
Abstract
This paper deals with the kinetics of CH4 dissociation on a model Pd/Al2O3 catalyst. The adsorption and conversion of CH4 over reduced or oxidized catalysts were studied by applying a temporal analysis of products (TAP) reactor. The experiments are discussed in the light of a selected mechanism involving CH4 decomposition into carbon and hydrogen that cannot be simplified to a global reaction because of the importance of CH2 ad-species. This mechanism was modified for oxidized catalysts including steps for the formation of water involving the recombination of two neighbor OHads species. TAP experiments over the oxidized catalyst confirm the involvement of the metal/support interface, with a spill-over effect previously characterized on the reduced catalyst. Optimized kinetics parameters were compared to theoretical values showing a relatively good agreement.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
Natural gas vehicles (NGVs), are good choices for high-mileage, centrally fuelled fleets that operate within a limited area (which can run on compressed natural gas), or for vehicles needing to travel long distances (which can run on liquefied natural gas) [1]. The advantages of natural gas as transportation fuel include its domestic availability and widespread distribution infrastructure, low cost, and inherently clean-burning qualities. In fact, atmospheric pollutant emissions are lowered compared to gasoline or diesel (no ash or particulate matter during combustion). However unburnt methane emissions contribute to the greenhouse effect. As a consequence efficient end-of-pipe technologies must be implemented, capable to oxidize methane in exhaust gas near stoichiometric conditions. In typical three-way conditions the gas composition oscillates with lean/rich cycles [2]. Up to now palladium-based catalysts exhibit a good efficiency [3, 4]. A previous investigation consisted in modelling the outlet flow rate curve of methane considering a first-order kinetic compared to the concentration of methane [5]. This investigation complete the previous one consisting in integrating elementary steps involved on reduced and oxidized surfaces. The main objective of this study is to elaborate a detailed representative mathematical model to simulate CH4 adsorption and conversion on reduced and oxidized surfaces stabilized in typical TWC conditions.
2 Materials and Methods
Methane adsorption over a Pd/Al2O3 catalyst was studied on a Temporal Analysis of Products reactor (TAP) between 400 and 550 °C. The catalyst was supplied by Umicore A.G. according to a wet impregnation of γ-Al2O3 (Sasol, 250 m2 g−1). The concentration of palladium nitrate solution was adjusted to obtain a precious metal loading of 2.5 wt%. The precursors thus obtained were successively calcined in air at 500 °C and then reduced in pure H2 at 500 °C. Thermal aging was performed at 980 °C in wet atmosphere with 10 vol. % H2O diluted in air [6, 7].
The TAP-1 instrument was equipped with two individual high speed pulse valves and enabled in situ pretreatments at atmospheric pressure. A description of the micro-reactor was reported elsewhere [5]. The micro-reactor was placed in the TAP reactor and heated in vacuum till 550 °C and cooled down to the selected temperature where the catalyst was reduced under H2 for 30 min. The single pulse experiments consisted of 160 pulses of a mixture containing CH4 in Ar (50 vol. %) to monitor m/z = 40 (Ar), 20 (Ne, Ar2+), 15 (CH4), 32 (O2), 44 (CO2), 28 (CO, CO2, N2), 2 (H2, CH4) and 18 (H2O). Other experiments were carried out over oxidized catalysts after heating the micro-reactor under flowing O2 at the selected temperature for 50 min.
3 TAP Modelling with Comsol
The reactor was composed of three zones running in isothermal conditions. As previously found, the Knudsen regime predominated. Accordingly, the partial differential equations (PDE) for the concentration of a component i, Ci, in a zone j can be written as follows:
with ε j representing the porosity of the zone j (0.34 for Quartz zone, 0.93 for catalyst zone), r k , the reaction rate of step k (only considered in the catalyst zone), and D i , the effective diffusion coefficient, related to D Ar or fixed to 0 for surface species.
At the inlet of the reactor, a flux is considered:
with N i , number of molecules in a single pulse, S, cross-section area, τ pulse , characteristic opening time of the valve. The outlet concentration was fixed as equal to 0. A 1D diffusion flow model based on COMSOL 4.4 was implemented to determine the diffusion coefficients.
4 Results and Discussion
4.1 Single CH4 Pulse Experiment on Reduced Pd/Al2O3
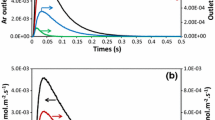
Pulses containing a small amount of reactant (≈ 1.1 × 10−9 mol of CH4 per pulse) were injected via a high-speed valve into the reactor, which contained the catalyst (with ~ 8.2 × 10−9 mol of accessible Pd sites). In Fig. 1, outlet flow rate curves of methane are compared to the reference Ar curve and reflect an irreversible adsorption of methane supported by a high methane conversion (from 52 % at 400 °C to 88 % at 500 °C). Correlatively, H2 production takes place. The strong tailing of the H2 response means a reversible interaction with the catalyst, whether from the noble metal active sites or their interaction with alumina because this behavior is not obtained on the support without Pd.

