
Abstract
A series of organic sulfur-functionalized trans-platinum(II) bis(alkenylarylalkynyl) complexes, having one anisolylthio group with general formula trans-[(PEt3)2Pt{C≡C–Ar–CH=CH(SC6H4–OCH3)}2], (2a-2d), (Ar = phenylene/biphenylene/2,5-dimethylphenylene/2,5-dimethoxyphenylene) was synthesized in excellent yields. All the new platinum(II) complexes have been fully characterized by physico-chemical and spectroscopic methods. Photophysical properties of the complexes were studied by absorption and emission spectroscopy. The lowest energy absorption band for the complexes, in the UV/Vis spectra, in THF solution, at room temperature, 2a-2d was observed in the range 355–391 nm, which depend on the spacers in the acetylide ligand e.g., the absorption of 2d is red shifted to 391 nm for the donor (OCH3) substituents in phenyl spacer. These absorptions originated predominantly from π–π*orbitals of acetylide ligand with significant contribution from the platinum dπ orbital as evident from the HOMO and LUMO, obtained from TD-DFT calculations. Fluorescence was observed in all complexes at room temperature in the range 400–428 nm with PLQY of 2–5%. At 77 k, the complexes 2a-2b only exhibited phosphorescence in the range 579–585 nm, but there is no phosphorescence at ambient temperature.
Graphical abstract

Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
There has been a lot of interest in the synthesis of small, medium, and large π-conjugated acetylenic materials due to the growing interest in the production of novel classes of materials with exciting optical properties and applications [1,2,3]. The potential uses of transition metal acetylide complexes and polymers in various areas of materials science, such as organic light emitting diodes [4, 5], photovoltaic cells [6,7,8], and field-effect transistors [9], have sparked significant interest concurrently. Over the last three decades, there has been a growing interest in the design of conjugated materials that exhibited a range of material properties such as linear and non-linear optics [10, 11], organo-gelators [12, 13], liquid crystals [14, 15], photovoltaic behavior [16,17,18], depending on the structure of the molecules, and the nature of the intermolecular interactions as well as their long-lived triplet excited states [19, 20]. The unique physical properties of transition metal acetylides are caused by their rigid rod structures and prominent π-electron conjugation involving the metal’s d-electrons and the π-system of the carbon–carbon triple bond [1,2,3]. On the other hand, the chemical and physical properties of carbon-rich organic oligomers often cannot be exploited due to their poor solubility, but this can be improved by incorporating alkyl substituent’s on the ligand as well as metal center on the oligomeric backbone [21]. The presence of platinum as a heavy metal induces strong spin–orbit coupling which in turn accelerates the inter system crossing by reducing the singlet–triplet energy gap [4, 5, 16]. Multi-functionalized platinum(II) bis(acetylide) complexes are well known in the field of photophysics due to their extensive photophysical characteristics [22,23,24].
In order to synthesize a wide variety of transition metal alkynyl complexes, several M–C≡C bond-forming reactions are available [25, 26]. Due to the π-unsaturated nature of alkynyl groups and their linear shape when connected to metal centers, metal alkynyls are easily coupled to create linear complexes, oligomers, and polymers with potential uses in materials science [1,2,3]. Particularly, extensive research has been done on the square planar geometry of the functionalized alkynyl system of platinum(II) complexes [16, 27,28,29,30].
Recently, we have reported the palladium catalyzed synthesis of novel organosulfur-functionalized trans-platinum(II) bis(acetylide) complexes having two phenylthio groups in each alkenyl backbone [31] and the photochemical synthesis of novel organoselenium-functionalized trans-platinum(II) bis(acetylide) complexes having two phenylseleno groups in each alkenyl backbone [32]. The radical facilitated thiolation of terminal alkyl/arylacetylenes is well-precedented in synthetic organic chemistry [33, 34], which is rare in organometallic chemistry. Our group only reported this type of reactions recently for the functionalization of platinum acetylides [35,36,37]. Among the π-conjugated organometallic materials, the conjugated polymers are promising for their ease of preparation, solution process ability and wide range of materials properties [1,2,3], but it has not been well regarded to small molecules [12, 16, 38, 39]. Therefore, researchers focused on the photochemical synthesis, spectroscopic characterization and optoelectronic properties of four new p-anisolylthiol-functionalized trans-platinum(II) bis(alkenylarylalkynyl) complexes, trans-[(Et3P)2Pt{C≡C–Ar–CH=CH(SC6H4OCH3-p)}2], (2) (where, Ar = phenylene/biphenylen/2,5-dimethylphenylene/2,5-dimethoxyphenylene), bearing one anisolylthio group in each alkenyl backbone, which are stabilized by the presence of monodentate ancillary phosphine ligands.
