gas chromatography (GC-HP-6890) using a HP-INNOWAX column (30 m × 0.32 mm × 0.25 μm) connected to a FID detector. Quantification of the products was performed using benzene as the internal standard. Reaction products were also identified by GC-MS with an Agilent Technologies 6890 Plus chromatograph (Palo Alto, CA, EE. UU.), equipped with a selective mass detector Agilent Technologies 5973 N (EI, 70 Ev, m/z 40–350) and a fused silica capillary column with 5% phenyl-poly(dimethylsiloxane) coating (DB-5ms, J&W Scientific, Folsom, CA, EE.UU.) (60 m × 0.25 mm × 0.25 μm).
3 Results and Discussion
Figure S1 (Supplementary material) shows the XRD patterns of the TiO2 nanotubes indicating that anatase is the only phase present with high crystallinity [15]. TEM images indicate that nanotubes are formed after hydrothermal reaction, see Fig. S2. It can be clearly seen that a large amount of randomly tangled NTs is produced with average outside diameter between 9 and 15 nm, with lengths up to 500 nm, in according to hydrothermal synthesis [14].
3.1 Spectroscopic Characterization of the Homogeneous Mo-complex
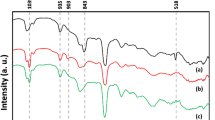
Figure 3 shows the FT-IR spectra of the 2-aminothiazole-4-carboxylic acid (free ligand), the trimethylsilyl-2-aminothiazole-4-carboxylate (silylated ligand), and the bis(trimethylsilyl-2-aminothiazole-4-carboxylato)dichlorodioxomolybdenum(VI) (complex). The characteristic thiazole ring signals corresponding to the symmetric and asymmetric C–S–C stretching are observed around 663 and 746 cm−1. The stretching signals of C–S (806 cm−1), C–N (1209 cm−1), C-O (1265 cm−1), C=C (1569 cm−1) and C=N (1626 cm−1) are observed [16, 17]. Additionally, the stretching signals of the C–H (3016 cm−1) of the thiazole ring, O–H (3148 cm−1) of the acid group and N–H (3407 cm−1) of the amino group in the free ligand are observed. The silylation of the 2-aminothiazole-4-carboxylic acid ligand is evidenced by the disappearance of the signal corresponding to the stretching of the OH group (3148 cm−1) and the appearance of new signals at 862 (–Si–CH3), 1036 (Si–O), 2910 and 2962 cm−1 (CH). Finally, the formation of the complex can be corroborated with the appearance of the doublet at 916 (asymmetric stretching) and 951 cm−1 (symmetric stretching) of the dioxo Mo (MoO2) and the redshift of the C=C and C=N signals. The N–H signal of the amino group is maintained once the complex is formed without shifting, indicating that the amine group is not part of the coordination of the ligand.

FT-IR spectrum of: (a) 2-aminothiazole-4-carboxylic acid, (b) trimethylsilyl-2-aminothiazole-4-carboxylate and (c) bis(trimethylsilyl-2-aminothiazole-4-carboxylato)dichlorodioxo molybdenum(VI)
Figure 4 displays the 13C NMR spectra of the sulfur compounds obtained using deuterated dimethyl sulfoxide (DMSO-d6) as solvent. The spectrum of the 2-aminothiazole-4-carboxylic acid ligand showed the four signals of the carbons present in the molecule (Fig. 4a). The C4 carbon signal (acid group) and the C2 carbon signal (amino group) of the thiazole ring are observed at 132.38 ppm and 159.29 ppm respectively [18, 19]. The C5 carbon signal of the thiazole ring without substituent appears at 117.75 ppm and finally, the carbonyl carbon signal of the acid group is observed at 169.98 ppm. For the silylated ligand (Fig. 4b), the appearance of the characteristic signals of the trimethylsilyl group (–Si–(CH3)3) close to 0 ppm and a slight shift in all signals towards high field is observed, being more important for the carbonyl carbon signal (169.98 ppm → 166.58 ppm). In the case of the bis(trimethylsilyl-2-aminothiazole-4-carboxylato) dichlorodioxo molybdenum(VI) complex (Fig. 4c) a general shift of the signals towards the low field and the splitting in two of each signal is observed. The 13C NMR information allows to establish that the complex obtained does not have high symmetry, therefore the structure of Fig. 2 is proposed. Similar implications were observed in 1H NMR using DMSO-d6 as solvent, see SI and Fig. S3.

13C NMR spectrum of a 2-aminothiazole-4-carboxylic acid, b trimethylsilyl-2-aminothiazole-4-carboxylato, c bis(trimethylsilyl-2-aminothiazole-4-carboxylate) dichloro dioxo molybdenum(VI)
3.2 Spectroscopic Characterization of the bis(2-aminothiazole-4-carboxylato)dichlorodioxomolybdenum(VI) Complex Anchored on TiO2 Nanotubes
The maximum amount of the complex supported on the TiO2 was determined by varying the amount used in the anchoring procedure (see Fig. S4). 0.78 mmol of dioxo-Mo complex per g of TiO2 nanotubes is the maximum amount anchored, and for quantities higher of Mo-complex, it was observed the presence of trimethylsilyl groups that do not react with the –OH groups on the surface of TiO2. In the anchoring process, the dioxo-molybdenum(VI) complex with the trimethylsilylated aminothiazole ligand was determined to be covalently linked to the TiO2 nanotubes.
