
Abstract
The kinetics of the oxidation of acetaldehyde by octacyanomolybdate(V) ions in aqueous alkaline medium has been studied. The reaction display zero-order kinetics in [Mo(CN) 3–8 ] and is first order in [CH3CHO]. An acetaldehyde-dependent reaction rate constant, \(k_{{{\text{CH}}_{3} {\text{CHO }}}}\) = 1.41 ± 0.03 × 10−5 s−1 has been observed at pH 9. A significant dependence on [OH−] has been observed, and a value pKb = − 0.40 ± 0.02 for the deprotonation of the hydrated form of acetaldehyde has been obtained. Activation parameters for the reaction have been obtained, and a reaction mechanism is proposed.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Potassium hexacyanoferrate(III) (E = 0.36 V [1] and potassium permanganate (E = 0.57 V) are commonly used as oxidants to oxidize organic functional groups in alkaline medium. Speakman and Waters [2] reported the oxidation of aldehydes, ketones and nitroparaffins with hexacyanoferrate(III) ions. Holluta and Mutshin [3] reported the oxidation of formaldehyde by a neutral permanganate solution and found the reaction to be base catalyzed.
Octacyanotungstate(V) ions (E = 0.54 V [4]) and octacyanomolybdate(V) ions (E = 0.80 V [4]) have approximately similar or slightly larger redox potentials compared to hexacyanoferrate(III) and permanganate. Furthermore, the M(CN) 3−8 (M = W, Mo) complex ions are relatively substitution inert in an alkaline medium. They could be a replacement for the above-mentioned K3Fe(CN)6 and KMnO4 in the oxidation of organic functional groups. This has prompted kinetic studies of the reactions of M(CN) 3−8 ions (M = Mo,W) with organic compounds.
The oxidation of formaldehyde (HCHO) with Fe(CN) 3−6 [5], W(CN) 3−8 [6] and Mo(CN) 3−8 [7] has been reported. In all of these studies, the reactions are first order in the metal cyanide complex, formaldehyde and the hydroxide ion and correspond to the rate law below (Eq. 1) with Kh the hydrolysis equilibrium constant of formaldehyde.
The dependence on the hydroxide ion is an indication of enolization of formaldehyde. The Mo(CN) 3−8 oxidation of formaldehyde has also been found to be alkali metal ion catalyzed. The effectivity of the alkali metal cation on the reaction rate is in the order Na+< K+< Rb+< Cs+ [7].
The carbonyl group is a structural component of a wide range of organic compounds. The difference in electronegativity between the carbon and the oxygen atoms is the main reason for the reactivity of the carbonyl functional group. Formaldehyde and acetaldehyde belong to this family of carbonyl-containing compounds. In aqueous medium, formaldehyde exists more than 99% in the hydrated form [8, 9]. Acetaldehyde also hydrolyzes in aqueous medium, but only to approximately 60% with a Kh value of 1.48 [10, 11].
Speakman and Waters [2] stated that an aldehyde cannot enolize if an α-carbon hydrogen is absent and also found that if an aldehyde cannot enolize, it cannot be oxidized by aqueous alkaline ferricyanide. Benson [12] concluded that unenolized or unhydrated aldehydes cannot be oxidized in an alkaline medium. Pandey et al. [13] reported the osmium tetroxide-catalyzed oxidation of acetaldehyde with alkaline ferricyanide. The reaction is first order in [CH3CHO] and [OH−], and a mechanism involving an enolized acetaldehyde, yielding a dimer, oxalic acid (COOH)2), is proposed. The enolization step is proposed to be rate determining. Kamble and Nandibewoor [14] reported the osmium(VIII)- and ruthenium(III)-catalyzed oxidation of acetaldehyde by periodate in an alkaline medium. The reaction is first order in periodate, acetaldehyde, hydroxide ions and the catalyst. The proposed mechanism proceeds initially via complex formation between the catalyst and hydroxide ion, followed by adduct formation between the catalyst complex and the substrate. This is then followed by a reaction of the newly formed adduct with the IO4−, yielding acetate and IO3− as products.
Experimental
Cesium octacyanomolybdate(V), Cs3Mo(CN)8.2H2O, was synthesized as described by Leipoldt et al. [15, 16] and was used as primary standard [17] after recrystallization. An aqueous acetaldehyde solution was prepared from Merck “pro analisi” standard CH3CHO and was standardized by the method of Siggia [18]. Buffer solutions consisting of sodium carbonate (0.10 M) and hydrogen carbonate (0.10 M) and also sodium chloride (0.10 M) and sodium hydroxide (0.10 M) were prepared and the pH was measured with a T and C model 1002 pH meter using a saturated sodium chloride calomel electrode. A stock solution of carbonate-free sodium hydroxide was prepared and then bubbled with nitrogen and stored in a nitrogen atmosphere. The solution was standardized with potassium phthalate [19, 20]. All reagents used were analytical grade, and redistilled water was used throughout.
Kinetic measurements were made under pseudo-first-order conditions, with CH3CHO more than tenfold in access, at constant ionic strength of 0.2 M (adjusted with NaCl) for all the reaction mixtures. Reactant concentrations are listed in Figs 1, 2, 3, 4, 5 and 6 and in the supplementary data Table 1S. This table also summarizes all the obtained rate constants for this study. Experiments were carried out in diffuse laboratory light as the Mo(CN) 3−8 ion is sensitive to light [21], although at the experimental conditions, the radiation effect is insignificant. Rate data were obtained by monitoring the absorbance decrease of the Mo(CN) 3−8 ion in the reaction mixture on a Pye Unicam SP 1700 double beam spectrophotometer using 1 cm path length cuvettes. For faster reactions, the rate data were obtained on a Durrum D110 stopped-flow spectrophotometer. Both spectrophotometers were fitted with a Fryka-Kaltetechnik KB 300 water bath with a Thermomix1440 thermostat for effective temperature control to within 0.1 °C.