Experimental
Material and methods
All reactions were performed under a nitrogen atmosphere. Solvents were dried, distilled by using appropriate drying agents and degassed before use [40]. All chemicals, except where stated, were purchased from commercial sources and used without further purification. The compounds HC≡C–C6H4–C≡CH [41], HC≡C–C6H4–C6H4–C≡CH [41], HC≡C–C6H2(p-CH3)2–C≡CH [41], HC≡C–C6H2(p-OCH3)2–C≡CH [41], trans-[(Et3P)2Pt{C≡C–C6H4–C≡CH}2] [25], trans-[(Et3P)2Pt{C≡C–C6H4–C6H4–C≡CH}2] [25], trans-[(Et3P)2Pt{C≡C–C6H2(p-CH3)2–C≡CH}2] [25], and trans-[(Et3P)2Pt{C≡C–C6H2(p-OCH3)2–C≡CH}2] [25] were prepared according to literature methods. NMR spectra were recorded on Bruker 400 MHz FT NMR spectrometer in CDCl3. 31P{1H} NMR spectra were referenced to external trimethylphosphite. 1H NMR spectra were referenced to internal TMS, and 13C{1H} NMR spectra were referenced to solvent resonances. Infrared spectra were recorded on Shimadzu FTIR prestige 21 spectrometer by using KBr pellets, and ESI-HR mass spectra were recorded on JEOL JMS-T100LC spectrometer. Microanalyses were performed on the analytical section of BCSIR, Dhaka. The UV visible absorption spectrum was recorded using a Shimadzu UV-1800 dual-beam spectrophotometer, and steady-state emission photoluminescence measurements were done using an Edinburgh FLS1000 spectrophotometer. Low temperatures emission was obtained using fingertip Dewar contained liquid nitrogen. The absolute quantum yield of the complexes was measured using an integrating sphere. Fluorescence lifetime decay was measured from a Pico Quant FT300 time correlated single photon correlation (TCSPC) instrument using Ti: Sapphire laser source. DFT and TD-DFT calculations were performed using Gaussian 16, with B3LYP functionality and SDD basis set. The calculations were conducted using the University of Texas HPC system. Column chromatography was performed on silica gel.
Synthesis of complex 2a
A mixture of trans-[(Et3P)2Pt{C≡C–C6H4–C≡CH}2] (1a) (0.068 g, 0.1 mmol) and p-anisolylthiol (0.035 g, 0.25 mmol) was added in chloroform (0.6 mL) and degassed under nitrogen atmosphere in a sealed tube. The resulting mixture was irradiated under tungsten lamp 500 W for 3 h (cool water was passed over the sealed tube to maintain room temperature). The completion of the reaction was examined by TLC and IR. The solvent was removed under reduced pressure. The resulting crude product was purified by silica column chromatography, eluting with hexane and dichloromethane, and the title complex 2a was isolated as a yellow solid in 98% yield (0.094 g), E/Z ratio: 60/40. IR (solid state, KBr): ν2099 (C≡C) cm−1; 1H NMR (400 MHz, CDCl3): (E/Z ratio: 60/40): δ 7.41–6.87 (m, 16H, Ar–H, SAr-H), 6.73 (d, 1.20 × 1H, JH–H = 15.2 Hz), 6.48 (d, 1.20 × 1H, JH–H = 15.2 Hz), 6.42 (d, 0.80 × 1H, JH–H = 10.8 Hz), 6.31 (d, 0.80 × 1H, JH–H = 11.2 Hz), 3.79 (s), 3.78 (s) {6H, SAr-OCH3-p}, 2.20–2.14 (m, 12H, CH2) and 1.26–1.16 (m, 18H, CH3); 13C{1H} NMR (100 MHz, CDCl3): δ 8.2, 16.2, 55.3, 109.2, 109.3, 109.6, 109.8, 114.7, 114.7, 124.1, 124.8, 125.4, 125.7, 126.9, 127.0, 127.4, 127.7, 128.2,129.4, 130.6, 131.0, 132.7, 133.0, 133.3, and 159.3; 31P{1H} NMR (161.83 MHz, CDCl3): δ11.6 (JPt–P = 2370 Hz); ESI-HRMS [M]+m/z = 962.2823 (100%), Calc. mass: 962.0932, Anal. Calc. for C46H56O2P2PtS2: C, 57.43; H, 5.87%. Found: C, 57.39; H, 5.98%.
Synthesis of complex 2b
The same procedure was followed for the synthesis of platinum(II) complex 2b as previously applied for the synthesis of 2a, but platinum(II) bis(acetylide) complex 1b, trans-[(Et3P)2Pt{C≡C–C6H4–C6H4–C≡CH}2] was used instead of trans-[(Et3P)2Pt{C≡C–C6H4–C≡CH}2] (1a), and the reaction was carried out for 5 h. The resulting crude product was purified by silica column chromatography, eluting with hexane and dichloromethane, and the title complex 2b was isolated as a yellow solid in 90% yield (0.100 g), E/Z ratio: 63/37. IR (solid state, KBr): ν 2097 (C≡C) cm−1;1H NMR (400 MHz, CDCl3): (E/Z ratio: 63/37): δ 7.62–7.32 (m, 20H, Ar–H, SAr-H), 6.92–6.83{5.26H (m, 4H, SAr-H; d, 1.26 × 1H), 6.53 (d, 1.26 × 1H, JH–H = 13.6 Hz), 6.50 (d, 0.74 × 1H, JH–H = 8.8 Hz), 6.40 (d, 0.74 × 1H, JH–H = 10.4 Hz), 3.81 (s), 3.80 (s) {6H, SAr-OCH3-p}, 2.21–2.16 (m, 12H, CH2) and 1.29–1.19 (m, 18H, CH3); 13C{1H} NMR (100 MHz, CDCl3): δ 8.3, 16.3, 55.3, 109.1, 109.4, 114.77, 114.8, 124.5, 125.3, 125.5, 126.1, 126.2, 126.4, 126.5, 126.7, 126.8, 127.9, 128.2, 128.5, 129.0, 131.2, 132.9, 133.3, 135.3, 135.3, 136.9, 137.0, 139.3, 139.7, 159.4, and 159.5;31P{1H} NMR (161.83 MHz, CDCl3): δ11.6 (JPt–P = 2396 Hz); ESI-HRMS [M]+m/z = 1114.3478 (100%), Calc. mass: 1114.2851, Anal. Calc. for C58H64O2P2PtS2: C, 62.52; H, 5.79%. Found: C, 62.24; H, 5.87%.
