
Abstract
This paper deals with the kinetics of CH4 dissociation on model Pd/Al2O3 catalysts. The adsorption and conversion of CH4 over oxidized catalysts, fresh or aged, were studied with a temporal analysis of products (TAP) reactor. Single CH4 pulse TAP experiments were performed on a stabilized surface. The experiments are discussed in the light of a selected mechanism involving CH4 decomposition into CO2 taking into account an interaction between the metallic active sites and the support. TAP experiments over the oxidized catalysts confirm the involvement of the metal/support interface, with a spill-over effect. Optimized kinetics parameters validate this interaction of OH species adsorbed on Pd sites with alumina, showing a relatively good agreement between experimental and calculated outlet gas composition. Both fresh and aged catalysts follow the chosen mechanism with the same kinetic constants at 400 °C only an alteration of the catalytic surface areas explain the loss of activity.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
Natural gas vehicles are good choices for high-mileage, centrally fueled fleets that operate within a limited area like buses [1]. Atmospheric pollutant emissions are lowered compared to gasoline or diesel, however unburnt methane emissions contribute to the greenhouse effect. As a consequence efficient end-of-pipe technologies must be implemented, capable to oxidize methane in exhaust gas near stoichiometric conditions [2]. Up to now palladium-based catalysts exhibit a good efficiency [3, 4]. Previous investigations consisted in modelling the outlet flux curve of methane considering elementary steps involved on reduced and oxidized surfaces [5] supposing ageing modifies the active sites and consequently the kinetic constants of one or more steps. Besides kinetic steps over metallic active sites, an interaction with the support is often necessary to explain H2O and CO2 behavior when alumina is present [6]. The main objective of this study is to elaborate a detailed representative mathematical model to simulate CH4 adsorption and conversion on oxidized surfaces, with the production of hydroxyl groups over metallic sites and an interaction with the support for these groups, validated on both fresh and aged catalysts. This model takes into account a spill over pathway for hydroxyl groups from a metallic active site to the support.
2 Materials and Methods
Methane adsorption over a Pd/Al2O3 catalyst was studied on a Temporal Analysis of Products reactor (TAP) at 400 °C. The catalyst was supplied by Umicore A.G. according to a wet impregnation of γ−Αλ2O3 (Sasol, 250 m2 g− 1). The concentration of palladium nitrate solution was adjusted to obtain a precious metal loading of 2.5 wt%. The precursors thus obtained were successively calcined in air at 500 °C and then reduced in pure H2 at 500 °C. Thermal aging was performed at 980 °C in wet atmosphere with 10 vol% H2O diluted in air [6, 7].
Metal dispersion was calculated from H2 chemisorption performed at 100 ◦C, to avoid the formation of palladium hydride, using a pulse technique on catalyst pre-reduced in pure H2 at 500 ◦C and assuming an atomic H/Pd ratio ∼ 1.
The TAP-1 instrument was equipped with two individual high speed pulse valves and enabled in-situ pretreatments at atmospheric pressure. A description of the micro-reactor was reported elsewhere [8]. The micro-reactor was placed in the TAP reactor and heated in vacuum until 550 °C and cooled down to the 400 °C where the catalyst was oxidized under O2 for 50 min. The single pulse experiments consisted of 160 pulses of a mixture containing CH4 in Ar (50 vol%) to monitor 8 AMU in a cycle : m/z = 40 (Ar), 20 (Ne, Ar2+), 15 (CH4), 32 (O2), 44 (CO2), 28 (CO, CO2, N2), 2 (H2, CH4) and 18 (H2O). Each separate cycle, corresponding to a specific AMU value, consisted of 0.1 s of base-line level collection and 3.9 s recording after the pulse, resulting in 20 pulses per AMU. The duration between two successive CH4/Ar pulses was 1 s.
