
Abstract
Two Co(II) coordination polymers (CPs), namely [Co(L1)(DCTP)]n (1) and [Co(L2)(DCTP)]n (2) [L1 = 1,4-bis(5,6-dimethylbenzimidazol-1-yl)butane, L2 = 1,5-bis(5,6-dimethylbenzimidazol-1-yl)pentane, H2DCTP = 2,5-dichloroterephthalic acid] were synthesized and characterized by single-crystal X-ray diffraction analysis, elemental analysis, powder X-ray diffraction (PXRD) and infrared spectroscopy. CP 1 has a 2D (4,4) corrugated sheet structure, which is further extended into a 2D double layer by C–H···O weak hydrogen bonding interactions, while CP 2 displays a 2D layer with hcb network, which is assembled into a 3D supramolecular framework through C–H···O hydrogen bonding. Both CPs exhibited promising photocatalytic activities for the degradation of methylene blue under UV irradiation. In addition, the thermal stabilities and the luminescence properties of both CPs have been investigated.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The self-assembly of coordination polymers (CPs) has emerged as an important research topic in recent years, not only because of their interesting and diverse topologies, but also due to their extensive applications in catalysis, electrochemistry, gas adsorption and magnetic materials [1,2,3,4,5,6,7]. The final architectures of new CPs remain hard to predict because of the many factors which can influence the self-assembly process, including temperature, solvent, pH value and choice of organic ligands [8,9,10,11,12,13]. Clearly, however, one of the most important factors in obtaining a desired molecular architecture is the careful selection of organic ligands as bridging or terminal groups, with transition metals as nodes [14,15,16,18]. Recently, flexible bis(benzimidazole) derivatives, which have remarkable coordination ability and versatile conformations, have attracted much interest as N-donor ligands. They can adopt diverse conformations by means of the torsional flexibility of their –(CH2)n– spacers, providing opportunities to meet different requirements of the metal centers in the assembly process [19, 20]. In addition, various weak intermolecular interactions may play a crucial role in the construction of supramolecular architectures with functional properties, such as hydrogen bonds and π–π stacking [21, 22]. Meanwhile, aromatic dicarboxylic acid ligands have also been widely used to synthesize coordination polymers, because of their versatile coordination modes and structural rigidity, chemical stability and appropriate connectivity [23, 24]. However, only a few metal–organic coordination polymers based on 2,5-dichloroterephthalic acid (H2DCTP) as a bridging ligand have been successfully prepared [25, 26].
In this work, we synthesized two CPs with flexible bis(benzimidazole) and 2,5-dichloroterephthalic acid ligands (scheme 1), namely, [Co(L1)(DCTP)]n (1), and [Co(L2)(DCTP)]n (2) (L1 = 1,4-bis(5,6-dimethylbenzimidazol-1-yl)butane, L2 = 1,5-bis(5,6-dimethylbenzimidazol-1-yl)pentane). CP 1 exhibits a 2D double-layer structure, while CP 2 has a 3D supramolecular framework. The photocatalytic activities of both CPs for the degradation of methylene blue (MB) were investigated.

Structural formulas of all ligands
Experimental
Materials and methods
The proligands L1 and L2 were synthesized according to the literature [27]. All other chemicals and reagents were purchased from Beijing InnoChem Science & Technology Co. and used without further purification. C, N and H contents were determined with a Perkin-Elmer 240C analyzer. FTIR spectra were recorded from KBr pellets in the range of 4000–400 cm−1 with an Avatar 360 (Nicolet) spectrophotometer. Powder X-ray diffraction (PXRD) investigations were carried out with a Rigaku D/Max-2500 diffractometer using Cu-Kα radiation (λ = 1.5418 Å) over the 2θ range from 5 to 50° at room temperature, using 40 mA and 40 kV. An Edinburgh instruments FS5 spectrophotometer was employed to record the spectra of powdered solid samples. Thermogravimetric analyses (TGA) were recorded on a NETZSCH STA 449F3 differential thermal analyzer at a rate of 10 °C/min, under an N2 atmosphere. Solid state UV/vis diffuse reflectance spectra were recorded on a UV–Vis spectrophotometer (Puxi, TU-1901), with a BaSO4 plate as the standard at room temperature.