Synthesis of complex 2c
The synthetic procedure was the same as for the synthesis of platinum(II) complex 2a, but platinum(II) bis(acetylide) complex 1c, trans-[(Et3P)2Pt{C≡C–C6H2(CH3–p)2–C≡CH}2], was used instead of trans-[(Et3P)2Pt{C≡C–C6H4–C≡CH}2], 1a, and the reaction was carried out for 3 h. The resulting crude product was purified by silica column chromatography, eluting with hexane and dichloromethane, and the title complex 2c was isolated as a pale yellow solid in 99% yield (0.101 g), E/Z ratio: 50/50. IR (solid state, KBr): ν 2091 (C≡C) cm−1; 1H NMR (400 MHz, CDCl3): (E/Z ratio: 50/50): δ 7.40–7.36 (m, 5H, SAr-H, Ar–H), 7.18 (s, 1H, Ar–H), 7.12 (s, 1H, Ar–H), 7.05 (s, 1H, Ar–H), 6.88 (d, 2H, SAr-H, JH–H = 6.4 Hz), 6.85 (d, 2H, SAr-H, JH–H = 6.4 Hz), 6.76 (d, 1 × 1H, JH–H = 15.6 Hz), 6.66 (d, 1 × 1H, JH–H = 15.2 Hz), 6.54 (d, 1 × 1H, JH–H = 10.4 Hz), 6.35 (d, 1 × 1H, JH–H = 10.8 Hz), 3.79 (s), 3.78 (s) {6H, SAr-OCH3-p}, 2.44 (s, 3H, Ar-CH3-p), 2.36 (s, 3H, Ar-CH3-p), 2.24 (s), 2.22 (s){6H, Ar-CH3-p)}, 2.18–2.11 (m, 12H, CH2) and 1.24–1.14 (m, 18H, CH3); 13C{1H} NMR (100 MHz, CDCl3): δ 8.2, 16.2, 19.1, 19.3, 20.7, 20.9, 55.2, 108.5, 108.7, 114.6, 114.7, 124.4, 124.5, 125.4, 125.5, 127.1, 127.1, 127.6, 127.6, 127.7, 127.8, 127.9, 128.0, 128.8, 131.6, 131.7, 131.7, 132.0, 132.0, 132.5, 132.6, 132.7, 132.9, 135.6, 136.1, and 159.1; 31P{1H} NMR (161.83 MHz, CDCl3): δ 12.2 (JPt–P = 2384 Hz); ESI-HRMS [M]+ m/z = 1018.3453 (100%), Calc. mass: 1018.1995, Anal. Calc. for C50H64O2P2PtS2: C, 58.98.89; H, 6.34%. Found: C, 58.84; H, 6.44%.
Synthesis of complex 2d
The same procedure as for the platinum(II) complex 2a was followed for the synthesis of 2d, but platinum(II) bis(acetylide) complex 1d, trans-[(Et3P)2Pt{C≡C–C6H2(OCH3-p)2–C≡CH}2], was used instead of trans-[(Et3P)2Pt{C≡C-C6H4-C≡CH}2], 1a, and the reaction was carried out for 3 h. The resulting crude product was purified by silica column chromatography, eluting with hexane and dichloromethane, and the title complex 2d was isolated as a deep yellow solid in 88% yield (0.096 g), E/Z ratio: 65/35. IR (solid state, KBr): ν 2094 (C≡C) cm−1; 1H NMR (400 MHz, CDCl3): (E/Z ratio: 65/35): δ 7.42–7.38 (m, 4H, SAr-H),7.18 (s, 0.70 × 1H, Ar–H), 6.90–6.73 [m, 10.6 × 1H, {SAr-H (4H), Ar–H (3.3 × 1H), –CH = CH– (3.3 × 1H)}], 6.34 (d, 0.70 × 1H, JH–H = 10.4 Hz), 3.85–3.75 (18H, SAr-OCH3-p, Ar-OCH3-p), 2.27–2.22 (m, 12H, CH2) and 1.27–1.17 (m, 18H, CH3); 13C{1H} NMR (100 MHz, CDCl3): δ8.4, 16.1, 55.3, 56.1, 56.2, 56.3, 105.4, 109.0, 111.8, 114.7, 115.2, 116.0, 117.9, 120.5, 123.1, 123.2, 123.2, 124.7, 124.8, 125.3, 125.4, 125.5, 126.9, 127.0, 132.7, 132.8, 150.3, 150.4, 154.0, 154.4, 159.2, and 159.3; 31P{1H} NMR (161.83 MHz, CDCl3): δ11.8 (JPt–P = 2395 Hz); ESI-HRMS [M]+ m/z = 1082.3258 (100%), Calc. mass: 1082.1971, Anal. Calc. for C50H64O6P2PtS2: C, 55.49; H, 5.96%. Found: C, 55.37; H, 6.03%.
