
Abstract
The present study is focused on the investigation of the low temperature Standard SCR reaction mechanism over Fe- and Cu-promoted zeolites. Different techniques are employed, including in situ DRIFTS, transient reaction analysis and chemical trapping techniques. The results present strong evidence of nitrite formation in the oxidative activation of NO and of their role in SCR reactions. These elements lead to a deeper understanding of the standard SCR chemistry at low temperature and can potentially improve the consistency of mechanistic mathematical models. Moreover, comprehension of the mechanism on a fundamental level can contribute to the development of improved SCR catalysts.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
Compliance with the current NOx emission limits for diesel vehicles requires highly efficient NH3-SCR systems. Investigation of the fundamental aspects of SCR chemistry is crucial for the development of improved catalysts and for the formulation of chemically and physically consistent mathematical models, which are essential tools for the design of SCR devices. Indeed, the catalytic mechanism governing low-temperature standard SCR activity is still debated: specifically, different hypotheses about the nature of the reaction intermediates have been proposed, including NO+/nitrites (formal N oxidation state = +3) [1–3] and nitrates (N = +5) [4]. According to recent in situ DRIFT studies, an NO+/nitrite species can be identified as a common reaction intermediate for both NO oxidation and standard SCR on Cu-zeolite catalysts [1]. In fact, we also observed formation of NO+ species in our previous in situ DRIFT study [5] of NOx (NO/NO + O2/NO2) adsorption on a Cu-CHA catalyst. Based on the IR spectroscopic evidence, we proposed a redox mechanism for NO oxidation implying NO+/nitrites species formation as a key step in the process. On the other hand, recent studies of the NO oxidative activation at low temperature on an Fe-ZSM-5 catalyst, based on chemical trapping techniques, provided clear evidence for the formation of nitrites, suggesting that such adspecies may play the role of reaction intermediates involved in both NO oxidation and standard SCR reactions over such catalysts [6, 7].
In this study, we focus on a commercial state-of-the-art Cu-CHA catalyst. Different techniques (transient reaction analysis, ex situ FTIR and in situ DRIFT measurements, chemical trapping techniques) are applied with the aim to conclusively identify the primary intermediates in the SCR reactions. In addition, we also compare the present results with data collected on an Fe-zeolite catalyst by chemical trapping in order to highlight possible analogies and differences in the mechanism of the SCR reactions over the two metal-promoted zeolite catalysts [2, 8].
2 Materials and Methods
In situ-DRIFT measurements were carried out on a slab cut from a commercial monolith catalyst wash coated with Cu-CHA. Nitric oxide adsorption experiments in the presence of 8 % v/v O2 were run on a pre-oxidized catalyst sample at 150 °C, both without and with pre-adsorbed NH3. The pre-oxidizing treatment, carried out prior to each experiment, consisted of feeding 8 % O2 in Ar for 1 h at 500 °C, followed by cool down of the sample to the desired temperature while keeping the O2 concentration constant. The experiments were run at 150 °C, due to the main interest in elucidating the low temperature NH3-SCR mechanism. Each DRIFT spectrum was collected by averaging 8 scans with a 2 cm−1 resolution: this ensured a high temporal resolution when following the surface species dynamics (averaged spectra recorded every 4 s), while maintaining a reasonable signal-to-noise ratio. In the case of runs with pre-adsorbed NH3, DRIFT measurements were coupled with gas phase analysis using a mass spectrometer to monitor N2 formation. More details concerning the equipment used for these experiments can be found in [5].
Adsorption of NOx (NO or NO2) + O2 was performed at 120 °C on mechanical mixtures comprising either an Fe-ZSM-5 or a Cu-CHA catalyst and BaO/Al2O3. The BaO, a well known NOx trap, was employed to capture unstable reaction intermediates resulting from the oxidation of NO over Fe or Cu or from NO2 adsorption. Reaction intermediates were then identified by the following methods: (i) analyzing their thermal decomposition products in TPD runs, (ii) ex situ FTIR analysis of the BaO/Al2O3 phase separated from the mixture, and (iii) probing their reactivity with gas phase NH3. Additional details on the procedure and reactor used during such chemical trapping experiments can be found in [9].