Experimental outlet flux over Pd/Al2O3 reduced catalyst at 400 °C for Ar (black line), CH4 (red line) and H2(blue line)
The dissociation of CH4 over metallic Pd sites can be described according to the sequence of elementary steps in Table 1 where some of them have already been discussed [8]. Pre-exponential factors, as well as activation energies, of the different steps were calculated thanks the UBI QEP method [9]. A sensitivity analysis restrained the number of variables to select the key-steps of the catalytic cycle. Only step (3), and its forward kinetic constant, k3, was optimized to fit experimental data obtained between 400 and 500 °C. The initial coverage of carbon ad-atoms, θC,ini, was calculated considering the average conversion over the 160 pulses. Even if θC,ini can be considered as negligible (≈ 0.08), the optimized values depend on the carbon coverage.
The computed outlet fluxes, for Ar and CH4, shown in Fig. 2 are in good agreement with the experimental values. However the outlet curve for H2 cannot be completely model from the mechanism reported in Table 1 involving only Pd sites, the support may also participate through H2 spillover effect from the metal to the alumina support as reported elsewhere [8]. In order to get a better description O’Connor et al. took into account these steps [10] on Pt/Al2O3:

Simulated outlet flux over Pd/Al2O3 reduced catalyst at 400 °C for Ar (black line), CH4 without reaction (red dotted line), CH4 with reaction (red line), H2(blue line)
The integration of Alumina as a reservoir for OH species in the model could be interesting for a better simulation of the experiments, but, in this case where the catalyst is reduced, another step may be required because no water formation is observed during these experiments:
This step would also explain the incomplete experimental mass balance in H2, where almost 50 % of expected H2 is lacking. In a previous study of partial oxidation of methane, at higher temperature, Buyevskaya et al. already pointed out the importance of the alumina interaction with water [11].
4.2 Single CH4 Pulse Experiment on Oxidized Pd/Al2O3
Methane conversion is not dependent upon the pre-treatment: 57 % over the oxidized catalyst at 400 °C, compared to 52 % for the reduced catalyst (80 % compared to 82 % at 550 °C). As expected, the products should be different due to the consumption of active oxygen leading to COx and water. In our conditions, no CO was detected and the production of H2O was difficult to monitor due to possible re-adsorption on alumina and spill-over from the support to noble metals further dissociating to produce OH ad-atoms. In Fig. 3, the experimental outlet curve for CO2 is similar to the H2 profile obtained on the reduced catalysts with a mass balance in CO2 incomplete.

Experimental outlet flux over Pd/Al2O3 oxidized catalyst at 400 °C for Ar (black line), CH4 (red line) and CO2(blue line)
When the surface is pre-covered by oxygen different elementary steps can be involved which account for the formation of CO2 and H2O (Table 2). The reaction mechanism was modified to neglect elementary steps that are not relevant to model the outlet flow curves. One of the most important finding is that the interaction of H2O with the catalyst cannot be omitted: methane conversion and long tailing of water signal depend on the forward reaction of steps (12) and (13). On the other hand, variations of the sticking coefficient of CO2 and the activation energies dedicated to CO and CO2 in a large extent do not significantly influence to optimized results. The problems arisen by water and carbon dioxide were also discussed by Burch et al. [12, 13], the steps (11) and (15) are responsible for such a behavior.
The initial coverage of O ad-atoms was calculated considering the average conversion over the 160 pulses. As a first attempt, the same kinetic constant for step (3) was optimized, leading to values divided by a factor 10 compared to the reduced catalyst.
Since water is not detected during the TAP experiments but continuously desorbs, because the baseline of m/z = 18 increases continuously along the pulses, a second attempt took into account the optimization of the initial O ad-atoms coverage assuming that the kinetic constant of step (3), k3, is identical whether the catalyst is reduced or oxidized.
The optimized parameters (kinetic constant of step (3) or initial coverage in O ad-atoms) of Table 3 were obtained using a Matlab procedure fitting the methane conversion over the different temperatures. The computed outlet fluxes, for Ar and CH4, shown in Fig. 4b are in better agreement with the experimental values than Fig. 4a, neglecting the outlet curve for CO2 and H2O which cannot be explained totally by the mechanism of Table 2.