Results and discussion
Syntheses
Under UV light irradiation, the platinum(II) complex, which has an extended ethynyl ligand trans-(Et3P)2Pt{C≡C–C6H4–C≡CH}2 (1a), was reacted with p-anisolylthiol [34, 35], and provided a yellow solid platinum(II) bis(alkenylarylalkynyl) complex (2a) in excellent yield (98%, isolated yield; Table 1, entry 1; Scheme 1). p-Anisolylthiol photolysis requires UV light with a wavelength between 300 and 250 nm, which was achieved using a 500 W tungsten lamp [42]. According to Oliver et al. (2012), the photodissociation of para-substituted thiophenols took place at ~ 280 nm. They discovered that UV irradiation in the range 305 > phot > 240 nm causes the fission of the S–H bond and the production of the p-YPhS˙ radical (Y = CH3/F/OCH3) [42]. The structure of the complex was identified by IR, multi-nuclear NMR spectroscopy, and ESI-HR mass spectrometry as well as elemental analysis.

Synthetic route of p-anisolylthiol-functionalized various trans-platinum(II) bis(alkenylarylalkynyl) complexes
With the analogous reaction conditions (Table 1, entry 1), various platinum(II) bis(acetylides), trans-(Et3P)2Pt(C≡C–Ar–C≡CH)2, (1) (Ar = biphenylene/2,5-dimethylphenylene/2,5-dimethoxyphenylene) were also examined (Scheme 1) by adding p-anisolylthiol under UV radiation. The outcome of these reactions is general, which gave expected p-anisolylthiol-functionalized complexes, 2b-2d (Table 1). This methodology tolerates trans-platinum(II) bis(acetylide) complexes, with extended ethynyl ligand of diterminal alkynes, containing aryl rings as well as substituted aryl rings.
All the reactants were converted to thiolated products within 3 to 5 h under irradiation with UV light (Table 1). The crude products were purified by silica column chromatography, eluting with hexane/dichloromethane. The highly pure E/Z isomers are inseparable mixtures which displayed a single spot on TLC plate. The pure compounds were isolated as yellow to deep yellow solids in 88–99% yields. The complexes are soluble in common organic solvents but are insoluble in hexane. The p-anisolylthiol-functionalized trans-platinum(II) bis(alkenylarylalkynyl) complexes are air-stable. In our previous study, the reaction of benzenethiol/p-tolylthiol with platinum(II) bis(acetylide) provided the thiolated complexes in 70–90% yields [35,36,37]. In the present study, methoxy substituted thiol gives much higher yields (88–99%). The trans-platinum(II) complexes (2) provided satisfactory elemental analyses. The products were also characterized by IR, 1H, 13C and 31P NMR spectroscopy and positive ESI-HR [M]+ mass spectrometry.
Stereochemistry
The newly synthesized complexes exhibited good regioselectivity. The isomeric nature (E/Z) of the complexes was ascertained using the measured values of the coupling constant of the vinylic protons, which are made obvious by 1H NMR spectroscopic analysis (Table 1). For instance, the observed values of the vinylic protons coupling constant for the trans-platinum(II) complex 2a are 10.8 and 11.2 Hz for Z isomers and 15.2 and 15.2 Hz for E isomers (Table 2) [36]. The E isomer is preferred over the creation of the Z isomer.
Characterization
The new organometallic complexes were characterized by using traditional methodologies such as elemental analysis, IR, 1H, 13C{1H} and 31P{1H} NMR spectroscopy and mass spectrometry (positive [M]+ ion in ESI-HR MS). The selected basic spectroscopic data for all the new compounds in this study are summarized in Table 2.
The platinum-alkyne carbon bond (Pt–C≡C) is confirmed to be sustained by a sharp single absorption band on the range of 2099–2091 cm−1 in the IR spectra of the p-anisolylthiol-functionalized trans-platinum(II) bis(alkenylarylalkynyl) complexes 2. In the IR spectra, the absence of the terminal ≡C–H absorption band confirms the completion of the reaction. As for example, platinum(II) complex 2a displayed a sharp single absorption band at 2099 cm−1, which is assigned to ν(C≡C) stretching vibration (Table 2) and showed no bands in the range 3200–3300 cm−1, which is the characteristic region for the ≡C–H stretching vibration [32]. It is clear that the anisolylthiol and terminal acetylenic group had a specific reaction.
The vinyl protons exhibit new peaks in the range of 6.31–6.92 ppm in the 1H NMR spectra of platinum(II) bis(alkenylarylalkynyl) complexes 2 (Fig. 1). For instance, platinum(II) complex 2a displayed two sets of doublets at 6.73 and 6.48 ppm for E isomer, and 6.42 and 6.31 ppm for Z isomer (Table 2) [36, 37, 43]. On the other hand, in the 1H NMR spectra of complexes 2, there was no signal at about 3 ppm, which is the characteristic region for the terminal alkyne proton ≡C–H. In the 1H NMR spectra of platinum(II) complex 2a, the methyl protons of two isomers (E/Z) of the p-anisolylthio group were seen as two singlets at 3.79 and 3.78 ppm. In each case, signals from the p-anisolylthio moiety, organic spacers, and ethyl phosphine protons likewise displayed peaks in the anticipated area. In 1H NMR spectra, platinum(II) complex 2b showed three vinyl proton doublets and one vinyl proton overlapped with the aromatic ring of the organic spacer, platinum(II) complex 2c showed four vinyl proton doublets, platinum(II) complex 2d showed one vinyl proton doublet and three vinyl protons overlapped with the aromatic ring of the organic spacers, and all other protons display peaks at the expected region.