3 Results and Discussion
3.1 In situ DRIFT Study
Figure 1a shows the temporal evolution of DRIFT spectra collected during NO + O2 adsorption at 150 °C on a pre-oxidized Cu-CHA catalyst sample. Already after 3 min of NO adsorption, bands located in the 1700–1400 and 2200–2100 cm−1 spectral regions appeared and then progressively grew in intensity. In the 1700–1400 cm−1 range, multiple bands can be distinguished, namely three distinct peaks centered at 1620, 1590 and 1570 cm−1, appeared after longer exposure times (15 min). A broad symmetric peak, centered at 2140 cm−1, was already visible after 3 min. Moreover, a shoulder at 2190 cm−1 appeared in the spectra after longer exposure times. These results are consistent with previous work [5], in which the results of NOx + O2 adsorption at 120 °C on a pre-oxidized or pre-reduced Cu-CHA catalyst evidenced the formation of NO+ and nitrates (bands at 2140 and 1620/1590/1570 cm−1, respectively, in Fig. 1a) as the dominant adspecies. However, different dynamic behaviors prevailed depending on the species present in the gas phase (NO2, NO + O2, NO on a pre-oxidized catalyst). In particular, in this case, nitrates were formed subsequently to NO+, as apparent in Fig. 1b showing the peak area evolution during NO + O2 adsorption. This points to the presence of an oxidized intermediate in the process of NO oxidation to surface nitrates, with a formal N oxidation state = + 3. Figure 1b shows that NO+ is relatively stable, since its presence was still detected on the catalyst surface once NO was removed from the gas phase. Due to its formal N oxidation state, NO+ can be regarded as a nitrite-like species.

a IR spectra during NO + O2 adsorption at 150 °C on a pre-oxidized Cu-CHA sample. b Peak area dynamics of NO+ (right y axis) and nitrate species (left y axis) during NO + O2 adsorption on a pre-oxidized Cu-CHA sample
The reactivity of surface species formed upon NO + O2 interaction with the catalyst surface has been also studied in the presence of pre-adsorbed NH3. Figure 2a shows selected spectra for an experiment in which NO + O2 were co-fed to the Cu-CHA catalyst after NH3 adsorption. The figure shows spectra after NH3 adsorption for 20 min, after a purge in Ar, during NO + O2 adsorption and after NO + O2 shut off. NH3 adsorption on the Cu-CHA catalyst resulted in typical features associated with NH3 bonded to Lewis acidic sites (peaks located at 1440 cm−1 and at high wavenumbers, 3340 and 3190 cm−1) and NH4 + (peak centered at 1630 and 3270 cm−1) [10–12]. Bands with low wavenumbers are determined by the N–H bending vibrations in the adsorbed NH3, whereas other bands located at 3340/3270/3190 cm−1 represent the stretching vibrations of the N–H bonds. In addition, as a result of NH3 adsorption, other negative peaks are detected at wavenumbers >3600 cm−1 and are assigned to Si–OH–Al (3600 cm−1) and to Si–OH bonds (3720 cm−1), suggesting displacement of OH groups during NH3 adsorption. By comparing the spectra obtained immediately after NH3 adsorption and after the purge in Ar, we can conclude that the species formed on the catalyst surface upon NH3 adsorption are stable: only a small decrease in the intensity of the bands corresponding to adsorbed NH3 and the Si–OH–Al bond can be noticed.

Reaction of NO + O2 with pre-adsorbed NH3 at 150 °C over Cu-CHA. a Resulting spectra after NH3 adsorption (20 min), after removing NH3 from the gas phase (40 min), feeding NO + O2 (53 min) and after removing NO + O2 (66 min); b gas phase concentration profiles during NO + O2 pulse
The situation changed completely when NO + O2 was fed to the catalyst; both the bands associated with Lewis bonded ammonia and Brønsted bonded NH3 decreased. Accordingly, also the 3340/3270/3190 cm−1 bands decreased. Moreover, the negative peak corresponding to the Si–OH–Al bond decreased in intensity. Overall, this suggests that the NH3 prestored on the catalyst was depleted by reaction with NO. This observation is supported by the gas phase analysis shown in Fig. 2b; as soon as the NO + O2 mixture was fed to the reactor, N2 was produced and detected by the mass spectrometer.
After NO + O2 shut off, all of the adsorbed NH3 was consumed, and nitrates were eventually detected (bands at 1570–1620 cm−1) at later times, as shown in Fig. 2a; finer temporal scanning was used to resolve the transition between these indicators of adsorbed surface species. It is particularly important to emphasize that nitrate species were detected only once all the NH3 pre-adsorbed on the catalyst was consumed, as this suggests that they do not play any role in the reaction which depletes NH3 from the catalyst surface forming the N2 detected in the gas phase. Indeed, nitrate species are reported to be stable [3–5] and thus, if they were formed during the reaction between NO + O2 and surface NH3, they would be easily detected.
3.2 Chemical Trapping Study
In order to better identify the role of nitrite and nitrate adspecies in SCR catalysis, additional chemical trapping experiments were performed on the Cu-CHA catalyst and compared to similar experiments over an Fe-ZSM-5 catalyst.
