
Abstract
Two ternary cobalt(II) coordination polymers (CPs), namely [Co(L1)(npht)] n (1) and {[Co2(L2)2(npht)2(H2O)]·H2O} n (2) (L1 = 4,4′-bis(benzimidazol-1-ylmethyl)biphenyl, L2 = 1,2-bis(5,6-dimethylbenzimidazol-1-ylmethyl)benzene, and H2npht = 4-nitrophthalic acid) have been synthesized and structurally characterized by X-ray crystallography. Both CPs feature similar 1D infinite chains containing two distinct loops. CP 1 further forms a 3D supramolecular network via weak C–H···O hydrogen bond interactions. CP 2 shows a 1D two-layer chain structure, assembled through π–π stacking interactions. The electrochemical, luminescence, and photocatalytic activities of the two CPs for the removal of methylene blue under visible or UV light were investigated. Possible photocatalytic mechanisms are discussed.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
With the development of manufacturing industry, many toxic contaminants have been discharged into the water system [1–3]. Heterogeneous photocatalysis offers an environmentally friendly, inexpensive, and highly efficient way to degrade such organic pollutants. Hence, research into coordination polymers (CPs) as functional materials has attracted considerable interest due to their high photocatalytic activities, tunable structures, and broad photoresponsing range in the solar spectrum [4–7]. Although much progress has been made into the design and fabrication of CPs, it is still a challenge to predict the structures of the resultant frameworks. These crystalline architectures are influenced by several factors, including ligands, the preferred coordination environment of the metal nodes, pH of the reaction, temperature, solvent, and so on [8–12]. Hydrogen bonding and π–π stacking interactions can be used as structure-directing tools for the assembly of CPs and may also be used to link the low-dimensional entities into higher dimensionalities [13–15]. Therefore, the prospect of tuning the architectures and properties of CPs through modification of organic ligands provides an impetus for further research into supramolecular coordination polymers [16–18].
Semirigid bis(benzimidazole) derivatives having sp 3 hybridization of their –CH2– spacers can freely twist to meet the coordination requirements of different metal centers in the assembly process, resulting in CPs with robust networks and useful properties, as well as allowing for extended N-donor frameworks [19–21]. Meanwhile, aromatic dicarboxylate ligands such as phthalic acid, terephthalic acid, and isophthalic acid are frequently used to fabricate CPs, due to their versatile coordination modes and strong coordinating abilities. The effect of counter anions on the assembly of dicarboxylate and metal centers may be mitigated [22–24]. Structures and properties of target CPs are also affected by the electronic and steric characteristics of ligand substituents. In particular, dicarboxylate ligand substituents are often involved in supramolecular assemblies because of their particular ability to shape hydrogen bonding [25–27].
To further investigate the photocatalytic properties of bis(benzimidazole)-based ternary cobalt(II) coordination polymers [9], herein we report the syntheses and characterization of two cobalt(II) mixed CPs, namely [Co(L1)(npht)] n (1) and {[Co2(L2)2(npht)2(H2O)]·H2O} n (2) (L1 = 4,4′-bis(benzimidazol-1-ylmethyl)biphenyl, H2npht = 4-nitrophthalic acid, and L2 = 1,2-bis(5,6-dimethylbenzimidazol-1-ylmethyl)benzene). The resulting CP 1 and CP 2 materials possess narrow band gaps (E g) and strong absorption in the visible and ultraviolet regions. The thermal analysis, luminescence properties and photocatalytic performances of CPs 1 and 2 for the degradation of methylene blue (MB) have been investigated.