The real amount of complex anchored in the TiO2 nanotubes was determined by C, H, N and S elemental analysis of and was correlated with thermogravimetric analysis. In this way, it was found that Mo is stoichiometrically coordinated to two molecules of the ligand and there is no uncoordinated Mo, in oxide form.
IR Spectrum of the complex anchored showed a shift towards higher wave numbers of the C=O stretching and the disappearance of the (–Si–CH3), (Si–O), and (C–H) characteristic vibration signals of the trimethylsilyl group. Furthermore, the IR ligand signals are maintained, see Fig. 5a. Raman spectrum of the Mo complex anchored on the TiO2 nanotubes displays the characteristic anatase phase peaks at 145 cm− 1 (symmetric stretching vibration of O–Ti–O), 390 cm−1 (symmetric bending vibration of O–Ti–O), 509 cm−1 (antisymmetric bending vibration of O–Ti–O) and 632 cm−1, corresponding to the Eg(1), B1g(1), [A1g + B1g(2)] and Eg(2) modes, respectively [20]. Besides, the peaks of the doublet corresponding to the symmetric and asymmetric stretching of the group Mo(=O)2 at 906 and 937 cm−1 is observed. The characteristic bands of the rocking, bending in the plane and scissoring vibrations of the –NH2 group are also observed at 582, 1409 and 1628 cm−1 [21]. The stretching of the NH2 group asymmetric (3414 cm−1) and symmetric (3404 cm−1) confirm that the coordination of the ligand to Mo occurs via the nitrogen atom of the thiazole ring instead of the –NH2 group. Likewise, the bending and stretching signals of the C–N bond are observed at 370 and 1207 cm−1. The symmetric and asymmetric stretching of the C–S bond is seen at 689 and 765 cm−1. The stretching of the C=N and C=C bonds is observed at 1497 and 1565 cm−1, respectively [22], see Fig. 5b, evidencing the presence of the Mo complex on the TiO2 nanotubes.

a FT-IR and b Raman spectra of the catalyst Mo(atca)2Cl2O2/TiO2-NT
The CP-MAS 13C NMR spectrum of the anchored Mo complex presents the same signals of the complex in solution (Fig. 6). Signal broadening is due to interactions such as chemical shift anisotropy, dipole couplings (homo and heteronuclear), and quadrupole coupling, indicating that the complex obtained does not have high symmetry. When the amounts of complex are greater than 0.78 mmol, the high field signals of the methyl carbons of the trimethylsilyl groups on the TiO2 surface are observed due to limitation in the immobilization of the complex.

CP-MAS NMR 13C spectra of Mo(atca)2Cl2O2/TiO2-NT catalyst
The electronic structure of the TiO2 nanotubes was characterized using diffuse reflectance (DR) UV–Vis spectroscopy (Fig, 7). It has shown a peak below 380 nm wavelength, which means that it exhibits good absorbance at UV region. The absorption threshold present in TiO2-NT spectra is attributed to a O2−:Ti4+ charge transfer of TiO2 [23]. There is no indication of reduced titania sites, because no significant absorption is observed in the visible region, characteristic of d–d transitions. On the other hand, in the the Mo(atca)2Cl2O2/TiO2-NT spectra low-energy absorptions recorded for all compounds between 500 and 600 nm are assigned to ligand-to-metal charge transfer (LMCT) transition arising between ligand molecular orbitals and empty d orbital of the molybdenum atom [8,9,10,11,12].

DRS spectra of TiO2 nanotubes support and Mo(atca)2Cl2O2/TiO2-NT catalyst
XPS analysis of TiO2-nanotube support and the dioxo-Mo(VI) complex anchored was made. Figure 8 shows that the TiO2 support showed Ti, O, C and Na elements on the surface, whereas dioxo-Mo(VI) complex is observed the following elements: Ti, O, C, Na, Mo, S and N. But Cl atoms were not found on the surface of the analyzed materials. Although MoO2Cl2 complex has two chlorine atoms per one molybdenum atom, it is known that Cl atoms in MoO2Cl2 can be displaced by water molecules present in the solvent used for the synthesis or atmospheric water [24], so, Cl atoms are not detected in the proportion expected.

Survey spectrum of a Mo(atca)2Cl2O2/TiO2-NT and b TiO2 nanotubes support
Figure 9a shows the Mo 3d signal recorded for Mo(atca)2Cl2O2/TiO2-NT catalyst. The binding energy found for Mo 2p3/2 was about 231.9 eV. This binding energy value suggest that Mo is present as Mo(VI), similar to the observed in other MoO2Cl2 complex studied in previously works [25]. The shape of Mo 3d signal does not suggest the presence of more Mo species. The proportion of Mo on the surface was about 6.6 Mo atoms per 100 Ti atoms. Besides Mo 3d signal, S 1s was also observed due to this signal is near to Mo 3d signal.

High resolution spectra for a Mo 3d, b Mo 3p and c S 2p recorded in Mo(atca)2Cl2O2/TiO2-NT catalyst
Figure 9b shows the Mo 3p signal which is overlapped with N 1s signal. Area constraints related to orbital spin coupling were used to fit the entire Mo 3p3/2 and Mo 3p1/2 signals, so that N 1s was assumed as the remainder area necessary to fit the observed signal. Figure 9c shows the S 2p signal, thus confirming the presence of the linker in the synthetized catalyst. S/Mo atomic ratio found was about 1.7, which is near to the expected value (2 Sulphur atoms per each Mo atom) according to the stoichiometry of the complex.