Synthesis of [Co(L1)(DCTP)]n (1)
A mixture of Co(OAc)2·4H2O (49.8 mg, 0.2 mmol), L1 (34.6 mg, 0.1 mmol), H2DCTP (23.5 mg, 0.1 mmol), NaOH (8 mg, 0.2 mmol) and H2O (10 mL) was sealed in a 25-mL Teflon-lined autoclave and heated continuously at 140 °C for 3 days under autogenous pressure. After cooling to room temperature at a rate of 5 °C/h, purple block-shaped crystals were obtained with 47.3% yield (based on Co). Anal. Calc. for C30H28Cl2CoN4O4: C 56.4; H 4.4; N 8.8%. Found: C 56.6; H 4.1; N 8.6%. IR (cm−1, KBr): 1631 (s), 1589 (s), 1511 (m), 1474 (m), 1376 (s), 1298 (w), 1076 (m), 842 (m), 524 (w).
Synthesis of [Co(L2)(DCTP)]n (2)
CP 2 was prepared by a procedure similar to CP 1, except that L1 was replaced with L2 (36.0 mg). Purple block crystals were obtained with 43.5% yield (based on Co). Anal. Calc. for C31H30Cl2CoN4O4: C, 57.1; H, 4.4; N, 8.3%. Found: C, 56.9; H, 4.6; N, 8.6%. IR (cm−1, KBr): 1617 (s), 1458 (m), 1377 (s), 1307 (m), 1217 (m), 1078 (m), 829 (w), 626 (w).
X-ray crystallography
Crystallographic data for a single crystal of CP 1 were collected at 100(2) K on an Agilent Technology SuperNova Atlas Dual System with a Cu microfocus source (λ = 1.54184 Å) and focusing multilayer optics. Data for CP 2 were collected on a Bruker Smart 1000 CCD area-detector diffractometer at 296(2) K using graphite-monochromated Mo-Kα radiation (λ = 0.71073 Å) with ω scan mode. Both structures were solved by direct methods and refined with full-matrix least-squares techniques based on F2 using the SHELXL-2014/7 program for CP 1 and SHELXL-2016/6 program for CP 2 [28]. All the non-hydrogen atoms were treated anisotropically. Hydrogen atoms of organic ligands were generated geometrically and refined isotropically using the riding model. The crystallographic data for CPs 1 and 2 are summarized in Table 1, and selected bond lengths and angles are listed in Table 2.
Photocatalytic experiments
The photocatalytic activities of CP 1 and CP 2 were evaluated for degradation of MB. Experiments were carried out under irradiation with a 500W high pressure halogen lamp. Before the reaction, 30 mg of the solid sample was added into a tube containing 100 mL of MB solution (10 mg/L). The reaction was stirred in a dark environment for 1 h in order to eliminate adsorption effects between the catalyst and solution. The solution was then exposed to UV irradiation. During the irradiation period, 3.5-mL aliquots were removed at 15-min intervals, and the liquid was separated by centrifugation. The resulting solutions were analyzed with a UV/vis spectrophotometer, to quantify the remaining MB in solution. A blank experiment was carried out under the same conditions, but without the catalyst. Considering the initial absorbance values of the dye solution as A0, and the absorbance values at time t as At, the degradation efficiency (D) of the dye is defined as follows:
Results and discussion
Crystal structure of CP 1
Single-crystal X-ray diffraction analysis revealed that CP 1 crystallizes in the triclinic space group Pī. The asymmetric unit contains one cobalt(II) center, one L1 ligand and two halves of DCTP2− ligands (Fig. 1a). Each Co(II) center is penta-coordinated by two nitrogen atoms (N1, N4A), (symmetry code: A = x, y, z + 1) from two different L1 ligands, plus three oxygen atoms (O2, O3, O4) from two DCTP2− ligands, assembling a slightly distorted trigonal bipyramid [CoN2O3] with a value of τ5 = 0.82 [29]. The Co–N bond lengths are 2.030(3) Å (Co–N1) and 2.029(3) Å (Co–N4A), and the Co–O bond lengths are 1.986(2) Å (Co–O2), 2.007(2) Å (Co–O3) and 2.448(2) Å. All these bond lengths are within the expected ranges [30].