A reaction trace of [Mo(CN) 3−8 ] versus time [Mo(CN) 3−8 ] = 6.0 × 10−4 M; [CH3CHO] = 0.0555 M; µ = 0.2 M; pH = 9.00; T = 25.0 ± 0.1 °C

kobsd versus [Mo(CN) 3−8 ] for the variation of [Mo(CN) 3−8 ] in the reaction mixture [CH3CHO] = 0.05463 M; µ = 0.2 M; pH = 9.00; T = 25.0 ± 0.1 °C. Insert: An inverse plot of 1/kobsd versus 1/[Mo(CN) 3−8 ] is linear confirming saturation kinetics

kobsd versus [CH3CHO] for the variation of [CH3CHO] in the reaction mixture. [Mo(CN) 3−8 ] = 5.0 × 10−4 M; pH = 9.00; µ = 0.2 M; T = 25.0 ± 0.1 °C

Observed rate constant versus the pH for the variation of [H+] in the reaction mixture. [Mo(CN) 3−8 ] = 5.0 × 10−4 M; [CH3CHO] = 0.0550 M; µ = 0.2 M; T = 25°C

Brønsted–Bjerrum plot of log k versus \(\frac{{I}^{1/2}}{{1+I}^{1/2}}\) [Mo(CN) 3–8 ] = 5.0 × 10−4 M; [CH3CHO] = 0.0550 M; µ = 0.2 M ; pH = 12; T = 25.0 °C