1H NMR spectra of Platinum(II) bis(alkenylarylalkynyl) complex 2a
The 31P{1H} NMR spectra of each platinum(II) bis(alkenylarylalkynyl) complexes 2 showed the expected signals consisting of three lines due to coupling with 195Pt, such as, platinum(II) complex 2c displayed a sharp singlet at 12.2 ppm along with two satellites positions on 19.5 and 4.8 ppm (Fig. 2). The trans geometry around the platinum-diphosphine centers was confirmed by 31P{1H} NMR spectroscopy based on the JPt–P coupling constant [36, 37]. The E/Z intimate mixtures mutually displayed a sharp singlet in 31P{1H} NMR (Table 2), because in both cases (E/Z), geometry is only associated with the terminal alkene, which is isolated from the phosphine ligands. The JPt–P values obtained, i.e., 2370, 2396, 2384, and 2395 Hz for the platinum(II) complexes 2a, 2b, 2c, and 2d, respectively, are in agreement by the values earlier reported for other square planar platinum(II) complexes which bearing trans geometry [44, 45]. 13C{1H} NMR spectrum was also performed for each complex and display signals in the expected region.

The 31P{1H} NMR spectra of platinum(II) bis(alkenylarylalkynyl) complexes 2c
The molecular formula of the newly synthesized complexes was also recognized by the intense molecular ion [M]+ peaks in the positive ion ESI-HR mass spectra observed at m/z 962.2823 for 2a, at m/z 1114.3478 for 2b, at m/z 1018.3453 for 2c, and at m/z 1082.3258 for 2d (Table 2).
Photophysical properties
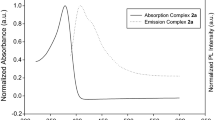
The absorption and emission properties of complexes 2a-2d were recorded in THF at room temperature and presented in Fig. 3a, b, c. The platinum(II) bis(alkenylarylalkynyl) complexes of 2a, 2b, and 2c showed the absorption maxima at around ~ 362 nm, whereas 2d displayed a red-shifted absorption peak at 491 nm due to the presence of an OMe substituent, which releases electrons. All complexes showed a relatively weak peak at around 300 nm. Compared to the absorption band of platinum(II) bis(alkynylarylalkynyl) complexes 1, we find that the position of the lowest energy absorption bands in the platinum(II) bis(alkenylarylalkynyl) complexes 2 are red-shifted, after the functionalization of p-anisolylthiol. The highest red-shift (20 nm) observed for our trans-platinum(II) complexes is recorded for complex 2c. The UV/Vis absorption maxima of complexes 1a, 1b, 1c, and 1d are observed at 345, 356, 335, and 377 nm, respectively, whereas those of their corresponding p-anisolylthiol functionalized complexes 2a, 2b, 2c, and 2d are observed at 362, 363, 355, and 391 nm, respectively. In each case, a small red-shift is observed, and the shifts are 17, 7, 20, and 14 nm for complexes 2a, 2b, 2c, and 2d, respectively as compared to complexes 1a, 1b, 1c, and 1d, respectively. These absorption bands are predominantly based on intra-ligand charge transfer consisting of acetylenic (C\(\equiv\)C) π–π*, and aromatic and aliphatic (C=C) π–π*transitions [46,47,48,49]. The assignment of UV/Vis absorption peaks is further supported by DFT optimized frontier molecular orbitals-HOMO and LUMO of 2a, 2b, 2c and 2d. The HOMO is delocalized along the acetylenic, phenyl and ethene π–π*orbitals with a small contribution from the platinum dπ orbital (Fig. 5). The delocalization of the HOMO and LUMO (ground and excited states) is the origin of the red shift of the absorption band of 2d compared to 2a, 2b and 2c. In addition, the excitation energies, along with their related transitions confirmed the presence of HOMO and LUMO in acetylene ligands.

a Absorption spectra in THF b room temperature emission spectra in THF c emission spectra at 77 K in Me-THF of complexes 2a-2d. d fluorescence lifetime decay of complexes 2a-2d
All of the complexes exhibited violet to blue fluorescence emission at room temperature. As complexes 2a, 2b, and 2c showed emission around 405 nm, while 2d appeared in the blue region (Fig. 3a, b, c). The complexes retained the same fluorescence emission in degassed THF solution, indicating no sign of phosphorescence at room temperature. Low temperature emission spectra were recorded in 2-Me THF to investigate the existence of any triplet state emission in the glassy state. Interestingly, 2a and 2b showed intense triplet state emission (phosphorescence) at 579, and 585 nm, respectively with a sharp peak due to the corresponding vibrational relaxation (Fig. 3a, b, c).
However, triplet emission did not show for complexes 2c and 2d. It is believed that the substitution of organic spacer with methyl and methoxy groups leading to rises the non-emissive decay from triplet state to ground state at low temperature (77 K), and thereby radiative decay of triplet state to ground state may not observe [50]. Yam VWW et al. (2011) reported that a series of multifunctional platinum(II) bipyridine complexes in which some of examples were alkoxy substituted bipyridine platinum(II) acetylides, that did not show triplet state emission at 77 K [50, 51], but those complexes showed fluorescence at room temperature. In going from room temperature to 77 K, non-radiative decay rate from the triplet state becomes suppressed by the restricted rotational and vibrational motion of the molecules in glassy state [52,53,54,55], hence the appearance of phosphorescence for the complexes 2a and 2b. The fluorescence lifetime of all four complexes were measured in THF at room temperature. The singlet state fluorescence lifetime for complexes 2a and 2c were found at 327 and 322 ps whereas a comparatively shorter lifetime of 309 ps was obtained for complex 2b (Fig. 3d). However, the excited singlet state lifetime for the complex 2d was shortened by around half (169 ps), which limits the intersystem crossing between singlet to triplet state significantly. Fluorescence quantum yields (Фfl) for the complex 2b and 2d was higher (~ 5%) than that of 2a and 2c (~ 2%). The photophysical data is summarized in Table 3.