3 Methodology for Kinetic Modelling
The reactor was composed of 5 zones running in isothermal conditions with an inner diameter of the silica insert of 4 mm. For experiments with the fresh catalyst, the micro-reactor comprised a 4 mm layer of quartz wool, a 3 mm layer of inert silica, a 4 mm layer of catalyst (approximately 20 mg), a 5 mm layer of inert silica, and a 4 mm layer of quartz wool. The temperature inside the reactor (TR) was supposed constant along its length, with TR = 0.89 × T, T as the reference temperature of the TAP reactor. As previously found [8], the Knudsen regime predominated. Accordingly, the partial differential equations (PDE) for the concentration of a component i, Ci, in a zone j can be written as follows:
with εj representing the porosity of the zone j (0.4 for silica and catalyst zone, 0.91 for Quartz wool zone), rk, the reaction rate of step k (only considered in the catalyst zone), \({\gamma }_{i,k}\), the stoichiometric number of component i in step k and Di,j, the effective diffusion coefficient of the zone j, related to DAr,j or fixed to 0 for surface species.
At the inlet of the reactor, a flux is considered through:
With Ni, number of molecules in a single pulse, S, cross-section area, τpulse, characteristic opening time of the valve. The outlet concentration was fixed as equal to 0. A 1D diffusion flow model based on COMSOL 5.3 was implemented to determine the pulse characteristics. Kinetic modelling was initially implemented applying first-principles micro-kinetic modeling for activation barriers and conventional transition state theory for pre-exponential factors [5, 8]. Then a sensitivity analysis restrained the number of variables to select the key steps of the catalytic cycle.
4 Results and Discussion
4.1 TAP Experiments
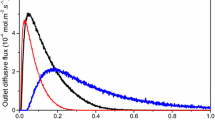
Pulses containing a small amount of reactant (≈ 1.1 × 10− 9 mol of CH4 per pulse) were injected via a high-speed valve into the reactor containing Pd/Al2O3 (with ∼9.2 × 10− 7 mol of accessible Pd sites). In Fig. 1, TAP pulse responses for AMU 15 are plotted as a function of the pulse number. The mean signal obtained over the 20 pulses is within a 10% margin of error considering the minimum and maximum integration value of the independent pulses. In Fig. 2, outlet flux curves of methane are compared to the reference Ar curve and reflect an irreversible adsorption of methane supported by a high methane conversion at 400 °C (from 57.7% for the aged catalyst to 87.9% for the fresh catalyst). Correlatively, CO2 production takes place. A strong tailing of the CO2 response appears on Pd catalyst which is not observed on the bare alumina support. Such a feature reflects reversible chemical processes at the surface including adsorption and diffusion process through the Pd/Support interface. Let us note that the outlet CO2 response underestimates the expected value from methane conversion.

Raw TAP signal for fresh Pd/Al2O3 oxidized catalyst at 400 °C for AMU 15 over the 20 cycles

Experimental outlet flux over Pd/Al2O3 oxidized catalyst at 400 °C for Ar (black line), CH4 (red line) and CO2 (blue triangle). a Fresh catalyst; b aged catalyst
About H2O formation, a first information from the AMU 18 signals indicate the absence of water formation suggesting the hydrogen containing species remain at the surface. However the signal baseline is statistically increased considering the beginning of the experiments and its end suggesting a slight desorption of water. For this work no water formation will be consequently envisaged, only hydroxyl groups storage is occurring. No other species were detected (H2 or CO).
4.2 TAP Diffusion Modelling
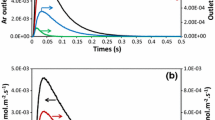
TAP modelling of Ar outlet flux with Comsol was previously discussed [8] and, for this paper, the parameter τpulse, characteristic opening time of the valve in (3), is optimized to fit the maximum value of Ar curve. In Fig. 3, the comparison between experimental and modelled Ar outlet flux for the aged Pd/Al2O3 catalyst matches for τpulse = 18.1 ms and DAr=2,34 cm2/s. Several assumptions, already discussed in [8], impact the modelling as the length of the 5 different zones and the diffusivity parameter of the quartz wool, which depends on its settlement in the reactor. Nevertheless, the implemented method gives a satisfactory agreement for further kinetic modelling.

Experimental (solid line) and calculated (dashed line) outlet flux over aged Pd/Al2O3 oxidized catalyst at 400 °C for Ar with τpulse = 18.1 ms
4.3 Kinetic Modelling of the Metal/Support Interface
1D kinetic modeling of surface reaction with Comsol necessitates the introduction of a concentration of active Pd sites: CPd (mol/m3), which will lead to the surface coverage of adsorbed species, for example with O ad-atoms \({\theta }_{O}= \frac{{C}_{O}}{{C}_{Pd}}\). To determine this concentration of active sites three parameters have to be considered:
-
The metallic Pd surface area, SPd (m2), calculated from H2 chemisorption performed at 100 °C.