Experimental
Materials and measurements
The proligands L1 and L2 were synthesized according to the previously reported procedure [28]. Other reagents were obtained from Jinan Henghua Sci. and Tec. Co. Ltd. and used without further purification. Elemental analysis for C, H, N was obtained on a Perkin-Elmer 240C elemental analyzer. Ultrasound was generated by a multiwave KQ2200DE instrument at a frequency of 40 kHz. The morphology and size of the samples before and after photodegradation of MB solutions were characterized with a scanning electron microscope (JSM-IT100) after gold coating. FTIR spectra (4000–400 cm−1) were recorded on KBr disks on an Avatar 360 (Nicolet) spectrophotometer. Thermogravimetric analysis (TGA) was conducted on a Netzsch TG 209 thermal analyzer under N2 with a heating rate of 10 °C min−1. Powder X-ray diffraction (PXRD) patterns were measured on a Rigaku D/Max-2500 diffractometer with Cu-Kα radiation (λ = 1.5418 Å). Luminescence spectra were collected with an FS5 fluorescence spectrophotometer at room temperature. Solid-state UV/Vis diffuse reflectance spectra were measured using a UV–Vis Puxi T9 UV–visible spectrophotometer, with BaSO4 as a reflectance standard. The electrochemical analysis was carried out using a CHI 660E electrochemical workstation (Chenhua Instrument, Shanghai, China) in 0.1 M Na2SO4 electrolyte solution.
Synthesis of CP 1
A mixture of Co(OAc)2·4H2O (0.1 mmol, 24.8 mg), L1 (0.1 mmol, 36.6 mg), H2npht (0.1 mmol, 21.1 mg), and H2O (12 mL) was transferred to a 25mL Teflon-lined stainless steel vessel and heated to 140 °C for 72 h. After cooling to room temperature at a rate of 5 °C h−1, pink block crystals were obtained by filtration and washed with distilled water. Yield (43.1 mg, 52.2%) based on Co. Elemental analysis (%) calcd for C36H25N5CoO6 (682.54): C, 63.4; H, 3.7; N, 10.3. Found (%): C, 63.6; H, 3.9; N, 10.5. IR (KBr, cm−1): 2920(w), 1644(s), 1514(s), 1463(w), 1440(m), 1383(s), 1345(s), 1091(m), 780(w), 743(s).
Synthesis of CP 2
The synthesis of CP 2 was similar to that of CP 1, except that L2 (0.1 mmol, 39.4 mg) was used in place of L1. Purple red block crystals of CP 2 were obtained. Yield (39.9 mg, 46.8%) based on Co. Elemental analysis (%) calcd for C68H62N10Co2O14 (1361.16): C, 60.0; H, 4.6; N, 10.3. Found (%): C, 60.2; H, 4.8; N, 10.6. IR (KBr, cm−1): 3445(s), 3110(w), 2920(w), 1624(s), 1515(s), 1442(s), 1210(w), 834(w), 622(w).
X-ray crystallography
Crystallographic diffraction data for CP 1 were collected on a Bruker Smart 1000 CCD diffractometer using graphite-monochromated Mo-Kα radiation (λ = 0.71073 Å) with multiscan mode at 273(2) K. A semiempirical absorption correction was applied using the SADABS program [29]. Crystallographic data for CP 2 were collected at 100(2) K on an Agilent SuperNova Dual diffractometer with a Cu microfocus source and focusing multilayer optics. CrysAlis PRO software was used to collect, index, scale, and apply analytical absorption corrections based on faces of the crystal [30]. Both structures were refined on F 2 by full-matrix least squares techniques using the SHELXL-2014 program [31]. The ligands in the structure of CP 1 were severely disordered, such that atoms C35, O3, O4, N5, O5, O6; C29, C30, C31, C32, C33, C34 in the npht2− ligand and atoms C16, C17, C18, C19 of the L1 ligand were refined using a split model. The corresponding site occupation factors were refined so that their sum was unity, 0.50/0.50, 0.44/0.56, 0.32/0.68, respectively. The corresponding bond distances in the disordered groups were restrained to be equal. The Uij parameters of these atoms were restrained to an approximate isotropic behavior. All non-hydrogen atoms were refined anisotropically. Hydrogen atoms of organic ligands were placed in calculated positions and included as riding atoms with isotropic displacement parameters. Residual hydrogen atoms of water molecules were located on a difference Fourier map. Crystallographic data and structure parameters for CP 1 and CP 2 are summarized in Table 1. Selected bond lengths and angles are listed in Table 2.