Electrochemistry
Cyclic voltammetry (CV) is widely used to obtain the highest occupied molecular orbitals (HOMO) and lowest unoccupied molecular orbital (LUMO) energy levels of redox active molecules/polymers [56]. The electrochemical measurements of the complexes 2a-2d were conducted in dichloromethane, in a three electrode system—glassy carbon as working electrode, platinum as auxiliary electrode, and Ag/Ag+ in acetonitrile as reference electrode [57]. In all measurements Fc/Fc+ (0.18 V vs. Ag/Ag+) were used as internal standard. The CV traces were presented in Fig. 4 and the oxidation potentials were summarized in Table 4. All the complexes exhibited one or more irreversible oxidation waves. The first oxidation waves of 2a, 2b, 2c, and 2d were observed at 0.47, 0.55, 0.45, and 0.25 V, respectively (Fig. 4 and Table 4). The TD DFT calculations (next section) revealed that the oxidations in the complexes were originated from the acetylide ligands of the complexes (Fig. 5). These oxidations are related to the HOMO energies of the complexes [30, 56, 58]. No reduction was observed within the potential windows (− 2 V to + 2 V). However, LUMO energies of the complexes were obtained from the difference between the HOMO energies and optical gap (Eopt). The HOMO and LUMO energies are listed in Table 4.

Cyclic voltammograms of trans-[(PEt3)2Pt{C≡C–Ar–CH=CH(SC6H4–OCH3)}2] (Ar = C6H4 (2a), Ar = C6H4–C6H4 (2b), Ar = C6H2(2,5-Me)2, (2c) and Ar = C6H2(2,5-OMe)2 (2d) in dichloromethane using Bu4NPF6(0.01 M) as supporting electrolyte

DFT optimized HOMO-LUMO of 2a, 2b, 2c, and 2d
Theoretical calculations
In order to investigate the electronic structures and energies of frontier molecular orbitals of the complexes (2a-d), we employed density functional theory (DFT) and time-dependent DFT (TD-DFT) calculations. These calculations utilized the Gaussian 16 program [59] and were based on the hybrid exchange correlation functional B3LYP [60] in conjunction with the Stuttgart–Dresden (SDD) basis set [61]. GaussView, the graphical user interface used with Gaussian software, was employed to visualize the frontier molecular orbitals using an isovalue of 0.02.
To simplify the computations and reduce the computational cost, the -Et groups on the phosphine ligands were substituted with -Me groups. Figure 5 displays the frontier molecular orbital diagrams for the geometry-optimized complexes (2a-d). A visual examination of the orbital plots reveals that the HOMO consists of p orbitals originating from each of the phenylacetylene units and a d orbital localized on the platinum atom. The LUMO consists of p orbitals on the phenylacetylene units and an empty d orbital on the platinum. Analyzing the LUMO images for complexes 2a and 2c from Table 5, it is evident that the electron density on one of the phenylacetylene units is notably less than the other, with reduced participation from the d orbital on the platinum atom. The TD-DFT results provided further insight into the basis of the complexes near-UV absorption bands, as demonstrated in Tables 1, 2, 3, and 4 in the SI section. These results indicate that the lowest energy transition exclusively involves the HOMO → LUMO transition. The lowest energy absorption, observed at 362 nm with a significant oscillation strength, precisely corresponds to the experimentally recorded maximum absorption at 362 nm for complex 2a. For complex 2c, the theoretically determined lowest energy absorption aligns closely with the experimental result of 355 nm. However, in the case of complexes 2b and 2d, the experimental absorptions are lower than the calculated values. Considering the distributions of the frontier orbitals, it is reasonable to conclude that the transition is predominantly π–π* in nature with a small degree of metal-to-ligand charge transfer character [62].
Conclusions
We have synthesized a series of p-anisolylthiol functionalized trans-platinum(II) bis(alkenylarylalkynyl) complexes in excellent yield (88–99%) by photochemical reaction. The optical absorption and emission spectra of the complexes are influenced by the nature of spacers in the acetylide ligand. All complexes showed emission bands in the violet-blue region of the electromagnetic spectrum at room temperature. Complexes without any substituents in the phenyl spacer displayed intense triplet state emission (phosphorescence) at low temperature, whereas complexes with substituted phenyl spacers (with CH3 and OCH3) did not. The TD-DFT calculations of the complexes shows that the absorption bands are originated from intra-ligand charge transfer (π–π*) with the participation of platinum dπ-orbital. Cyclic voltammetry (CV) and the TD DFT calculations revealed that the oxidations in the complexes were originated from the acetylide ligands of the complexes. These oxidations are related to the HOMO energies of the complexes.