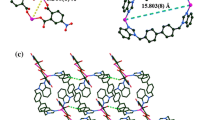
a Coordination environment around the Co(II) centers in CP 1. Hydrogen atoms are omitted for clarity (symmetry code: A: x, y, 1 + z, B: −x, −y 1 − z, C: x, y, 1 + z). b Schematic view of 2D layer constructed by ligands DCTP2−, L1, and Co(II) centers. c The schematic of the 2D layer topology in CP 1
In CP 1, the DCTP2− ligands adopt alternating (κ1 − κ0) − (κ1 − κ0) − μ2 and (κ1 − κ1) − (κ1 − κ1) − μ2 coordination modes, forming a 1D straight [Co–DCTP–Co]n chain with a contiguous Co···Co distance of 10.867(9) Å. The ligand L1 shows a trans-coordinated conformation in which the nitrogen atoms from two benzimidazole rings concatenate neighboring Co(II) centers to build a 1D [Co-L1-Co]n straight chain. The dihedral angle between the two benzimidazole rings within a complete L1 ligand is 82.34(1), and the neighboring Co···Co distance is 13.278(1) Å. These chains are further connected by sharing of the Co(II) centers along different directions to form a 2D (4,4) network. This arrangement gives rise to a tetranuclear unit, constructed from two L1 ligands, two DCTP2− ligands and four Co(II) centers (Fig. 1b). The network consists of rhombuses, which can be regard as a four-connected sql corrugated network; the size of each rhomboid, defined by Co–Co distances, is 13.277 × 10.860 Å (Fig. 1c). The sheet structure is further developed into a 2D double layer through hydrogen bonding between C–H groups of the L1 ligands and oxygen atoms of the DCTP2− ligands [C10–H10A···O3, C12–H12A···O1] with H···O distances of 2.490(2) and 2.448(3) Å, respectively.
Crystal Structure of CP 2
CP 2 crystallizes in the triclinic space group Pī. The unit cell contains one cobalt(II) center, one L2 ligand and two halves of DCTP2− ligands in the asymmetric unit (as shown in Fig. 2a). The Co(II) center is penta-coordinated by two nitrogen atoms (N1, N4A), (symmetry code: A = 2 − x, − y + 1, − z + 1) from two L2 ligands, plus three oxygen atoms (O1, O2, O3) from two DCTP2− ligands, again giving a distorted trigonal bipyramid [CoN2O3], for which τ5 = 0.43 [29]. The Co–N bond lengths are 2.021(2) and 2.050(2) Å, while the Co–O bond lengths are in the range of 1.960 (2) to 2.854(2) Å. These values are all in the normal ranges, and comparable to those found in CP 1.

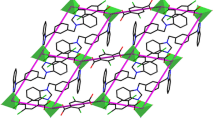
a Coordination environment around the Co(II) centers in CP 2. Hydrogen atoms are omitted for clarity (symmetry code: A: − x + 2, − y + 1, − z + 1, B: − x, − y + 1, − z + 1, C: − x, − y + 2, − z + 2). b A [Co2(L2)2] unit in CP 2. c Schematic view of 2D layer constructed by ligands DCTP2−, L2, and Co(II) centers. d Schematic view of the 3D supramolecular framework by C–H···O stacking interactions in CP 2
In CP 2, two L2 ligands are located on both sides of two Co(II) centers, adopting a bis(monodentate) coordination mode to bridge adjacent Co(II) centers in a 24-membered macrocycle [Co2(L2)2] (Fig. 2b). The dihedral angle between the two benzimidazole rings of each L2 ligand is 88.57(7). Meanwhile, the DCTP2− ligands also adopt the alternating (κ1 − κ0) − (κ1 − κ0) − μ2 and (κ1 − κ1) − (κ1 − κ1) − μ2 coordinating mode seen in CP 1, forming a 1D wave-like [Co-DCTP-Co] chain with the Co···Co distances of 11.165(6) and 11.103(5) Å. In this way, the [Co2(L2)2] units lie on the crests and troughs of the 1D [Co-DCTP-Co] chains, such that Co···Co is 13.080(7) Å, forming a 2D layered hcb structure with the 3-connect point symbol {63} (Fig. 2c). The 2D hcb layers are further connected by C–H···O hydrogen bonds, with C16–H16···O4 angle of 150° and H16···O4 distance of 2.56 Å, leading to a 3D supramolecular framework (Fig. 2d).