In particular, we studied the interaction of NO + O2 with mechanical mixtures of either Cu-CHA and BaO/Al2O3 or Fe-ZSM-5 and BaO/Al2O3, as well as with the individual components of the powder mixtures. Figure 3a illustrates the adsorption dynamics of NO + O2 onto the Cu-CHA + BaO/Al2O3 mechanical mixture at 120 °C. A delay in the outlet NO concentration profile is apparent with respect to the feed, suggesting storage of surface species. Notably, no significant storage took place on the individual components of the mechanical mixture. Figure 3a indicates also NO2 production, ascribable to NO oxidation to NO2. In an analogous experiment carried out on the Fe-ZSM-5 + BaO/Al2O3 mechanical mixture, NO outlet traces similarly showed first a delay and then dynamic behavior consistent with a significant quantity of stored surface species [7].

a NO + O2 adsorption dynamics at 120 °C on a pre-oxidized Cu-CHA mixed with BaO/Al2O3, b TPD run in He following NO + O2 adsorption at 120 °C on Cu-CHA + BaO/Al2O3 and on Fe-ZSM-5 + BaO/Al2O3 physical mixtures
Identification of the species formed on the catalyst upon NO + O2 adsorption was initially performed by analysis of their decomposition profiles during TPD. Figure 3b shows the TPD profiles obtained in the case of the Cu-CHA + BaO/Al2O3 (solid lines) and the Fe-ZSM-5 + BaO/Al2O3 (dashed lines) mechanical mixtures after isothermal adsorption of NO + O2 at 120 °C; in both cases NO and NO2 were desorbed in equimolar amounts with a maximum at 200 °C, in line with the thermal decomposition of nitrites/HONO [7]. Again, the TPD performed after isothermal adsorption of NO + O2 on the individual components of the mechanical mixtures at the same conditions confirmed no appreciable storage of NOx in these cases. This suggests that nitrites are initially formed on the metal active sites of either the Cu-CHA or the Fe-ZSM-5 catalysts, and then trapped and stabilized on the barium phase. The interaction between the two phases may proceed via gaseous HONO (in equilibrium with nitrites) or via direct solid phase interaction. Indeed, the TPD profiles obtained for the two catalysts are quite similar, suggesting that the decomposition behavior is dominated by the BaO phase, which is identical in the two mechanical mixtures. In the case of the Cu-CHA catalyst, however, we can observe a slightly higher NO2 release between 250 and 400 °C, which can be ascribed to the presence of small amounts of nitrates formed due to the presence of gas phase NO2, formed by NO oxidation to NO2. This observation is confirmed: (i) by the evolution of gaseous NO2, though only in trace quantities, observed during the isothermal adsorption of NO + O2 on the Cu-CHA + BaO/Al2O3 mechanical mixture (Fig. 3a); (ii) by the in situ DRIFT data in Fig. 1, which show nitrate formation on the Cu-CHA catalyst under the presence of NO + O2. Moreover, in situ DRIFT experiments clearly point out that nitrates are formed sequentially to NO+/nitrites, and this strongly suggests that nitrates could be formed as a consequence of further oxidation of NO+ species, e.g. after production of gas phase NO2 and its disproportionation [5, 13].
Presence of nitrites on the BaO component of the mechanical mixtures was also confirmed by ex situ FTIR analyses of the BaO/Al2O3 phases separated from the physical mixtures after exposure to NO + O2 (1090, 1220, 1270, 1350 cm−1 in Fig. 4a and 4b for the Cu-CHA and the Fe-ZSM-5 containing mixture, respectively). As previously discussed, small amounts of nitrates were present on the Cu-CHA + BaO/Al2O3 mechanical mixture (1640–1540–1380 cm−1), as indicated by the TPD profile. On the contrary, Fig. 4b shows that almost negligible amounts of nitrates were detected on the BaO component of the Fe-ZSM-5 containing mixture. This is consistent with the Fig. 3b TPD results, and such a difference can be related to the greater stability of nitrite/nitrate species on Cu- than on Fe-zeolite catalysts, as reported in the literature [12, 13].