Results and discussion
Crystal structure of CP 1
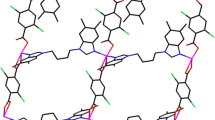
X-ray crystallographic analysis reveals that CP 1 crystallizes in the triclinic crystal system, space group Pī with one Co(II) center, one L1 ligand, and one npht2− ligand in the asymmetric unit. As depicted in Fig. 1a, the Co(II) center adopts a tetrahedral coordination geometry, of which the four vertexes are O1, O3A, N4, and N1B (symmetry codes: A: − x + 2, − y + 1, − z + 2; B: − x + 1, − y + 2, − z + 1). The two nitrogen and two oxygen atoms are provided by two L1 ligands and two npht2− ligands, respectively. The Co–O/N bond lengths vary from 1.945(2) to 2.032(3) Å.

a Coordination environment of Co(II) centers in CP 1 with 30% thermal ellipsoids. Hydrogen atoms are omitted for clarity (symmetry codes: A: − x + 2, − y + 1, − z + 2; B: − x + 1, − y + 2, − z + 1). b The 1D chain structure in CP 1. c The 2D supramolecular architecture extended through C5–H5···O2C hydrogen bonding interactions (bright green dashed line, symmetry codes: C: x + 1, y, z; − x + 2, − y + 1, − z + 2). d The 3D supramolecular architecture further extended by C17–H17···O2D hydrogen bonding interactions (bright yellow dashed line, symmetry codes: D: − x, − y + 1, − z + 1)
The L1 ligands adopt a trans-conformation, with Ndonor–N–Csp3–Csp3 torsion angles of 69.02(6)°. Each L1 ligand acts as a μ 2-bridging linker, bridging adjacent Co centers to form a [Co2(L1)2] loop with a non-bonding Co···Co distance of 15.803(8) Å. The npht2− ligands adopt a (κ 1)-(κ 0)-(κ 1)-(κ 0)-μ 2 coordination mode, connecting neighboring Co centers to generate a [Co2(npht)2] ring with a non-bonding Co···Co separation of about 5.300(6) Å. Two of these different loops cross-link each other to obtain a 1D chain (Fig. 1b). In addition, adjacent 1D loop-like chains are further extended into a 2D supramolecular architecture through C5–H5···O2C hydrogen bonding interactions between the L1 ligands and npht2− ligands (H5···O2 = 2.36 Å, C5–H5···O2 = 149°, symmetry code: C = x + 1, y, z; Fig. 1c). In addition, the 2D structure is further extended into a 3D supramolecular framework via C17–H17···O2D hydrogen bonding interactions between L1 and npht2− ligands (H17···O2 = 2.50 Å, C17–H17···O2 = 148°, symmetry code: D = − x, − y + 1, − z + 1; Fig. 1d).
Crystal structure of CP 2
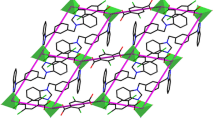
CP 2 crystallizes in the triclinic space group Pī. The asymmetric unit comprises two individual Co(II) centers, two npht2− ligands, two L2 ligands, one-coordinated water ligand, and one lattice water molecule. As illustrated in Fig. 2a, the Co1 center is six-coordinated by two nitrogen atoms (N3, N10A, symmetry codes: A: x + 1, y − 1, z) from two L2 ligands, plus four oxygen atoms (O1, O7, O8, O1W) from the carboxyl groups of two npht2− ligands and one water molecule, giving a distorted octahedral geometry. Dissimilar to Co1, the Co2 center is four-coordinated by two oxygen atoms (O3, O10) of two independent npht2− ligands and two nitrogen atoms (N6B, N7, symmetry codes: B: x − 1, y + 1, z) of two L2 ligands, in a distorted tetrahedral environment.