References
Haque A, Al-Balushi RA, Al-Busaidi IJ, Khan MS, Raithby PR (2018) Chem Rev 118:8474–8597
Wong WY, Ho CL (2006) Coord Chem Rev 250:2627–2690
Zhou GJ, Wong WY (2011) Chem Soc Rev 40:2541–2566
Xu LJ, Zeng XC, Wang JY, Zhang LY, Chi Y, Chen ZN (2016) ACS Appl Mater Interfaces 8:20251–20257
Bullock JD, Xu Z, Valandro S, Younus M, Xue J, Schanze KS (2020) ACS Appl Electron Mater 2:1026–1034
Li Y, Tong H, Xie Z, Wang L (2013) Polym Chem 4:2884–2890
Holt ED, Wang J, Winkel RW, Younus M, Schanze KS (2021) Photo Chem Photo Bio 8:100060. https://doi.org/10.1016/j.jpap.2021.100060
Wong WY, Ho CL (2010) Acc Chem Res 43:1246–1256
Yan L, Zhao Y, Wang X, Wang XZ, Wong WY, Liu Y, Wu W, Xiao Q, Wang G, Zhou X, Zeng W, Li C, Wang X, Wu H (2012) Macromol Rapid Commun 33:603–609
Durand RJ, Gauthier S, Achelle S, Kahlal S, Saillard JY, Barsella A, Wojcik L, Poul NL, Guen FRL (2017) Dalton Trans 46:3059–3069
Goswami S, Cekli S, Alarousu E, Winkel RW, Younus M, Mohammed OF, Schanze KS (2020) Macromolecules 53:6279–6287
Tam AYY, Wong KMC, Yam VWW (2009) J Am Chem Soc 131:6253–6260
Camerel F, Ziessel R, Donnio B, Bourgogne C, Guillon D, Schmutz M, Lacovita C, Bucher JP (2007) Angew Chem Int Ed 46:2659–2662
Takahashi S, Murata E, Kariya M, Sonogashira K, Hagihara N (1979) Macromolecules 12:1016–1018
Spencer M, Santoro A, Freeman GR, Díez Á, Murray PR, Torroba J, Whitwood AC, Yellowlees LJ, Williams JAG, Bruce DW (2012) Dalton Trans 41:14244–14256
Younus M, Köhler A, Cron S, Chawdhury N, Al-Mandhary MRA, Khan MS, Long NJ, Friend RH, Raithby PR (1998) Angew Chem Int Ed 37:3036–3039
Goswami S, Hernandez JL, Gish MK, Wang J, Kim B, Laudari AP, Guha S, Papanikolas JM, Reynolds JR, Schanze KS (2017) Chem Mater 29:8449–8461
Hilderbrandt A, Kovalski E, Korb M (2021) Eur J Inorg Chem 2021(25):2523–2532
Beljonne D, Wittmann HF, Köhler A, Graham S, Younus M, Lewis J, Raithby PR, Khan MS, Friend RH, Brédas JL (1996) J Chem Phys 105:3868–3877
Silverman EE, Cardolaccia T, Zhao X, Kim KY, Haskins-Glusac K, Schanze KS (2005) Coord Chem Rev 249:1491–1500
Hagihara N, Sonogashira K, Takahashi S (1981) Adv Poly Sci 41:149–179
Liu L, Zhang C, Zhao J (2014) Dalton Trans 43:13434–13444
Lara R, Lalinde E, Moreno M (2017) Dalton Trans 46:4626–4641
Mullin WJ, Qin H, Mani T, Muller P, Panzer MJ, Thomas SW (2020) Chem Commun 56:6854–6857
Sonogashira K, Fujikura Y, Yatake T, Toyoshima N, Takahashi S, Hagihara N (1978) J Organomet Chem 145:101–108
Khan MS, Davies SJ, Kakkar AK, Schwartz D, Lin B, Johnson BFG, Lewis J (1992) J Organomet Chem 424:87–97
Scattergood PA, Delor M, Sazanovich IV, Bouganov OV, Tikhomirov SA, Stasheuski AS, Parker AW, Greetham GM, Towrie M, Davies ES, Meijer AJHM, Weinstein JA (2014) Dalton Trans 43:17677–17693
Wong HL, Tao CH, Zhu N, Yam VWW (2011) Inorg Chem 50:471–481
Nishijo J, Uchida M, Enomoto M, Akita M (2021) Transit Met Chem 46:373–380
Younus M, Valandro S, Gobeze HB, Ahmed S, Schanze KS (2023) J Photochem Photobiol, A 435:114303
Rahman MM, Nomoto A, Younus M, Ogawa A (2014) Eur J Inorg Chem 2014(16):2613–2617
Rahman MM, Younus M, Ogawa A (2015) Eur J Inorg Chem 2015(8):1340–1344
Ichinose Y, Wakamatsu K, Nozaki K, Birbaum JL, Oshima K, Utimoto K (1987) Chem Lett 16(8):1647–1650
Wille U (2013) Chem Rev 113:813–853
Rahman MM, Younus M, Ogawa A (2014) RSC Adv 4:25389–25392
Rahman MM, Younus M, Naher M, Masud MK, Nomoto A, Ogawa A, Rudnick A, Köhler A (2016) J Organomet Chem 818:185–194
Rahman MM, Pramanik SK, Paul D, Sarkar M, Ahmed MJ, Saha R, Ogawa A (2019) Transit Metal Chem 44:247–252
Cardolaccia T, Li Y, Schanze KS (2008) J Am Chem Soc 130:2535–2545
Dai FR, Chen YC, Lai LF, Wu WJ, Cui CH, Tam GP, Wang XZ, Lin JT, Tian H, Wong WY (2012) Chem Asian J 7:1426–1434
Armarego WLF, Perrin DD (1996) Purification of Laboratory Chemicals, 4th edn. Butterworth-Heinemann, Guildford
Takahashi S, Kuroyama Y, Sonogashira K, Hagihara N (1980) Synthesis 1980(8):627–630
Oliver TAA, King GA, Tew DP, Richard ND, Ashfold MNR (2012) J Phys Chem A 116:12444