3.3 Monoterpenes Photo-Oxidation
The effect of solvent and oxidant agent were evaluated in order to improve the reaction conditions. OAT photo-induced of α-pinene was conducted in various solvents using Mo(atca)2Cl2O2/TiO2-NT as a catalyst in a continuous reaction during 18 h with light and O2.
3.3.1 Effect of Solvent and Oxidant Agent in the α-pinene Photooxidation
The α-pinene conversion at different solvents decreased in the following order: acetonitrile > ethanol > chloroform > dichloromethane, as can be seen in Fig. S5a. Due to the increasing coordination ability of the solvent [26]. Despite obtaining a high conversion with ethanol as solvent, oxidation products of ethanol as acetaldehyde and acetic acid are obtained in smaller quantities. Secondly, when molecular oxygen was changed by tert-butyl hydroperoxide (TBHP) and peroxide hydrogen there was an increase in the conversion of α-pinene. however, the selectivity towards formation of α-pinene oxide decreased (Fig. S5b). When the activity of the Mo(atca)2Cl2O2/TiO2-NT catalyst was evaluated with light and 70 °C with alquildydroperoxides, allylic oxidation products were obtained in higher quantities with the ketone as the main product. Campholenic aldehyde was also observed among the other by-products and the selectivity to the epoxide was as low as 11%. The favored formation of verbenone (and other allylic oxidation products) under radiation in the presence of the Mo(atca)2Cl2O2/TiO2-NT catalyst and H2O2 can be explained by the generation of h+ in TiO2-NT and their further reaction with H2O2, thereby favoring the separation of charge (e-/h+) and allowing the generation of HO• [27]. This results in a poor selectivity to α-pinene oxide and in the preferred formation of oxidized products in alpha position to the double bond, primarily verbenone and verbenol. In addition to the epoxide and verbenone, verbenol and others oxidation products were also detected. Such behavior may be explained by the formation of stable Mo-complex peroxides during the oxidation reaction with H2O2 due to the large positive charge of Mo [28]. Hydrogen peroxide can be linked to the molybdenum in the supported dioxo-Mo(VI) complex leading to an intermediary hydroxido hydroperoxido intermediate that is able to perform the oxygen atom transfer to the olefin to produce the corresponding epoxide. However, the system is not selective under these reaction conditions, thereby explaining such variety in the oxidation products and the resulting decrease in the selectivity to the epoxide.
3.3.2 A Long-Term α-pinene Photooxidation Reaction During 18 h Under O2 Atmosphere and UV Light
Previously, control reactions with α-pinene were performed as follows: (1) catalyst + O2 (without light); (2) light + O2 (without catalyst); (3) Mo(atca)2Cl2O2 (homogeneous complex) + light + O2; (4) Bare TiO2-NT + light + O2. As seen in the Fig. S6, in all cases less than 10% conversion were obtained after 18 h. The results show that the free complex is not able to activate the O2 to carry out the OAT reaction. When the bare TiO2-NT was used the selectivity increased towards the formation of verbenone and verbenol. Plus, the catalytic activity of the Mo(atca)2Cl2O2/TiO2-NT evaluated during 18 h in the presence of molecular oxygen and UV light evidences a photo-stimulated OAT reaction for the Mo-complex anchored.
The 18 h photo-OAT activity with Mo(atca)2Cl2O2/TiO2-NT catalyst with molecular oxygen and UV light shows conversion increased in the following order: β-pinene < camphene < (R)-limonene < α-pinene (Fig. 10) (using equimolar amounts of the supported complexes 1 × 10− 5 moles of dioxo-Mo complex/g-TiO2)), due to reactivity differences of the terpenes and the position of their double bonds, in agreement with the level of substitution of olefinic double bonds, the carbons skeleton and the accessibility of vinyl and allyl hydrogens (for allylic oxidation) [29]. The α-pinene was observed to be the most reactive monoterpene in the series [30]. Epoxidation is favored when the oxygen source has electron accepting groups and when the alkene has electron donating groups. α-pinene is favored compared to the double bond with lower electron density of β-pinene.

Conversion of α-pinene, β-pinene, camphene and (R)-limonene by photostimulated the OAT reactions using Mo(atca)2Cl2O2/TiO2-NT (1 × 10−5 moles of dioxo-Mo complex per gram of TiO2), with UV light and O2 for 18 h of reaction
The more substituted olefinic terpene (character richer in electrons) are more reactive than the less substituted terminal olefins, sometimes the electronic effects may be more important than the steric effects, this may explain the higher conversion obtained for the (R)-limonene and α-pinene with the Mo(atca)2Cl2O2/TiO2-NT catalyst. Likewise, it was found that camphene exhibited intermediate reactivity with respect to β-pinene and (R)-limonene, which may be partially related to the presence of the C=C terminal exocyclic bond. Therefore, it is a less substituted olefin than the other two monoterpenes that contain an endocyclic double bond [31].
The elevated selectivity of epoxides evidences that the Mo(atca)2Cl2O2/TiO2-NT catalyst promotes the OAT photo-induced reaction, see Fig. 11. The formation of allylic oxidation products (ketones and alcohols) is related to the activation of O2 mediated by TiO2 and some traces of OH groups in the organic solvent, but they were obtained in relatively minor amounts for all the terpene olefins studied.