Influence of the N-donor ligands on the structures
In this work, we selected the carboxylic acid H2DCTP− and two alternative bis(benzimidazole) ligands to synthesize cobalt(II) CPs. From the structure descriptions above, ligands L1 and L2, which have different spacer lengths, act as linkers to connect the Co(II) centers in CPs 1 and 2. The DCTP2− ligands in both CPs also act as linkers; they are fully deprotonated and adopt the same bis(monodentate) and bis(chelating) coordination modes. In CP 1, the L1 ligands adopt a trans-coordinated mode to connect Co(II) centers, to give infinite 1D [Co(L1)]n chains which are further assembled with DCTP2− ligands, ultimately giving a 2D (4,4) layer structure. In CP 2, the L2 ligands again adopt a trans-coordinated mode to connect neighboring Co(II) centers, with a 24-membered macrocycle [Co2(L2)2] which is further connected by DCTP2− ligands give a 2D (6,3) layered structure. Consequently, the spacer lengths of the bis(benzimidazole) ligands have a significant influence on the structures of the resulting ternary Co(II) CPs.
Spectroscopic and PXRD characterization
The asymmetric and symmetric carboxylate vibrations of DCTP2− ligands in both CPs are evident as strong bonds at 1589, 1474 and 1376 cm−1 for CP 1 and at 1617, 1458 and 1377 cm−1 for CP 2. The values of ∆ν[νas(COO)–νs(COO)] are thus 115 and 213 cm−1 for CP 1 and 159 and 240 cm−1 for CP 2, indicative of both the monodentate and chelating coordination modes of the DCTP2− ligands in CPs 1 and 2. Bands in the region of 1500 cm−1 for both CPs are assigned to C=N stretching vibrations of the N-donor ligands, while bands around 640 nm−1 can be attributed to the C–Cl group of the DCTP2− ligands in both CPs.
In order to confirm the purities of both CPs, their PXRD patterns were checked at room temperature. The peak positions obtained from experiment were almost identical to the simulated PXRD crystal data (Fig. 3), confirming the phase purities of both bulk samples.

Simulated and experimental patterns of X-ray powder diffraction of CP 1 (a) and CP 2 (b)
The solid state photoluminescence properties of these CPs were investigated at room temperature. The H2DCTP ligand has a negligible contribution to the luminescence emission. Therefore, the emission of the title CPs is mainly associated with the presence of the bis(benzimidazole) ligands. The free N-donor proligands exhibit maxima at 313 nm (λex = 280 nm) for L1 and 361 nm (λex = 216 nm) for L2, while emission bands are observed at 522 nm (λex = 498 nm) for CP 1 and 364 nm (λex = 354 nm) for CP 2 (Fig. 4). Hence, compared with the free N-containing proligands, the emission peaks of both CPs are both blue-shifted, which may be due to the fact that the coordinated ligands are not allowed to relax by torsional modes on photoexcitation [31].