Linear relationship ln (kobsd/T) versus 1/T from the linearized Eyring equation. [Mo(CN) 3−8 ] = 5.0 × 10−4 M; [CH3CHO] = 0.0550 M; pH = 9.0; µ (NaCl) = 0.2 M
The extinction coefficients of Mo(CN) 3−8 and Mo(CN) 4−8 at 390 nm are 1400 M−1cm−1 and 140 M−1cm−1, respectively [22]. Both the reagent, Mo(CN) 3−8 , and the product, Mo(CN) 4−8 , absorbed light in the visible spectrum at this wavelength. The Beer–Lambert law thus cannot be applied directly to determine the concentration of the reagent Mo(CN) 3−8 at a time t during the experiment. The concentration of the Mo(CN) 3−8 was thus calculated by using Eqs. 2 and 3. If the initial concentration of the Mo(CN) 3−8 equals “a” mol dm−3, then the total absorbance at a time t is given by
These equations were used throughout to calculate [Mo(CN) 3−8 ] at a time t.
Results and discussion
Reaction traces of absorbance versus time indicated that the reaction is zero order in the reagent Mo(CN) 3−8 at the concentrations used. This indicates that the oxidation of acetaldehyde by Mo(CN) 3−8 differs completely from the oxidation of formaldehyde [5,6,7]. The zero order in Mo(CN) 3−8 was confirmed with a zero-order plot of [Mo(CN) 3−8 ] versus time (Fig. 1). The reaction is zero order for more than 65% depletion of Mo(CN) 3−8 . Variation of the concentration of Mo(CN) 3−8 in the reaction mixture shows a slight increase in the reaction rate with increasing [Mo(CN) 3−8 ]. A leveling-off in reaction rate at the higher [Mo(CN) 3−8 ] was observed, indicating saturation kinetics [23] (Fig. 2, Supplementary data Table 1S, entries 1–11). Saturation kinetics was confirmed by a linear inverse plot of 1/kobsd versus 1/[Mo(CN) 3−8 ] [23] (Fig. 2 insert). Variation of the [CH3CHO] in the reaction mixture yields a first-order dependence on the reagent CH3CHO (Fig. 3, Supplementary data Table 1S, entries 15–21) with a \(k_{{{\text{CH}}_{ 3} {\text{CHO}}}} = 1.41 \pm 0.03 \times 10^{ - 5}\) s−1 (Fig. 3). The zero intercept of Fig. 3 is an indication that the alkaline and radiation decomposition of Mo(CN) 3−8 [21, 24] is not significant at the reaction conditions.
Variation of the pH of the reaction mixture indicates a significant hydroxide ion dependence (Fig. 4). The observed experimental data (Supplementary data Table 1S, entries 22–38) fitted to Eq. 4 [25, 26]
yield a value of Ka = 4 ± 1 × 10−15 (pKa = 14.4 ± 0.1) for the deprotonation of the hydrated form of acetaldehyde. Since Kw = 10−14 = KaKb, this translates into a calculated value for Kb of 2.5 ± 0.1 (pKb = − 0.40 ± 0.02).
Variation of [Mo(CN) 4−8 ] in the reaction mixture (Supplementary data Table 1S, entries 6, 12–14) had no effect on the reaction rate. No intermediate free radicals have been detected by adding vinyl acetate to the reaction mixture. Variation of [Na+] in the reaction mixture (Supplementary data, entries 39−43) shows no alkali metal ion catalysis while variation of the ionic strength (Supplementary data Table 1S, entries 44–49) has a slight effect on the reaction rate.
During attempts to determine the stoichiometry of the reaction, a brownish coloring developed in the reaction mixture soon after mixing of the reagents. This was not observed during the kinetic experiments as the concentration of the solutions was very dilute. A yellowish brown powder, insoluble in water, was obtained when the mixture was allowed to react to completion. Elemental analysis of the yellowish brown product (Table 2S supplementary data) indicates that the expected cyano product seemed to decompose. This issue was not considered further as it is considered a separate study in the total project regarding the cyano complexes of Mo and W. An oxidation product of the acetaldehyde could also not be identified. This could be because of the precipitate identified in the reaction mixture. Literature evidence from several studies of the oxidation of acetaldehyde indicates acetic acid or the acetate ion as oxidation product [14].
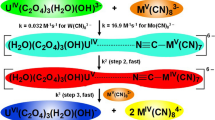
The experimental kinetic results could thus be explained by the reaction mechanism shown in Scheme 1.