-
The catalytic active site density, ΓPd (mol/m2) deriving from the atomic radius.
-
The volume of the reactor, V (m3), restricted to the catalytic bed volume in this case.
Considering two different sites with their concentration, CPd and CSupport, for the metallic Pd sites and for the Support sites respectively, the reverse diffusion of an adsorbed hydroxyl group initially adsorbed on a Pd site to a Support free site requires to take into account 2 adsorbed species (OHads and Al2O3OH for hydroxyl groups adsorbed on Pd and Al2O3 respectively), 2 free sites (Pd(s) and Al2O3) and an adaptation of the rate of reaction as described in Table 1.
The support area is deduced from XPS analysis [7] and is adjusted pondering literature about the interaction between alumina and hydroxyl groups [9, 10] without considering interactions between gaseous H2O and Al2O3. The support area was about 32,000 and 30,000 cm2 for our model corresponding to fresh and aged calculations. Considering Support active sites, the initial surface coverage is supposed to be equally partitioned between Al2O3 and Al2O3OH, with \(\theta _{{A{l_2}{O_3}OH,~~ini}}^{{support}}=0.2\).
Considering Pd active sites, 3 species (amongst 10 detailed in Table 2) are supposed to be initially present:
-
Oxygen ad-atoms, θO,ini, is calculated considering the average conversion over the 160 pulses.
-
Hydroxyl group, θOH,ini, is supposed to be at equilibrium with the support.
-
Pd free sites, θs,ini, so that θO,ini + θOH,ini + θs,ini = 1
4.4 Overall Kinetic Modelling
The dissociation of CH4 over oxidized Pd sites in interaction with the support can be described according to the sequence of elementary steps in Table 2 where some of them have already been discussed [11, 12]. This mechanism consumes O ad-species that will be regenerated once the catalyst is in lean conditions [6]. The production of OH ad-species is favored to hydrogen ad-atoms or Pd free sites to enhance the metal/support interaction with hydroxyl groups. C and CH ad-species were not considered due to the coverage in O and OH of the catalytic surface. Kinetics constants at 400 °C are calculated taking into account pre-exponential factors in respect with the Transition State Theory and activation energies calculated thanks the UBI QEP method [13].
A sensitivity analysis restrained the number of variables to select the key-steps of the catalytic cycle; this explains that backward reactions for steps (S2) to (S7) are neglected. This analysis consists mainly in determining the impact of modifying a parameter (increase and decrease of 10% in the first run) to the conversion of methane.
Experimentally, CO is not detected as a product. Different mechanisms were tested considering CO, for example with a backward reaction for (S7) or with the addition of an adsorption/desorption step, but no difference on CO2 outlet curve was observed.
Only step (S9), and its kinetic constants, k9 and k− 9, were optimized to fit experimental data obtained at 400 °C.
Modelling attempts without step (S9) depended on the initial coverage of OH species on Pd active sites; leading to a total conversion of CH4 if θOH,ini = 0, because the availability of free sites enables the adsorption of methane by step (S1), or to an absence of conversion if hydroxyl groups poison the catalyst. Step (S9) provides an equilibrium that guarantees the reproducibility of the CH4 response over the different experimental pulses.
The computed outlet fluxes, for Ar and CH4, shown in Figs. 4 and 5 were obtained using a Matlab procedure fitting the methane conversion by an in-house modified fminsearch routine with a precision below 1%. For the fresh catalyst, this method gives interesting results, however for the aged catalyst, kinetics parameters should be improved to correct some misfits. Ageing of the catalyst can be modelled considering only a diminution of the active site surface from 455 cm2 for the fresh catalyst to 228 cm2 for the aged one. The modelled outlet curve for CO2 exhibit a total conversion of CH4 into CO2, the metal support interaction modifies the forecast coverage but further work should explain if CO ad-species remains at the surface of the metallic active site or determine the carbonaceous compounds. The formation of H2O has already been discussed [5] and is difficult to monitor experimentally for the selected Pd/Al2O3 catalysts in TAP conditions; in the proposed mechanism, hydroxyl compounds are stored into the support. Water formation has to be envisaged for steady state reactors [14].