Selectivity observed after the 18 h of reaction of α-pinene, β-pinene, camphene and (R)-limonene with Mo(atca)2Cl2O2/TiO2-NT (1 × 10−5 moles of dioxo-Mo complex per gram of TiO2) using UV light and O2
The notorious catalytic results evidence that the oxidation of monoterpenes using O2 requires the joint participation of semiconductor support and the active center MoO2Ln with an electro-donating ligand, which is activated by UV light to give rise to the effect synergistic or photoinduced TAO that allows increasing the activity of the proposed hybrid catalyst [12].
3.4 Formation of Intermediate Reoxidized Species Mo(O)(O2)
Nam’s group have studied the formation of the oxo-M, peroxo-M, superoxo-M, hydroperoxo-M species under O2 modulated by the ligand structure in the complexes and their amphoteric properties in the electrophilic and/or nucleophilic reactions [32,33,34,35,36,37,38,39]. We have studied the species formed during the reoxidation of the reduced Mo-Complex with O2. Nucleophilic character of peroxo-Mo (electron donor) was evaluated with 2-methyl-1,4-naphthoquinone. The electrophilic character of superoxo-Mo (electron acceptor) complex was evaluated with the oxidation of xanthene [39].
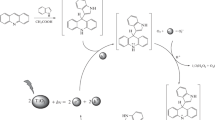
Formation and reactivity of intermediary reoxidized species {Mo(O)(O2)} were evaluated with a solution (1 × 10−2 M) of 2-methyl-1,4-naphthoquinone (MNQ) (electrophilic) and xanthene (nucleophilic) by means a reaction under light + N2 and reoxidation of the catalyst with O2 in darkness. MNQ are formed in a higher proportion than the oxidation products of xanthene. Figure 12 shows 0.5 moles of naphthoquinone oxide formed per mole of Mo complex under UV light and a N2 atmosphere (Step 1). In this case, the stoichiometric OAT occurs for the poor-electron olefins MNQ and the formation of 0.25 moles of xanthone per Mo complex. In second step 2 (O2 + darkness), reoxygenation of reduced oxo-Mo(IV) by O2 occurs. Third step 3 (N2 + light) shows the transfer of two oxygen atoms from the peroxo-oxo-Mo to produce 1 mol of the naphtoquinone oxide per mole of the Mo complex by nucleophilic oxo-peroxo -Mo mediated reaction and 0.5 mol of xanthone per mole of the Mo complex by electrophilic superoxo-Mo mediated reaction. The sequence (N2 + light) and (O2 + darkness) was repeated and it was observed newly the transference of two oxygen atoms to model molecules used, see Fig. 12d and e.

Formation and reactivity of intermediary reoxidized species {Mo(O)(O2)}during the model reaction model of solution (1 × 10− 2 M) of 2-methyl-1,4-naphthoquinone and xanthene with the Mo(atca)2Cl2O2/TiO2-NT: (a) UV light under N2, (b) dark under O2, (c) UV light under N2, (d) dark under O2, and (e) UV light under N2
Previously, Rappé and Goddard [40,41,42] proposed an intrinsic polarity effect on one of the bonds Moδ+–Oδ−, where the second metal-oxo geminal bond partially transfers a charge density giving a triple bond character of the type Mo≡O+. This fact supports the ability of the dioxo-Mo(VI) unit to catalyze nucleophilic and electrophilic reactions in the first step in N2 with light. Reoxidation regenerates the catalysts to form the oxo-peroxo-Mo(VI) unit.
3.5 Spectroscopic Characterization of Reduced and Reoxidised Catalyst
IR and Raman spectroscopic characterization of the Mo(atca)2Cl2O2/TiO2-NT catalyst was performed at the end of each step of the reaction: (N2 + light) and (O2 + darkness). Figure 13 indicates the IR spectra of the following samples: (1)—fresh catalyst, dioxo-Mo(VI), (2)—reduced catalyst, oxo-Mo(IV) (after 5 h of reaction under N2 atmosphere and UV), (3)—reoxidated catalyst, oxo-peroxo-Mo(VI) (after 12 h in the dark under O2), (4)—catalyst samples (4–7) were taken every two hours, after the second step of reoxygenation, step 3 (N2 + UV light).
By comparing the intensity of the Mo(=O)2 bands of the catalysts “fresh” (1) and “reduced” (2), the symmetric stretching vibration at 919 cm−1 disappears due to the oxygen transfer to the α-pinene. Hence, reduced catalyst (2) exhibits a single band of the Mo=O at 951 cm−1 [7,8,9,10,11,12,13]. In the reoxidized catalyst (3) appears the signals of the peroxo group at 524 and 862 cm−1, which corresponds to the (Mo–O2) and (O–O) vibrations respectively [43, 44]. In the case of samples (4–7), it is noted that the peroxo signal decreases (524 and 862 cm−1), and a new signal appears at 1048 cm−1 associated with superoxo group according to Chiang et al. [45] Then, superoxo signal increases up to 4 h of reaction (sample 5) and then decreases (sample 6), and finally, both the peroxo and superoxo signals disappear (sample 7). In addition, the reduced sample (7) is similar to reduced sample (2). Asymmetric (951 cm−1) and symmetric (919 cm−1) peaks of the dioxo unit (Mo(=O)2) are presented in the samples (4), (5) and (6). These IR spectroscopic results explain the pattern observed of nucleophilic to electrophilic oxidation product ratio of the Fig. 12.