a Coordination environment of Co(II) centers in CP 2 with 30% thermal ellipsoids. Hydrogen atoms and the free water molecule are omitted for clarity (symmetry codes: A: − x + 2, − y + 1, − z + 2; B: − x + 1, − y + 2, − z + 1). b View of the 1D loop-like chain in CP2. c 1D two-layer chain formed by π–π stacking interactions
As shown in Fig. 2b, two distinct L2 ligands serve as bridges linking pairs of Co(II) centers to build a rectangular [Co2(L2)2] ring, in which the dihedral angles between the mean planes of the two benzimidazole rings of the two L2 ligands are 85.72(6)° and 86.94(6)°, and the Co1···Co2 distance is 10.552(8) Å. The npht2− ligands show two coordination modes, specifically (κ 1)-(κ 0)-(κ 1)-(κ 0)-μ 2 and (κ 1)-(κ 1)-(κ 1)-(κ 0)-μ 2, and connect adjacent Co(II) centers to shape another [Co2(npht)2] loop, with a Co1···Co2 distance of 5.501(7) Å. The neighboring two loops are further cross-linked with each other to generate a 1D chain. In addition, through π–π stacking interactions between the benzene rings of two L2 ligands (centroid-to-centroid distance of 3.752 Å, interplanar angle α, slipping angles β and γ of 0°, 17.78° and 17.78°, respectively; Cg1: C44–C45–C46–C47–C48–C49, Cg1C: C44C–C45C–C46C–C47C–C48C–C49C, symmetry codes: C = − x, − y + 1, − z + 1; Fig. 2c), these 1D loop-like chains are extended into a 1D two-layer chain structure, which is further consolidated by O2W–H2WA···O10D hydrogen bonding interactions between water ligands and npht2− ligands (H2WA···O10 = 2.14 Å, O2W–H2WA···O10D = 151°, symmetry code: D = x, y + 1, z).
In comparison with the present work, two related coordination polymers have been reported previously, namely {[Co(L1)(1,3-bdc)(H2O)]·1.5H2O} n (3) and {[Co2(L1)2(nip)2]·2H2O} n (4) (1,3-H2bdc = 1,3-benzenedicarboxylic acid and H2nip = 5-nitroisophthalic acid) [32]. Thus, two semirigid bis(benzimidazole) derivatives (L1 and L2) with different spacers gave four cobalt(II) CPs. CP 1 and CP 2 exhibit 1D chain structures, while CP 3 and CP 4 are 2D layered frameworks. In four complexes, the N-donor ligands all adopt the bidentate bridging mode, linking the Co(II) centers with Co···Co separations of 15.803(8) Å for CP 1, 10.552(8) Å for CP 2, 17.921 Å for CP 3, and 13.803 and 16.778 Å for CP 4. The four Co(II) centers show different coordination geometries (tetrahedral for CP 1, distorted octahedral and tetrahedral for CP 2, distorted octahedral for CP 3 and CP 4, Tables 3 and 4). In CP 1, the npht2− ligand adopts the bis-monodentate coordination mode, while the npht2− ligands employ the bis-monodentate and chelating-monodentate coordination modes for CP 2. Further, the 1,3-bdc2− ligands of CP 3 adopt a bidentate–monodentate coordination mode to act as connectors in a 2D 4-connected (44,62) sql net, while the nip2− ligands of CP 4 exhibit bis-bidentate coordination, resulting in a 2D uninodal 4-connected (43.63) network. Finally, CP 1 and CP 4 are self-assembled into 3D supramolecular structures via hydrogen bonding interactions, while CP 2 is further formed into a 1D bilayer chain through π–π stacking interactions. Overall, the different dimensionalities of CPs 1–4 may be attributed to their different aromatic carboxylates and the resulting supramolecular interactions.