Possible reaction mechanism for the oxidation of acetaldehyde by Mo(CN) 3−8 in an alkaline medium
A general theory for the influence of the ionic strength of the reaction medium on the reaction rate has been formulated by Brønsted and Bjerrum [27]. The logarithmic form of the equation, where ∂ is the Debye–Hückel solvent constant, is
The Debye–Hückel solvent constant (∂) for water is ~ 0.51 at 25 °C which simplifies the equation to
The increase in the ionic strength of the reaction medium resulted in a slight increase in the reaction rate (Supplementary data Table 1S, entries 44–49). If the slope of log k versus \(\left(\frac{I^{1/2}}{1+I^{1/2}}\right)\) has a positive value, it follows that ZA and ZB have similar nonzero charges (i.e., both positive or both negative). The data fitted to Eq. 6 (Fig. 5) yield a gradient of ZAZB = 0.37 ± 0.01. This value for ZAZB is almost zero which is a clear indication that at least one reactant in the rate determining step has a zero charge. This criterion is met in the second step of Scheme 1 (reaction 6) where the reactants are CH3CH(OH)2 and OH−, indicating the second step of Scheme 1 (reaction 6) to be the rate determining step.
By applying the steady-state theory to the intermediate CH3CH(OH)(O−) and by recognizing \(K_{\text{h}} = \frac{{{\text{CH}}_{ 3} {\text{CH(OH)}}_{ 2} }}{{\left[ {{\text{CH}}_{ 3} {\text{CHO}}} \right]\left[ {{\text{H}}_{ 2} {\text{O}}} \right]}}\), the rate law Eq. 13 can be derived. (See supplementary information for derivation)
The rate law (Eq. 13) is consistent with the observed saturation kinetics (Fig. 2). At the higher concentrations of Mo(CN) 3−8 , k2[Mo(CN) 3−8 ] >> k−1[H20] and k−1[H20] become negligible and the reaction is zero order in Mo(CN) 3−8 . At the lower concentrations of Mo(CN) 3−8 , k−1[H20] >> k2[Mo(CN) 3−8 ] and k2[Mo(CN) 3−8 ] become negligible and the reaction is first order in Mo(CN) 3−8 .
The activation parameters ΔH# and ΔS* have been obtained by varying the temperature of the reaction mixture between 14.8 and 39.9 °C (Supplementary data Table 1S, entries 50–55) and by application of the Eyring equation Eq. 14.
Equation 14 converted to the logarithmic form yields the linearized Eyring equation [28], Eq. 15.
A plot of ln \(\left( {\frac{k}{T}} \right)\) versus \(\frac{1}{T}\) should be linear and ΔH# can be obtained from the slope, \(\frac{{ - \Delta H^{{\# }} }}{R}\) and ΔS* from the intercept \(\frac{\Delta S*}{R}\) + \(\ln \left( {\frac{{k_{B} }}{h}} \right)\). Nonlinear least-squares fits of the data (Fig. 6, Supplementary data Table 1S, entries 50–55) to Eqs. 14 and 15 respectively yield an activation enthalpy, ΔH# = 53 ± 2 kJ mol−1 and an activation entropy, ΔS* = 183 ± 7 J K−1 mol−1. The obtained activation parameters have been validated by using Eq. 16 as explained in Ref. [28]. From the experimental results Tav = {(14.8 + 19.8 + 25.0 + 30.0 + 35.1 + 39.9)/6} + 273.1 = 300.5 K. From the activation parameters.
References
Dobos D (1975) Electrochemical data. Elsevier, New York
Speakman PT, Waters WA (1955) J Chem Soc Part 1:40
Halluta J, Mutschin A (1930) Z Phys Chem 150:381
Campion RJ, Purdie N, Sutin N (1964) Inorg Chem 3:1091
Singh VN, Gangwar MC, Saxena BBL, Singh MP (1969) Can J Chem 47:1051
Basson SS, Leipoldt JG, Grobler SR (1977) J Inorg Nucl Chem 39:376
Dennis CR, Potgieter IM, Basson SS (2011) React Kinet Mech Catal 104:1
Sutton HC, Downes TM (1972) J Chem Soc Chem Commun 1:1
Bell RP (1966) Adv Phys Org Chem 4:1
Bell RP, Clunie JC (1952) Trans Faraday Soc 48:439
Kurtz JL, Coburn JI (1967) J Am Chem Soc 89:3528
Benson D (1976) Mechanism of oxidation by metal ions, 1st edn. Elsevier Scientific Publishing Co, Amsterdam
Pandey PC, Singh VN, Singh MP (1971) Indian J Chem 9:430
Kamble DL, Nandibewoor ST (1998) J Phys Org Chem 11:171
Leipoldt JG, Bok LDC, Cilliers PJ (1974) Z Anog Allg Chem 409:343
Dennis CR, Van Wyk AJ, Basson SS, Leipoldt JG (1992) Transit Met Chem 17:471
Basson SS, Bok LDC, Grobler SR (1974) Z Anorg Anal Chem 268:287
Siggia S, Maxcy W (1947) Anal Chem 19:1023
Meitz L (1963) Handbook of analytical chemistry, 1st edn. McGraw-Hill, New York
Vogel AI (1972) A textbook of quantitative inorganic analysis, 3rd edn. Longman, London
Gray GW, Spence JT (1971) Inorg Chem 10:2751
Leipoldt JG, Bok LDC, Dennis CR (1975) J Inorg Nucl Chem 38:1655
Swarts JC, Aquino MAS, Han JY, Lam KY, Sykes AG (1995) Biochim et Biophys Acta 1247:215
Dennis CR, Potgieter IM, Basson SS (2010) React Kinet Mech Catal 99:63
Wilkens RG (1991) The study of kinetics and mechanism of reactions of transition metal complexes, 2nd edn. Allyn and Bacon, Boston, p 41
Han J, Swarts JC, Sykes AG (1996) Inorg Chem 35:4629
Laidler KJ (1963) Reaction kinetics, vol 2. Pergamon Press Ltd, London
Lente G, Fabian I, Poe J (2005) New J Chem 29:759
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Dennis, C.R., Potgieter, I.M., Langner, E.H.G. et al. The oxidation of acetaldehyde by the octacyanomolybdate(V) ion in an aqueous alkaline medium. Transit Met Chem 44, 161–165 (2019). https://doi.org/10.1007/s11243-018-0280-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11243-018-0280-y