FT-IR spectrum of (1)—fresh catalyst, dioxo-Mo(VI), (2)—reduced catalyst, oxo-Mo(IV) (after 5 h of reaction under N2 atmosphere and UV light), (3)—reoxidated catalyst, oxo-peroxo-Mo(VI) (after 12 h in the dark under O2), (4)—the catalyst under N2 atmosphere and UV light: samples (4–7) were taken every two hours, after the second step of reoxygenation, step 3 (N2 + light)
Figure 14 shows the Raman spectra of solids taken at the end of each reaction step described above: the fresh sample (1) presents antisymmetric and symmetric stretching of the dioxo group (Mo(=O)2) at 937 cm−1 and a “shoulder” at 906 cm−1. Sample (2) has only one band at 906 cm−1 due to the loss of an oxygen atom after first OAT reaction under N2, correlated with formation of reduced Oxo-Mo(IV). Sample (3) after reoxidation displays oxo-peroxo-Mo(VI) bands at 584 cm−1 (stretching M–O2) and 861 cm−1 (stretching O–O) [46]. During the OAT under N2, in the samples (4–7), intensity of peaks associated with peroxo-Mo decrease slowly and finally disappeared. After 2 h of reaction, the appearance of a signal between 1110 and 1140 cm−1 is associated with the superoxo-Mo(V) group, with an intensity maximum at 4 h. Finally, bands of the peroxo-Mo(VI) and superoxo-Mo(V) disappear after 8 h of reaction, these Raman results are correlated with the IR characterization of the solids and are evidence of the reactivity observed with the model reaction.

Raman spectra of (1)—fresh catalyst, dioxo-Mo(VI), (2)—reduced catalyst, oxo-Mo(IV) (after 5 h of reaction under N2 atmosphere and UV light), (3)—reoxidated catalyst, oxo-peroxo-Mo(VI) (after 12 h in the dark under O2), (4)—the catalyst under N2 atmosphere and UV light: samples (4–7) were taken every two hours, after the second step of reoxygenation, step 3 (N2 + UV light)
These results allow evidence of the formation of the intermediate species of the catalyst. These species play an important role in the oxidation of monoterpenes since the nucleophilic character of the oxo-peroxo-Mo(VI) species favors the epoxidation reaction. As the terpenic olefin is the nucleophilic reagent, the oxygen transfer step is more favorable in the case of the more substituted, electron richer endocyclic olefins (case of α-pinene and R-limonene) than for the less substituted exocyclic olefins (camphene and β-pinene). Terpenic epoxide formation is based on the OAT to the investigated monoterpenes from the Mo(VI)(atca)2O2Cl2 complex anchored on the TiO2 nanotubes under molecular O2 (as the direct oxidant) and UV irradiation. The oxygen atom transfer process is based on the (nucleophile) olefin attack on an empty (electrophile) Mo=O π* orbital [47] as depicted in Fig. 15 (part A), in agreement with the Dewar–Chatt–Duncanson model for the OAT. Additionally, in this process, the amount of epoxide produced corresponds to the transfer of one oxygen atom from the MoO2Ln (Ln = Bipy) unit (stoichiometric amount of the OAT reaction), thereby resulting in the formation of the reduced anchored oxo-Mo species {MoO(IV)}. Then the Mo = O(IV) specie is reoxidized by molecular oxygen to form an intermediary oxo-peroxo Mo(VI) entity exhibiting a high nucleophilic character [48] (Fig. 15 part B), as could be corroborated in the nucleophilic and electrophilic reactions with MNQ and xanthene. Then, oxo-peroxo Mo(VI) unit reacts with the double bond of the terpenic olefins to form the corresponding epoxides. At this stage, a dioxo-Mo(VI) entity is formed, which is once again able to transfer one oxygen atom to the monoterpenes, thereby leading to the reduced Mo=O(IV) entity. This sequence can be repeated at will in reactions with light and O2.

Illustration of the Oxygen Atom Transfer to the investigated monoterpenes from the Mo(VI)O2Ln entity of the Mo(atca)2Cl2O2/TiO2-NT catalyst under O2 atmosphere and UV irradiation, implying the formation of peroxo-Mo species
3.6 Stability of the Mo(atca)2Cl2O2/TiO2-NT Catalyst
In order to assess the stability and reusability of the Mo(atca)2Cl2O2/TiO2-NT catalyst, a series of three successive runs was performed under O2 and UV–Vis light with (R)-limonene as substrate. The catalyst was separated from the reaction medium by filtration after a catalytic run of 18 h, then washed with acetone and finally dried before being subjected to another cycle of reaction. A new (R)-limonene solution was employed every time. Results presented in Table S1 (SI) indicated that the activity of the dioxoMo(VI) complex immobilized on the TiO2 nanotubes was preserved after the first (R)-limonene photo-epoxidation reaction. However, the selectivity to the limonene-1,2-epoxide was modified and the selectivity towards 1,2-limonene oxide decreases.
Furthermore, the stability of the catalysts was verified by different analyzes regarding their Mo content and the possible leaching of Mo in the supernatant solution. After three consecutive reactions with different (R)-limonene solutions the Mo leaching was about 11%. For this, UV–Vis analysis of the supernatant solution (after filtration of the catalyst, Fig. S7) was performed. The spectra of the solution, once the third reaction is finished, show a band around 200 nm corresponding to the π → π* type transitions of the limonene double bonds [49]. Additionally, the spectrum of the supernatant solution shows the same characteristic band around 600 nm of the LMCT transitions of the complex. At the end of the first reaction, the catalyst was removed and the reaction was continued under irradiation and an O2 atmosphere. In the first hour of reaction, a moderate increase in the conversion of (R)-limonene is observed. These results suggest that Mo leaching occurs in the company of the ligand, forming complexes in solution that allow the OAT process to continue in a homogeneous phase. However, the homogeneous complex is inactivated by the rapid formation of µ-oxo-Mo dimers.