Ex situ IR analysis of a BaO/Al2O3 component from the Cu-CHA containing mixture, b BaO/Al2O3 component from the Fe-ZSM-5 containing mixture
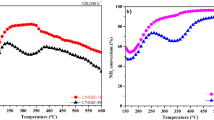
Storage of nitrites on the BaO phases upon NO + O2 adsorption, as well as their role in the SCR mechanisms, was eventually confirmed by their reactivity with NH3 to give standard SCR products (N2 and H2O), as shown in Fig. 5. Initially, NO + O2 was fed to the mechanical mixture at 120 °C, in order to activate NO to surface nitrites and store them on the BaO phase of the mechanical mixture. Then, NH3 was fed to the mechanical mixture and after an isothermal steady state phase, an NH3-TPSR was performed in order to investigate the reactivity of the more stable surface species. In the case of the Cu-CHA + BaO/Al2O3 mixture (Fig. 5a), no N2 production was observed at 120 °C. However, upon increasing the temperature, NH3 consumption (and release) and N2 production were observed in the range 120–140 °C. Such a low-temperature reactivity can be ascribed to nitrite species; indeed, at these temperatures, nitrates are known to be nonreactive with NH3. Similarly, the reactivity of nitrites with NH3 was studied also in the case of the Fe-ZSM-5 + BaO/Al2O3 mixture, as shown in Fig. 5b. Again, a significant N2 production was observed in the range 120–140 °C. In this case, however, N2 production was already active during isothermal NH3 feed at 120 °C. This difference with respect to the Cu-CHA containing mixture could be due to a different stability of nitrite species on the Cu- and Fe-zeolite catalysts. This indicates that, contrary to what occurs during the thermal decomposition of nitrites (Fig. 3a), their reactivity with ammonia is controlled not by the BaO/Al2O3 phase, but by the catalytic component (Cu-CHA or Fe-ZSM-5).

NH3 TPSR after NO + O2 adsorption on: a Cu-CHA + BaO/Al2O3, b Fe-ZSM-5 + BaO/Al2O3 mechanical mixture
4 Conclusions
In the present study, in situ DRIFT measurements are coupled with transient reaction analysis and complemented by chemical trapping experiments with the aim to identify elusive unstable SCR reaction intermediates resulting from the activation of NO over Cu-CHA, and to compare the results with previous studies over an Fe-ZSM-5 catalyst. Based on a variety of experiments, we present evidence of nitrite formation in the oxidative activation of NO over both metal-promoted zeolites, and therefore propose their general role in the mechanism of SCR reactions. The collected experimental evidence also appears to indicate that nitrates do not have a role as primary intermediates in the NO oxidation reaction nor as key reactive adspecies in the standard SCR reaction. The present data therefore support a standard SCR mechanism based on a “nitrite route”. Moreover, differences noted in the stability of nitrites could possibly account for the different activity of Fe- and Cu-zeolite catalysts. The improved understanding of the standard SCR chemistry at low temperature will play a crucial role in the formulation of a coherent mechanism and in the development of improved metal exchanged zeolite catalysts.
References
Kwak JH, Lee JH, Burton SD, Lipton AS, Peden CHF, Szanyi J (2013) Ang Chem Int Ed 52:9985–9989
Ruggeri MP, Nova I, Tronconi E (2013) Top Catal 56:109–113
Doronkin DE, Casapu M, Günter T, Müller O, Frahm R, Grunwaldt J-D (2014) J Phys Chem C 118:10204–10212
Wang D, Zhang L, Kamasamudram K, Epling WS (2013) Acs Catal 3:871–881
Ruggeri MP, Nova I, Tronconi E, Pihl JA, Toops TJ, Partridge WP (2015) Appl Catal B 166:181–192
Ruggeri MP, Selleri T, Colombo M, Nova I, Tronconi E (2015) J Catal 328:258–269
Ruggeri MP, Selleri T, Colombo M, Nova I, Tronconi E (2014) J Catal 311:266–270
Colombo M, Nova I, Tronconi E (2010) Catal Today 151:223–230
Wallin M, Karlsson C-J, Palmqvist A, Skoglundh M (2004) Top Catal 30:107–113
Sjövall H, Blint RJ, Olsson L (2009) Appl Catal B 92:138–153
Zhu H, Kwak JH, Peden CHF, Szanyi J (2013) Catal Today 205:16–23
Colombo M, Nova I, Tronconi E (2012) Appl Catal B 111:433–444
Grossale A, Nova I, Tronconi E, Chatterjee D, Weibel M (2009) Top Catal 52:1837–1841
Acknowledgments
This manuscript has been co-authored by UT-Battelle, LLC, under Contract No. DE-AC0500OR22725 with the U.S. Department of Energy. The United States Government retains and the publisher, by accepting the article for publication, acknowledges that the United States Government retains a non-exclusive, paid-up, irrevocable, world-wide license to publish or reproduce the published form of this manuscript, or allow others to do so, for the United States Government purposes. The Department of Energy will provide public access to these results of federally sponsored research in accordance with the DOE Public Access Plan (http://energy.gov/downloads/doe-public-access-plan).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Ruggeri, M.P., Selleri, T., Nova, I. et al. New Mechanistic Insights in the NH3-SCR Reactions at Low Temperature. Top Catal 59, 907–912 (2016). https://doi.org/10.1007/s11244-016-0567-1
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11244-016-0567-1