Infrared spectra, PXRD and TGA
The IR spectra of CP 1 and CP 2, respectively, show characteristic absorption bands at 1644 and 1383 cm−1 for CP 1, 1624 and 1442 cm−1 for CP 2, attributed to the asymmetric and symmetric stretching vibrations of the carboxylic groups. The benzimidazole ν(C–N) structures are observed at 1514 and 1515 cm−1 for CP 1 and CP 2, respectively (Fig. S1). Powder X-ray diffraction (PXRD) experiments were carried out to confirm the phase purities of the bulk samples. The experimental PXRD patterns well matched the simulations, confirming the phase purity of the bulk samples (Fig. S2).
The TGA curve of CP 1 shows an initial weight loss above 185 °C, assigned to decomposition of the organic ligands. The residue corresponds to CoO (11.2% obsd; 11.0% calcd). For CP 2, the TGA curve shows an initial weight loss of 2.5% from 130 to 225 °C, which is in good agreement with the removal of two water molecules (calcd. 2.6%). The second sharp weight loss is observed from 260 to 720 °C corresponding to decomposition of the organic ligands. The remaining residue of 5.7% corresponds to the formation of CoO (calcd 5.5%) (Fig. S3).
Luminescence properties
The luminescence spectra of both CPs and their corresponding free ligands were recorded in the solid state (Fig. 3). The proligands L1 and L2 show emission peaks at 370 nm (λ ex = 325 nm) and 347 nm (λ ex = 300 nm), respectively, which may be ascribed to π → π* and/or n → π* transitions [33, 34]. CPs 1 and 2 give luminescence emissions bands at 381 nm (λ ex = 304 nm) and 383 nm (λ ex = 290 nm), respectively, which show different degrees of redshift compared with the free proligands (ca. 11 nm for CP 1 and ca. 36 nm for CP 2). These redshifts may be related to intraligand luminescence emissions [35].

Luminescence spectra of CPs 1–2, free ligands L1 and L2
In order to investigate the potential of the two CPs for sensing organic solvent molecules, 3 mg of the powder of CP (1 or 2) was immersed in 3 mL of organic solvent, specially methanal, acetonitrile, ethanol, pentanone, dichloromethane, acetone, DMSO, dimethylacetamide (DMAc), ethylene glycol (EG), and formaldehyde. Each suspension was treated by ultrasonication for 1.0 h. After aging for over 24 h, shaking, filtering, and drying in air, the corresponding fluorescence emission spectra were recorded at room temperature, with the results shown in Fig. 4. It is clear that only the luminescence intensity of CP 1@HCHO was slightly enhanced. For the other CP 1@solvents, different degrees of decreased luminescence were observed. In particular, acetone shows a significant quenching effect. For CP 2, only the luminescence intensity of CP 2@HCHO and CP 2@CH3CN was slightly enhanced; the other CP 2@solvents showed decreases in luminescence, especially CP 2@Acetone. In addition, the measured PXRD patterns of CP 1@Acetone and CP 2@Acetone well matched the corresponding simulated patterns, and the SEM images of CP 1@Acetone and CP 2@Acetone are similar to those of pure CP 1 and CP 2 powders, indicating that the structures of both CPs are well preserved (Figs. 5 and S2). Hence, both CP 1 and CP 2 can serve as potential luminescence sensors for detecting acetone.