4 Conclusions
The Mo(atca)2O2Cl2/TiO2-NT system with UV light is able to promote the photostimulated OAT reaction allowing the selective oxidation of α-pinene, β-pinene, (R)-limonene and camphene, and permitting the use of O2 as the main oxidizing agent. The selective formation of the epoxide is due to the transfer of oxygen from the dioxo-Mo(VI) unit to the respective monoterpene stimulated by UV light. The role of molecular oxygen is to reoxidize the unit {Mo(IV)=O}, forming oxo-peroxo-Mo which, in the presence of UV radiation, transfers two oxygen atoms to the monoterpene according to our schematic representation of the photostimulated OAT proposed previously [13].
Data Availability
This work was presented to The 6th Latin-American congress of Photocatalysis, Photochemistry and Photobiology LACP3, from 22 to 28 of October of 2021 at Ciudad de México (http://eventos.iingen.unam.mx/7thLACP3/about.html). The results are part of the doctoral thesis of HM.
Code Availability
Not applicable for that section.
References
Hossain MK, Schachner JA, Haukka M, Richmond MG, Mösch-Zanetti NC, Lehtonen A, Nordlander E (2021) Oxygen atom transfer catalysis by dioxidomolybdenum(VI) complexes of pyridyl aminophenolate ligands. Polyhedron 205:115234
Heinze K (2015) Bioinspired functional analogs of the active site of molybdenum enzymes: intermediates and mechanisms. Coord Chem Rev 300:121–141
Eglence-Bakır S, Sacan O, Sahin M, Yanardag R, Ülküseven B (2019) Dioxomolybdenum(VI) complexes with 3-methoxy salicylidene-N-alkyl substituted thiosemicarbazones. Synthesis, characterization, enzyme inhibition and antioxidant activity. J Mol Struct 1194:35–41
Frija L, Pombeiro A, Kopylovich M (2016) Coordination chemistry of thiazoles, isothiazoles and thiadiazoles. Coord Chem Rev 308:32–55
Moradi-Shoeili Z, Zare M, Bagherzadeh M, Kubicki M, Boghaei D (2015) Dioxomolybdenum(VI) complexes with pyrazole based aryloxide ligands: synthesis, characterization and application in epoxidation of olefins. J Coord Chem 68:548–559
Ghorbanloo M, Alamooti AM (2017) Encapsulation of molybdenum(VI) complex withthiazole-hydrazone ligand in zeolite Y: an efficient reusable catalyst for oxidation of primary alcohols and hydrocarbons. J Porous Mater 24:769–777
Páez CA, Castellanos NJ, Martínez F, Ziarelli F, Agrifoglio G, Páez-Mozo EA, Arzoumanian H (2008) Oxygen atom transfer photocatalyzed by molybdenum(VI) dioxodibromo-(4,4’-dicarboxylate-2,2’-bipyridine) anchored on TiO2. Catal Today 133:619–624
Páez CA, Lozada O, Castellanos NJ, Martínez F, Ziarelli F, Agrifoglio G, Páez-Mozo EA, Arzoumanian H (2009) Arylalkane photo-oxidation under visible light and O2 catalyzed by molybdenum(VI)dioxo-dibromo (4,4’-dicarboxylato-2,2’-bipyridine) anchored on TiO2. J Mol Catal A 299:53–59
Castellanos NJ, Martínez F, Lynen F, Biswas S, Van Der Voort P, Arzoumanian H (2013) Dioxygen activation in photooxidation of diphenylmethane by a dioxomolybdenum(VI) complex anchored covalently onto mesoporous titania. Trans Met Chem 38:119–127
Martínez H, Cáceres MF, Martínez F, Páez-Mozo EA, Valange S, Castellanos NJ, Molina D, Barrault J, Arzoumanian H (2016) Photo-epoxidation of cyclohexene, cyclooctene and 1-octene with molecular oxygen catalyzed by dichloro dioxo-(4,4’-dicarboxylato-2,2’-bipyridine) molybdenum(VI) grafted on mesoporous TiO2. J Mol Catal A 423:248–255
Martínez H, Amaya AA, Páez-Mozo EA, Martínez F (2018) Highly efficient epoxidation of α-pinene with O2 photocatalyzed by dioxoMo(VI) complex anchored on TiO2 nanotubes. Micropor Mesopor Mater 265:202–210
Martínez H, Páez-Mozo EA, Martínez F (2021) Selective photo-epoxidation of (R)-(+)- and (S)-(–)-limonene by chiral and non-chiral dioxo-Mo(VI) complexes anchored on TiO2-nanotubes. Top Catal 64:36–50
Martinez H, Alvaro A, Amaya AA, Paez-Mozo EA, Martinez F, Valange S (2021) Photo-assisted O-atom transfer to monoterpenes with molecular oxygen and a dioxoMo(VI) complex immobilized on TiO2 nanotubes. Catal Today 375:441–457
Fen LB, Han TK, Nee NM, Ang BC, Johan MR (2011) Physico–chemical properties of titania nanotubes synthesized via hydrothermal and annealing treatment. Appl Surf Sci 258:431–435
Leong KH, Monash P, Ibrahim S, Saravanan P (2014) Solar photocatalytic activity of anatase TiO2 nanocrystals synthesized by non-hydrolitic sol–gel method. Sol Energy 101:321–332