Emission spectra of CPs 1–2 and the free ligands
The absorption features of the CPs were revealed through their diffuse reflectance UV/Vis spectra (Fig. 5a). The main absorption peaks at 251 nm for CP 1 and 267 nm for CP 2 can be assigned to π*–π transitions of the ligand and/or ligand-to-metal charge transfer (LMCT) [32]. An additional absorption broad observed in the visible for CP 1, from 526 to 592 nm, and for CP 2 from 528 to 589 nm, could originate from d → d spin-allowed transitions of the metal centers [33]. The band gap energies (Eg) of the CPs can be calculated by extrapolation of the linear portion of the absorption edges, giving values of 3.09 eV for CP 1 and 3.58 eV for CP 2 (Fig. 5b) according to the formula. The energy band gap sizes suggest that these CPs may exhibit potential capability for photocatalytic degradation under UV irradiation. Where R is the reflectance at a given energy, the formula is as follows:

UV/Vis absorption spectra of CPs 1–2 with BaSO4 as background. b Diffuse reflectance spectra of Kubelka–Munk function versus energy of CPs 1–2
Thermogravimetric analysis
In order to assess the thermodynamic stabilities of these CPs, TGA experiments were carried out (Fig. 6). For CP 1, there is one weight loss in the temperature range from 290 to 590 °C, attributed to the decomposition of all organic ligands. The residue is identified as CoO (calcd. 11.7%, found. 12.1%). The thermogravimetric analysis of CP 2 showed one weight loss stage between 280 and 550 °C, again assigned to decomposition of all organic ligands. The residue is again CoO (calcd. 11.5%, found. 11.7%).

TG curves of CPs 1–2
Photocatalytic properties
In this work, the organic dye MB was selected as a target pollutant in order to study the photocatalytic properties of CPs 1 and 2. The change in concentration of MB was followed by the characteristic absorption bond at 664 nm (Fig. 7). The absorption peak did not show any significant change when the solution was kept in the dark. However, the absorption bands of MB decreased with increasing time of irradiation from 0 to 120 min in the presence of both photocatalysts CP 1 and CP 2. The degradation efficiency for MB reached nearly 86.6% for CP 1 and 83.1% for CP 2 after 120 min, while the degradation efficiency of a blank experiment without photocatalyst was 22.2% after 120 min under the same conditions. Hence, both CPs are effective photocatalysts for the degradation of MB, with very similar performances.

Absorption spectra of the MB solution during the decomposition reaction under UV irradiation with the presence of photocatalysts CPs 1–2(left for CP 1, right for CP 2)
Conclusion
Two Co(II) coordination polymers, [Co(L1)(DCTP)]n and [Co(L2)(DCTP)]n based on DCTP2− and different N-donor ligands were synthesized under hydrothermal conditions. CP 1 shows a 2D double-layer structure, while CP 2 exhibits a 3D supramolecular framework involving C–H···O hydrogen bond interactions. These results demonstrate that the spacer length of flexible bis(5,6-dimethylbenzimidazole) ligands plays an important role in construction of coordination polymers with diverse structural features. Both CPs displayed high photocatalytic activity for the degradation of MB.
Supplementary material
CCDC 1565136 and 1565137 contain the supplementary crystallographic data for the CPs 1–2. These data can be obtained free of charge via http://www.ccdc.cam.ac.uk/conts/retrieving.html, or from the Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; fax: (+44) 1223-336-033; or e-mail: deposit@ccdc.cam.ac.uk.