Solid-state luminescence spectra of CP 1 (a) and CP 2 (b) after immersing the as-synthesized powders into various pure solvents

a SEM images of nanopore structures of CP 1 (left), CP 1@Acetone (middle), and CP 1 after the photodegradation of MB solutions under UV light irradiation (right). b SEM images of cubelike nanostructures of CP 2 (left), CP 2@Acetone (middle), and CP 2 after the photodegradation of MB solutions under UV light irradiation (right)
Electrochemical behaviors
A method of preparation of CP bulk-modified carbon paste electrodes (CPE) is given in a previous paper [19]. To study the redox properties of the present CPs, the electrochemical properties of 1-CPE and 2-CPE were investigated in 0.1 M Na2SO4 at room temperature. As shown in Fig. 6, compared to the bare CPE, both 1-CPE and 2-CPE showed one quasireversible redox peak, such that the mean peak potential E 1/2 = (E pa + E pc)/2 was approximately 390 mV for 1-CPE and 386 mV for 2-CPE; these can be attributed to the CoIII/CoII redox couple [36]. The influence of varying scan rates on the electrochemical activities of 1-CPE and 2-CPE was also studied. As depicted in Fig. 6, the peak potentials changed gradually: the cathodic peak potentials shifted to negative direction and the corresponding anodic peak potentials shifted to positive direction. The anodic and cathodic peak currents are proportional to the scan rates, suggesting that the redox processes are close to being surface-controlled for both CPEs [37].

Cyclic voltammograms of 1-CPE and 2-CPE at different scan rates (from inner to outer): 20, 50, 80, 110, 140, 170, 200 mV s−1 in 0.1 M Na2SO4 aqueous solution. The inset shows the plots of the anodic and the cathodic peak currents versus scan rates
Photocatalytic activities
The UV/Vis diffuse reflectance spectra were recorded in order to obtain the band gaps (E g) of the two CPs [38, 39]. As depicted in Fig. 7a, CP 1 and CP 2 exhibit intense absorption bands at 260 and 320 nm, respectively, which can be attributed to π* → π or π* → n transitions of the ligand or metal-to-ligand charge transfer. An additional broad adsorption band in the visible region for both CPs may originate from the d → d spin-allowed transition of the Co(II) centers [40]. The band gaps (E g) were determined as the intersection point between the energy axis and the line extrapolated from the linear portion of the absorption edge in a plot of Kubelka–Munk function F versus energy E. The Kubelka–Munk function is given by:
where R is the reflectance of an infinitely thick layer at a given wavelength. Plots of F versus E for both CPs are shown in Fig. 7b, and E g values for CP 1 and CP 2 were obtained as 2.9 and 2.8 eV, respectively.

a UV–vis absorption spectra of CP 1 and CP 2. b Diffuse reflectance spectra of Kubelka–Munk functions versus energy (eV) for CP 1 and CP 2
Photocatalytic experiments for the degradation of methylene blue (MB) were carried out, and the detailed reaction process is given in the supporting information. The photodegradation of MB without any catalyst was investigated under the same conditions, as a control experiment. As shown in Figs. 8 and S4, the photodegradation efficiency of the control experiment under UV irradiation was only 13.9% after 105 min. CP 1 showed significant catalytic behavior, with photodegradation efficiencies of 55.4 and 90.0% under visible and UV light irradiations, respectively. Similarly, CP 2 also exhibited good catalytic behavior, with photodegradation efficiencies of 56.9 and 93.8% under visible/UV light irradiations, respectively. Hence, both CPs are promising candidates for the degradation of dyes, especially under UV irradiation. The slightly different catalytic behaviors of the two CPs may be due to the different coordination environments about the Co(II) centers [41]. However, the previously described CPs 3 and 4 showed higher photocatalytic activities than CPs 1 and 2 under UV irradiation (Table 4).

Experimental results of the photocatalytic degradation of methyl blue
To verify the possible photocatalytic reaction mechanism, trapping experiments for the active species of both CPs were conducted in the presence of tertiary butyl alcohol (TBA, a quencher of ·OH), benzoquinone (BQ, a quencher of ·O2 −), and ammonium oxalate (AO, a quencher of holes, h+). As shown in Fig. 9 and Fig. S5, the photodegradation of MB was affected by the addition of TBA, BQ, and AO; BQ in particular had the most significant effect on the photocatalytic removal of MB by both CPs. These observations suggest that among the photogenerated species ·O2 −, h+ and ·OH, the ·O2 − radical is the main active species in these experiments [42].