Dehbanipour Z, Moghadam M, Tangestaninejad S, Mirkhani V, Mohammadpoor-Baltork I (2017) Copper(II) bis-thiazole complex immobilized on silica nanoparticles: preparation, characterization and its application as a highly efficient catalyst for click synthesis of 1,2,3-triazoles. Polyhedron 138:21–30
Almalki S, Bawazeer T, Asghar B, Alharbi A, Aljohani M, Khalifa M, El-Metwaly N (2021) Synthesis and characterization of new thiazole-based Co(II) and Cu(II) complexes; therapeutic function of thiazole towards COVID-19 in comparing to current antivirals in treatment protocol. J Mol Struct 1244:130961–130972
El-Metwaly N, Althagafi I, Khedr A, Al-Fahemi J, Katouah H, Hossan A, Al-Dawood A, Al-Hazmi G (2019) Complexes-dyes and their usage in dyeing cotton to be special bandage for cancerous wounds. J Mol Struct 1194:86–103
Abu-Dief A, El-Metwaly N, Alzahrani S, Alkhatib F, Abualnaja M, El-Dabea T, El Aleem M, El-Remaily A (2021) Synthesis and characterization of Fe(III), Pd(II) and Cu(II)-thiazole complexes; DFT, pharmacophore modeling, in-vitro assay and DNA binding studies. J Mol Liq 326:115277–115291
Hearne GR, Zhao J, Dawe AM, Pischedda V, Maaza M, Nieuwoudt MK, Kibasomba P (2004) Effect of grain size on structural transitions in anatase TiO2: a Raman spectroscopy study at high pressure. Phys Rev 70:134102–134110
Miao SB, Ji BM, Zhou L (2013) Synthesis and crystal structures of a new thiazole-hydrazone ligand and its Co(III) complex. Synth React Inorg Metal Org Nano Metal Chem 43:1296–1300
Chen X, Hu Y, Gao J (2013) Tautomers of 2-aminothiazole molecules in aqueous solutions explored by Raman, SERS and DFT methods. J Mol Struct 1049:362–367
Oja I, Mere A, Krunks M, Nisumaa MR, Solterbeck CH, Es-Souni M (2006) Structural and electrical characterization of TiO2 films grown by spray pyrolysis. Thin Solid Films 515:674–677
Ishikawa S, Maegawa Y, Waki M, Inagaki S (2018) Immobilization of a molybdenum complex on bipyridine-based periodic mesoporous organosilica and its catalytic activity for epoxidation of olefins. ACS Catal 8:4160–4169
Martínez H, Farinha Valezi D, Di Mauro E, Páez-Mozo E, Martínez F (2021) Characterization of peroxo-Mo and superoxo-Mo intermediate adducts in photo-oxygen atom transfer with O2. Catal Today 375:441–457
Niakan M, Asadi Z, Masteri-Farahani M (2019) Immobilization of salen molybdenum complex on dendrimer functionalized magnetic nanoparticles and its catalytic activity for the epoxidation of olefins. Appl Surf Sci 481:394–403
Lachheb H, Guillard C, Lassoued H, Haddaji M, Rajah M (2017) Photochemical oxidation of styrene in acetonitrile solution in presence of H2O2, TiO2/H2O2 and ZnO/H2O2. J Photochem Photobiol A 346:462–469
Jimtaisong A, Luck RL (2006) Synthesis and catalytic epoxidation activity with TBHP and H2O2 of dioxo-, oxoperoxo-, and oxodiperoxo molybdenum(VI) and tungsten(VI) compounds containing monodentate or bidentate phosphine oxide ligands: crystal structures of WCl2(O)2(OPMePh2)2, WCl2(O)(O2)(OPMePh2)2, MoCl2(O)2dppmO2·C4H10O, WCl2(O)2dppmO2, Mo(O)(O2)2dppmO2, and W(O)(O2)2dppmO2. Inorg Chem 45:10391–10402
Carari DM, da Silva MJ (2014) Fe(NO3)3-catalyzed monoterpene oxidation by hydrogen peroxide: an inexpensive and environmentally benign oxidative process. Catal Lett 144:615–622
Bakhvalov OV, Fomenko VV, Salakhutdinov NF (2008) Modern methods for the epoxidation of α- and β-pinenes, 3-carene and limonene. Chem Sustain Dev 16:633–691
Neves P, Gago S, Pereira CCL, Figueiredo S, Lemos A, Lopes AD, Goncalves IS, Pillinger M, Silva CM, Valente AA (2009) Catalytic epoxidation and sulfoxidation activity of a dioxomolybdenum(VI) complex bearing a chiral tetradentate oxazoline ligand. Catal Lett 132:94–103
Cho J, Woo J, Han JE, Kubo M, Ogura T, Nam W (2011) Chromium(v)-oxo and chromium(iii)-superoxo complexes bearing a macrocyclic TMC ligand in hydrogen atom abstraction reactions. Chem Sci 2:2057–2062
de Visser SP, Rohde JU, Lee YM, Cho J, Nam W (2013) Intrinsic properties and reactivities of mononuclear nonheme iron-oxygen complexes bearing the tetramethylcyclam ligand. Coord Chem Rev 257:381–393
Annaraj J, Cho J, Lee YM, Kim SY, Latifi R, de Visser SP, Nam W (2009) Structural characterization and remarkable axial ligand effect on the nucleophilic reactivity of a nonheme manganese(III)-peroxo complex. Angew Chem Int Ed 48:1–5
Yokoyama A, Cho KB, Karlin KD, Nam W (2013) Reactions of a chromium(III)-superoxo complex and nitric oxide that lead to the formation of chromium(IV)-oxo and chromium(III)-nitrito complexes. J Am Chem Soc 135:14900–14903