References
Hu JM, Blatov VA, Yu BY, Van Hecke K, Cui GH (2016) Dalton Trans 45:2426–2429
Van de Voorde B, Bueken B, Denayer J, De Vos D (2014) Chem Soc Rev 43(16):5766–5788
Liu J, Chen L, Cui H, Zhang J, Zhang L, Su CY (2014) Chem Soc Rev 43(16):6011–6061
Cui JW, Hou SX, Van Hecke K, Cui GH (2017) Dalton Trans 46:2892–2903
McKinlay AC, Eubank JF, Wuttke S, Xiao B, Wheatley PS, Bazin P, Lavalley JC, Daturi M, Vimont A, De Weireld G, Horcajada P, Serre C, Morris RE (2013) Chem Mater 25(9):1592–1599
Dhakshinamoorthy A, Asiri AM, Garcia H (2014) Chem Commun 50(85):12800–12814
Wang XX, Yu B, Van Hecke K, Cui GH (2014) RSC Adv 4(106):61281–61289
Fang DL, Mo SY, Wu KF, Huang ZJ (2017) Transit Met Chem 42:273–283
Li SL, Lan YQ, Ma JF, Yang J, Wei GH, Zhang LP, Su ZM (2008) Cryst Growth Des 8(2):675–684
Liu J, Zhang HB, Tan YX, Wang F, Kang Y, Zhang J (2014) Inorg Chem 53(3):1500–1506
Zhu X, Sun PP, Ding JG, Li BL, Li HY (2012) Crystal Growth Des 12:3992–3997
Wang DZ, Li JP, Fan JZ, Jia DZ (2016) Polyhedron 111:123–131
Hao SY, Li YF, Li YH, Cui GH (2017) Polyhedron 133:169–178
Deria P, Mondloch JE, Karagiaridi O, Bury W, Hupp JT, Farha OK (2014) Chem Soc Rev 43(16):5896–5912
Hao SY, Hao ZC, Liu YG, Dong GY (2017) Chin J Struct Chem 36(1):118–126
Du DY, Qin JS, Li SL, Su ZM, Lan YQ (2014) Chem Soc Rev 43(13):4615–4632
Qin L, Xiao SL, Ma PJ, Cui GH (2013) Transit Metal Chem 38(6):627–633
Li CP, Chen J, Mu YH, Du M (2015) Dalton Trans 44(24):11109–11118
Guo XG, Yang WB, Wu XY, Zhang QK, Lu CZ (2013) CrystEngComm 15(46):10107
Liu GX, Zhu K, Xu HM, Nishihara S, Huang RY, Ren XM (2010) CrystEngComm 12:1175–1185
Dong YM, Fan RQ, Wang XM, Wang P, Zhang HJ, Wei LG, Chen W, Yang YL (2016) Cryst Growth Des 16:3366–3378
Zhang DP, Bian YZ, Qin J, Wang P, Chen X (2014) Dalton Trans 43:945–949
Wang XL, Sha XT, Liu GG, Chen NL, Tian Y (2015) CrystEngComm 17:7290–7299
Liu L, Ding J, Li M, Lv X, Wu J, Hou H, Fan Y (2014) Dalton Trans 43:12790–12799
Hao SY, Li YH, Hao ZC, Cui GH (2017) Ultrason Sonochem 37:414–423
Carter KP, Kalaj M, Cahill CL (2017) Inorg Chem Frontiers 4(1):65–78
Fairley TA, Tidwell RR, Donkor I, Naiman NA, Ohemeng KA, Lombardy RJ, Bentley JA, Cory M (1993) J Med Chem 36:1746–1753
Sheldrick GM (2015) Acta Crystallogr Sect A 71:3–8
Yang L, Powell DR, Houser RP (2007) Dalton Trans 0(9):955–964
Meng XM, Fan CB, Bi CF, Zong ZA, Zhang X, Fan YH (2016) CrystEngComm 18(16):2901–2912
Ganesan P, Ranganathan R, Chi Y, Liu XK, Lee CS, Liu SH, Lee GH, Lin TC, Chen YT, Chou PT (2017) Chem-Eur J 23(12):2858–2866
Dai M, Su XR, Wang X, Wu B, Ren ZG, Zhou X, Lang JP (2014) Cryst Growth Des 14:240–248
Liu L, Ding J, Huang C, Li M, Hou HW, Fan YT (2014) Cryst Growth Des 14:3035–3043
Acknowledgments
The work was supported by the National Natural Science Foundation of China (51474086), Natural Science Foundation—Steel and Iron Foundation of Hebei Province (B2015209299).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Kang, WC., Li, YH., Qin, ZB. et al. Synthesis, structures and characterization of two cobalt(II) coordination polymers with 2,5-dichloroterephthalic acid and flexible bis(benzimidazole) ligands. Transit Met Chem 43, 563–570 (2018). https://doi.org/10.1007/s11243-018-0242-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11243-018-0242-4