Trapping experiment of active species during the photocatalytic reaction for CP 1
Accordingly, a possible photocatalytic reaction mechanism for two CPs may be deduced. Under light irradiation, L1, L2, and/or the H2npht ligands encourage N and/or O to Co center charge transfer and it promote electrons from the HOMO to the LUMO. This concomitantly results in the same amount of holes (h+). The hydroxyl radical (·OH) is then generated by the combination of H+ with ·O2 − generated from the reduction of O2 by e−. In order for the CP to return to its stable HOMO state, one electron is captured from water, resulting in the formation of ·OH radicals. These ·OH active species can efficiently decompose the MB, giving the photocatalytic effect.
In addition, successive cycle experiments have been carried out, as shown in Fig. S6. The photodegradation efficiencies of both CPs as catalysts show no obvious decline over several experiments. PXRD and SEM images of both CPs were recorded after the photocatalytic reactions (Figs. 5 and S2). The PXRD patterns are nearly identical to those of the original CPs, while the SEM images indicate that the surfaces of the CPs before and after photocatalysis were quite similar. Hence, both CP 1 and CP 2 possess good stability toward photocatalysis and may be used repeatedly.
Conclusion
Two Co(II) coordination polymers based on semirigid bis(benzimidazole) ligands have been synthesized and characterized. CP 1 exhibits a 1D chain structure, further extended into a 3D supramolecular framework through C–H···O hydrogen bonding interactions, while CP 2 also shows a 1D loop-like chain, with a bilayer structure formed by π–π stacking interactions. Both of these CPs act as selective fluorescent sensors for acetone and also show photocatalytic behaviors for the decomposition of MB solutions under visible or UV light irradiation, although their efficiencies are lower than those of some related CPs, which were reported previously.
Supplementary materials
CCDC 1560789 and 1561077 contain the supplementary crystallographic data for CPs 1–2. These data can be obtained free of charge via http://www.ccdc.cam.ac.uk/conts/retrieving.html, or from the Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB21EZ, UK; fax: (+44) 1223-336-033; or e-mail: deposit@ccdc.cam.ac.uk.
References
Wan Z, Zhang G, Wu XY, Yin S (2017) Appl Catal B 207:17–26
Sun M, Wang Y, Shao Y, He YH, Zeng Q, Liang HK, Yan T, Du B (2017) J Colloid Interface Sci 501:123–132
Ghanbari F, Moradi M (2017) Chem Eng J 310:41–62
Zhang JW, Kan XM, Li XL, Luan J, Wang XL (2015) CrystEngComm 17:3887–3907
Wu XY, Qi HX, Ning JJ, Wang JF, Ren ZG, Lang JP (2015) Appl Catal B 168:98–104
Meng W, Xu ZQ, Ding J, Wu DQ, Han X, Hou HW, Fan YT (2014) Cryst Growth Des 14:730–738
Chen YQ, Li GR, Qu YK, Zhang YH, He KH, Gao Q, Bu XH (2013) Cryst Growth Des 13:901–907
Zhou HF, He T, Yue KF, Liu YL, Zhou CS, Yan N, Wang YY (2016) Cryst Growth Des 16:3961–3968
Hao SY, Hou SX, Van Hecke K, Cui GH (2017) Dalton Trans 47:1951–1964
Cui JW, An WJ, Van Hecke K, Cui GH (2016) Dalton Trans 45:17474–17484