Simulated outlet flux over Pd/Al2O3 reduced catalyst at 400 °C for Ar (black line), CH4 (red line), CO2(blue line) and H2O (gray line). a considering the optimisation of the pre-exponential factor of step (3). b considering the optimisation of the initial coverage of O-adatoms
The calculated activation energy for step (3) of 44.7 kJ/mol can be compared with the UBI-QEP method estimate of 80 kJ/mol. As observed significant difference are noticeable with a much lower experimental value. Such an apparent divergence can be related to the fact that the UBI-QEP calculations account for a Pd(111) surface at zero coverage which could not be representative of polycrystalline Pd/Al2O3 catalyst. Indeed, single TAP experiments were performed on a stabilized surface after 160 methane pulse. Even, if carbon coverage is likely low, carbon contamination could induce a surface coverage dependency of the activation energy as previously discussed. Finally, as argued, the metal/support interaction likely play a key role and could lower this value as previously demonstrated [8].
5 Conclusions
Numerical simulation of the TAP reactor, including a detailed reaction mechanism for the dissociation of CH4 over a reduced and oxidized catalyst, has been used to refine kinetics steps.
The dissociation of CH4 is affected by the degree of surface reduction of the catalyst. Over reduced catalyst, H2 is formed as primary product by dehydrogenation of CH4 with carbon containing species deposition on the surface of the catalyst. The mechanism implemented in this paper shows the important role of CHx species to adjust experimental responses to simulated outlet fluxes.
In the temperature range of 400–550 °C, the kinetic steps of the catalyst have been considered unchanged according to the thermal pretreatment in reductive or oxidative conditions. Over oxidized catalysts, the formation of CO2 is not influenced by the kinetic parameters which is not the case for H2O.
The discussion refers to a strong support/noble metal interaction, whether for reduced or oxidized catalyst. Consequently further work should take into account hydroxyl groups from alumina, so that there will be a competition for adsorbed hydroxyl groups between reacting to form water or migrating to alumina. This competition will lead to a change in the H2O outlet flux curve.
References
EU Project—BRPR960213 (1999) Use of natural gas in passenger cars—components for bifuel vehicles and concepts to handle varying gas compositions
Klingstedt F, Neyestanaki AK, Byggningsbacka R, Lindfors LE, Lundén M, Petersson M, Tengström P, Ollonqvist T, Väyrynen J (2001) Appl Catal A 209:301–316
Farrauto RJ, Hobson MC, Kennelly T, Waterman EM (1992) Appl Catal A 81:227–237
Renème Y, Dhainaut F, Granger P (2009) Top Catal 52:2007–2012
Renème Y, Dhainaut F, Pietrzyk S, Chaar M, Van Veen AC, Granger P (2012) App Catal B 126:239–248
Renème Y, Dhainaut F, Frère M, Gengembre L, Granger P, Dujardin C, De Cola P (2010) Surf Interface Anal 42:530–535
Dhainaut F, Reneme Y, Pietrzik S, Schuurman Y, Mirodatos C, Granger P (2013) Top Catal 56:279–286
Renème Y, Dhainaut F, Schuurman Y, Mirodatos C, Granger P (2014) App Catal B 160–161:390–399
Shustorovich E, Sellers H (1998) Surf Sci Rep 31:1–119
O’Connor AM, Schuurman Y, Ross JRH, Mirodatos C (2006) Catal Today 115:191–198
Buyevskaya OV, Wolf D, Baerns M (1994) Catal Lett 29:249–260
Burch R, Urbano F, Loader P (1995) Appl Catal A 123:173–184
Burch R, Loader P, Urbano F (1996) Catal Today 27:243–248
Author information
Authors and Affiliations
Corresponding author
Additional information
To the memory of Stan, died in December, 2014.
Rights and permissions
About this article

Cite this article
Dhainaut, F., van Veen, A.C., Pietrzyk, S. et al. CH4 Dissociation Mechanisms on Aged Three-Way Natural Gas Vehicle Pd/Al2O3 Catalyst. Top Catal 60, 295–299 (2017). https://doi.org/10.1007/s11244-016-0614-y
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11244-016-0614-y