Riduan SN, Ying JY, Zhang Y (2012) Org Lett 14:1780–1783
Grim SO, Keiter RL, McFarlane W (1967) Inorg Chem 6:1133–1137
Rieger AL, Carpenter GB, Rieger PH (1993) Organometallics 12:842–847
Khan MS, Al-Mandhary MRA, Al-Suti MK, Feeder N, Nahar S, Kohler A, Friend RH, Wilson PJ, Raithby PR (2002) J Chem Soc Dalton Trans 12:2441–2448
Liu Y, Jiang S, Glusac K, Powell DH, Anderson DF, Schanze KS (2002) J Am Chem Soc 124:12412–12413
D’Amato R, Furlani A, Colapietro M, Portalone G, Casalboni M, Falconieri M, Russo MV (2001) J Organomet Chem 627:13–22
Ahmad MF, Rahman MM, Khan MAAT, Siddique AB, Ara MH, Biswas MK, Bhoumik NC, Ghosh S, Jagadesan P, Khan MMR, Younus M (2021) J Organomet Chem 950:121970
Li Y, Tam AYY, Wong KMC, Li W, Wu L, Yam VWW (2011) Chem Eur J 17:8048–8059
Ko CC, Wu L, Wong KMC, Zhu N, Yam VWW (2004) Chem Eur J 10:766–776
Baroncini M, Bergamini G, Ceroni P (2017) Chem Commun 53:2081–2093
Reineke S, Baldo MA (2014) Sci Rep 4:3797
Amin MK, Rahman MM, Naher M, Islam T, Ahmad MF, Khan MA, Khan MMR, Alam MA, Younus M, Biswas MK, Haque Y (2017) Synth Met 232:96–102
Ahmed MK, Rahman MM, Naher M, Mehdi SS, Khan AR, Khan MMR, Islam SMS, Younus M, Wedler S, Bagnich S, Hofmann AL, Rudnick A, Köhler A (2019) Macromol Chem Phys 220:1800494
Cardona CM, Li W, Kaifer AE, Stockdale D, Bazan GC (2011) Adv Mater 23:2367–2371
Elgrishi N, Rountree KJ, McCarthy BD, Rountree ES, Eisenhart TT, Dempsey JL (2018) J Chem Educ 95:197–206
Dai F-R, Zhan H-M, Liu Q, Fu Y-Y, Li J-H, Wang Q-W, Xie Z, Wang L, Yan F, Wong W-Y (2012) Chem Eur J 18:1502–1511
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Petersson GA, Nakatsuji H, Li X, Caricato M, Marenich AV, Bloino J, Janesko BG, Gomperts R, Mennucci B, Hratchian HP, Ortiz JV, Izmaylov AF, Sonnenberg JL, Williams-Young D, Ding F, Lipparini F, Egidi F, Goings J, Peng B, Petrone A, Henderson T, Ranasinghe D, Zakrzewski VG, Gao J, Rega N, Zheng G, Liang W, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Throssell K, Montgomery JA Jr, Peralta JE, Ogliaro F, Bearpark MJ, Heyd JJ, Brothers EN, Kudin KN, Staroverov VN, Keith TA, Kobayashi R, Normand J, Raghavachari K, Rendell AP, Burant JC, Iyengar SS, Tomasi J, Cossi M, Millam JM, Klene M, Adamo C, Cammi R, Ochterski JW, Martin RL, Morokuma K, Farkas O, Foresman JB, Fox DJ (2016) Gaussian 16, Revision C.01. Gaussian Inc, Wallingford
Becke AD (1993) J Chem Phys 98:5648–5652
Andrae D, Häußermann U, Dolg M, Stoll H, Preuß H (1990) Theoret Chim Acta 77:123–141
Cooper TM, Krein DM, Burke AR, McLean DG, Rogers JE, Slagle JE (2006) J Phys Chem A 110:13370–13378
Acknowledgements
Rahman MM, Islam T, and Khan MAAT, contributed equally to this work. Corresponding author (Rahman MM) e-mail: mostafiz-che@sust.edu. We would like to thank the Research Center of Shahjalal University of Science & Technology for its generous support for funding (PS/2018/2/15). We also acknowledge the funding from Higher Education Quality Enhancement Project (HEQEP) of the University Grants Commission of Bangladesh for purchasing the FT-IR and UV–vis spectrometer. We are grateful to Dr. Kirk Schanze of the University of Texas at San Antonio, San Antonio, Texas, USA for his generous support, providing us with instrumental facilities for low temperature PL measurement and excited state lifetime measurements.
Author information
Authors and Affiliations
Contributions
MR: Methodology, writing—review & editing, Writing—original draft, Conceptualization. TI: Methodology & writing (DFT & CV). AATK: Methodology & writing (Photophysical). MY: Review & editing. DP: Review & editing. SJ: Methodology. AO: Review & editing.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Rahman, M.M., Islam, T., Khan, M.AAT. et al. Synthesis, spectroscopic characterization, and photophysical properties of new p-anisolylthiol-functionalized platinum(II) bis(alkenylarylalkynyl) complexes. Transit Met Chem 48, 377–387 (2023). https://doi.org/10.1007/s11243-023-00550-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11243-023-00550-x