Jo Y, Annaraj J, Seo MS, Lee YM, Kim SY, Cho J, Nam W (2008) Reactivity of a cobalt(III)-peroxo complex in oxidative nucleophilic reactions. J Inorg Biochem 102:2155–2159
Cho KB, Kang H, Woo J, Park YJ, Seo MS, Cho J, Nam W (2014) Mechanistic insights into the C–H bond activation of hydrocarbons by chromium(IV) oxo and chromium(III) superoxo complexes. Inorg Chem 53:645–652
Ray K, Pfaff FF, Wang B, Nam W (2014) Status of reactive non-heme metal–oxygen intermediates in chemical and enzymatic reactions. J Am Chem Soc 136:13942–13958
Sankaralingam M, Lee Y, Nam W, Fukuzumi S (2018) Amphoteric reactivity of metal–oxygen complexes in oxidation reactions. Coord Chem Rev 365:41–59
Rappé AK, Goddard WA (1980) Bivalent spectator oxo bonds in metathesis and epoxidation alkenes. Nature 285:311–312
Rappé AK, Goddard WA (1982) Olefin metathesis-a mechanistic study of high-valent group VI catalysts. J Am Chem Soc 104:448–456
Rappé AK, Goddard WA (1982) Hydrocarbon oxidation by high-valent group VI oxides. J Am Chem Soc 104:3287–3294
Bagherzadeh M, Zare M, Amani V, Ellern A, Keith WL (2013) Dioxo and oxo-peroxo molybdenum(VI) complexes bearing salicylidene 2-picoloyl hydrazone: structures and catalytic performances. Polyhedron 53:223–229
Wolk AB, Leavitt CM, Fournier JA, Kamrath MZ, Wijeratne GB, Jackson TA, Johnson MA (2013) Isolation and characterization of a peroxo manganese (III) dioxygen reaction intermediate using cryogenic ion vibrational predissociation spectroscopy. Int J Mass Spectrom 354:33–38
Chiang CW, Kleespies ST, Stout HD, Meier KK, Li PY (2014) Characterization of a paramagnetic mononuclear nonheme iron-superoxo complex. J Am Chem Soc 136:10846–10849
Woertink JS, Tian L, Maiti D, Lucas HR, Himes RA, Karlin KD, Neese F, Würtele C, Holthausen MC, Bill E, Sundermeyer J, Schindler S, Solomon E (2010) Spectroscopic and computational studies of an end-on bound superoxo-Cu(II) complex: geometric and electronic factors that determine the ground state. Inorg Chem 49:9450–9459
Rayati S, Rafiee N, Wojtczak A (2012) Inorg Chim Acta 386:27–35
Sisemore MF, Selke M, Burstyn JN, Valentine JS (1997) Inorg Chem 36:979–984
Smialek MA, Hubin-Franskin MJ, Delwiche J, Duflot D, Mason NJ, Vrønning-Hoffmann S, de Souza GGB, Ferreira Rodrigues AM, Rodrigues FN, Limao-Vieira P (2012) Limonene: electronic state spectroscopy by high-resolution vacuum ultraviolet photoabsorption, electron scattering, He(I) photoelectron spectroscopy and ab initio calculations. Phys Chem Chem Phys 14:2056–2064
Acknowledgements
This research was partially supported by Ministerio de Ciencia, Tecnología e Innovación, Ministerio de Educación Nacional, Ministerio de Industria, Comercio y Turismo, and ICETEX, Programme Ecosistema Científico-Colombia Científica from Fondo Francisco José de Caldas; Grant No. RC-FP44842-212-2018. HMQ is grateful to COLCIENCIAS (Doctorados Nacionales 647 program) for his scholarship doctoral. The authors are especially grateful to Laboratorio central de Ciencia de Superficies (SurfLab) of Vicerrectoría de Investigación y extensión at Universidad Industrial de Santander for the XPS analyses.
Funding
The authors thank funding from the Colombian Ministry of Science, Technology and Innovation, the Ministry of Education, the Ministry of Industry, Commerce and Tourism, and ICETEX, Program Ecosistema Científico-Colombia Científica, from the Francisco José de Caldas Fund, Grant No. RC-FP44842-212-2018. H. M. author is grateful to COLCIENCIAS (Doctorados Nacionales 647 program) for the scholarship program.
Author information
Authors and Affiliations
Contributions
All authors contributed equally to this work. HM—prepared, characterized the catalysts, and performed the photo-catalytic measurements and interpretation. AA—analyzed XPS results of the catalysts and contributed to the interpretation of the experimental results. EP and FM—designed the experiments and contributed to the interpretation of the experimental results. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflicts of interest to reveal.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Martinez Quiñonez, H., Amaya, Á., Paez-Mozo, E.A. et al. Aminothiazole Ligand-Type Dioxo-Mo(VI) Complex Anchored on TiO2 Nanotubes for Selective Oxidation of Monoterpenes with Light and O2. Top Catal 65, 1088–1101 (2022). https://doi.org/10.1007/s11244-022-01656-9
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11244-022-01656-9