Nath JK, Mondal A, Powell AK, Baruah JB (2014) Cryst Growth Des 14:4735–4748
Kang YF, Liu JQ, Liu B, Zhang WT, Liu Q, Liu P, Wang YY (2014) Cryst Growth Des 14:5466–5476
Li SB, Ma HY, Pang HJ, Zhang L (2014) Cryst Growth Des 14:4450–4460
Nobakht V, Beheshti A, Proserpio DM, Carlucci L, Abrahams CT (2014) Inorg Chim Acta 414:217–225
Wang X, Zhang J, Liu G, Lin H (2011) J Solid State Chem 184:280–288
Li X, Zhou P, Dong Y, Liu H (2015) J Inorg Organomet Polym Mater 4:650–656
Zhang MD, Qin L, Yang HT, Li YZ, Guo ZJ, Zheng HG (2013) Cryst Growth Des 13:1961–1969
Erer H, Yeşilel OZ, Arıcı M (2015) Cryst Growth Des 15:3201–3211
Wang XL, Le M, Lin HY, Luan J, Liu GC, Sui FF, Chang ZH (2015) Inorg Chem Front 2:373–387
He X, Lu XP, Li MX, Morris RE (2013) Cryst Growth Des 13:1649–1654
Liu L, Ding J, Li M, Lv XF, Wu J, Hou HW, Fan YT (2014) Dalton Trans 43:12790–12799
Shen Y, Fan CC, Wei YZ, Du J, Zhu HB, Zhao Y (2016) Cryst Growth Des 16:5859–5868
Wang XL, Gao Q, Liu GC, Lin HY, Tian AX, Li J (2011) Inorg Chem Commun 14:745–748
Liu L, Huang C, Xue X, Li M, Hou H, Fan Y (2015) Cryst Growth Des 15:4507–4517
Du M, Zhang ZH, You YP, Zhao XJ (2008) CrystEngComm 10:306–321
Abourahma H, Moulton B, Kravtsov V, Zaworotko MJ (2002) J Am Chem Soc 124:9990–9991
Abourahma H, Coleman AW, Moulton B, Rather B, Shahgaldian P, Zaworotko MJ (2001) Chem Commun 22:2380–2381
Li LL, Li HX, Ren ZG, Liu D, Chen Y, Zhang Y, Lang JP (2009) Dalton Trans 40:8567–8573
Sheldrick GM (1996) SADABS. University of Göttingen, Göttingen
Pro CA (2012) Data collection and data reduction software package. Agilent Technologies, Santa Clara
Sheldrick GM (2015) Acta Crystallogr Sect C 71:3–8
Hu JM, Guo R, Liu YG, Cui GH (2016) Inorg Chim Acta 450:418–425
Zhang X, Hou L, Liu B, Cui L, Wang YY, Wu B (2013) Cryst Growth Des 13:3177–3187
Yang YQ, Yang J, Kan WQ, Yang Y, Guo J, Ma JF (2013) Eur J Inorg Chem 2013:280–292
Pu AT, Yang J, Kan WQ, Yang Y, Ma JF (2013) Polyhedron 50:556–570
Hu JM, Blatov VA, Yu BY, Van Hecke K, Cui GH (2016) Dalton Trans 45:2426–2429
Cui GH, He CH, Jiao CH, Geng JC, Blatov VA (2012) CrystEngComm 14:4210–4216
Wen LL, Zhao JB, Lv KL, Wu YH, Deng KJ, Leng XK, Li DF (2012) Cryst Growth Des 12:1603–1612
Huan DH, Zhao YQ, Hao ZC, Cui GH (2016) Z Anorg Allg Chem 642:804–811
Sarma D, Ramanujachary KV, Lofland SE, Magdaleno T, Natarajan S (2009) Inorg Chem 48:11660–11676
Zhang X, Zhao YQ, Wang FS, Dong GY (2016) Chin J Struct Chem 35:765–773
Yu C, Li G, Kumar S, Yang K, Jin R (2014) Adv Mater 26:892–898
Acknowledgements
The project was supported by the National Natural Science Foundation of China (51474086), Natural Science Foundation – Steel and Iron Foundation of Hebei Province (B2015209299).
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Chang, H.N., Li, Y.H., Hao, Z.C. et al. Synthesis, crystal structures, luminescence, and photocatalytic properties of two 1D Co(II) coordination polymers constructed with semirigid bis(benzimidazole) and dicarboxylate ligands. Transit Met Chem 42, 783–793 (2017). https://doi.org/10.1007/s11243-017-0186-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11243-017-0186-0