Modelled outlet flux over fresh Pd/Al2O3 oxidized catalyst at 400 °C for Ar (black line) and CH4 (blue line) considering kinetic constants of Table 2 and experimental CH4 outlet flux (red line)

Modelled outlet flux over aged Pd/Al2O3 oxidized catalyst at 400 °C for Ar (black line) and CH4 (blue line) considering kinetic constants of Table 2 and experimental CH4 outlet flux (red line)
These results depend on the initial coverage of hydroxyl groups on alumina, assumed at 0.2 in this paper, which is low considering the pre-treatment of the catalyst. The support is not supposed to be free of hydroxyl groups since the heat of adsorption of water on alumina is relatively important which leads to an important storage of hydroxyl groups at the surface [15]. However this coverage is also far from 1 otherwise water would exhibit a negative signal for AMU 18 while reacting with methane [8]. Experiments with water are always complex to set up in a TAP apparatus but a further work with bare alumina should elucidate this point.
5 Conclusions
Numerical simulation of the TAP reactor, including a detailed reaction mechanism for the dissociation of CH4 over an oxidized catalyst at 400 °C, has been used to refine kinetics steps highlighting the mobility of hydroxyl groups.
CO2 formation is modelled considering metallic sites only, further work should include the interaction with the support to explain the strong tailing observed.
H2O formation is excluded towards a reversible storage of hydroxyl groups upon the support, which will limit the metallic free sites available for CH4 adsorption.
The discussion refers to a strong support/noble metal interaction enabling hydroxyl species to spill over the support. The kinetics constants are validated whether the catalyst is fresh or aged, the conversion is only limited by the sintering of the catalyst and the reduction of its reactive surface.
References
EU Project - BRPR960213 (1999) Use of natural gas in passenger cars - components for bifuel vehicles and concepts to handle varying gas compositions
Klingstedt F, Neyestanaki AK, Byggningsbacka R, Lindfors LE, Lundén M, Petersson M, Tengström P, Ollonqvist T, Väyrynen J (2001) Appl Catal A 209:301–316
Farrauto RJ, Hobson MC, Kennelly T, Waterman EM (1992) Appl Catal A 81:227–237
Renème Y, Dhainaut F, Granger P (2009) Top Catal 52:2007–2012
Dhainaut F, Van Veen AC, Pietrzyk S, Granger P (2017) Top Catal 60:295–299
Dhainaut F, Reneme Y, Pietrzik S, Schuurman Y, Mirodatos C, Granger P (2013) Top Catal 56:279–286
Renème Y, Dhainaut F, Frère M, Gengembre L, Granger P, Dujardin C, De Cola P (2010) Surf Interface Anal 42:530–535
Renème Y, Dhainaut F, Pietrzyk S, Chaar M, Van Veen AC, Granger P (2012) Appl Catal B 126:239–248
Peri JB (1965) J Phys Chem 69:211–219
De Boer JH, Fortuin JMH, Lippens BC, Meijs WH (1963) J Catal 2:1–7
Renème Y, Dhainaut F, Schuurman Y, Mirodatos C, Granger P (2014) Appl Catal B 160–161:390–399
Van den Bossche M, Grönbeck H (2015) J Am Chem Soc 137:12035–12044
Shustorovich E, Sellers H (1998) Surf Sci Rep 31:1–119
Granger P, Renème Y, Dhainaut F, Schuurman Y, Mirodatos C (2017) Top Catal 60:289–294
Zamora M, Cordoba A (1978) J Phys Chem 82:584–588
Acknowledgements
Chevreul institute (FR 2638), Ministère de l’Enseignement Supérieur et de la Recherche, Région Nord – Pas de Calais and FEDER are acknowledged for supporting and funding this work.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Dhainaut, F., Granger, P. Kinetic Modeling of the Metal/Support Interaction for CH4 Reaction over Oxidized Pd/Al2O3. Top Catal 62, 331–335 (2019). https://doi.org/10.1007/s11244-018-1125-9
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11244-018-1125-9