
Abstract
A series of iminophosphineplatinum(II) complexes have been prepared from pro-ligands derived from aniline derivatives containing electron-donating methoxy groups or electron-withdrawing fluorides and [PtCl2(η 2 − coe)]2 (coe = cis-cyclooctene). All new pro-ligands and metal complexes have been fully characterized, including an X-ray diffraction study for compound 11 (derived from para-methoxyaniline). Additionally, the molecular structure of a di-iminophosphineplatinum dication 11a has been determined. The platinum compounds showed no appreciable cytotoxic properties against two glioma cell lines using the MTT method.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The discovery that cis-diamminedichloroplatinum(II) (cisplatin, or cis-DDP) inhibits cellular division in Escherichia coli spawned an enormous amount of research into the design and testing of platinum complexes for their potential ability to treat various cancers [1,2,3,4,5,6,7]. The mechanism of action of cisplatin is believed to result from the initial loss of the chloride ligands which are readily replaced by a molecule of water to give either cis-[(H3N)2PtCl(OH2)]+ or cis-[(H3N)2Pt(OH2)2]2+. The aqua ligands can readily be substituted with an N-heterocyclic base of DNA such as guanine or purine. The cytotoxic mode of action of cisplatin results from these interactions with DNA, especially intrastrand crosslink adducts, which activate signal transduction pathways such as those involving ATR, p53, p73 and MAPK, whereupon the end result is activation of apoptosis [8, 9]. Serious problems such as cisplatin resistance and side effects arising from cisplatin’s poor solubility in physiological media and lack of selectivity for cancer cells have limited its therapeutic use as an anticancer agent. As such, much research has focused on designing and testing the second- and third-generation platinum complexes by varying either the inert amine groups of the labile chloride ligands. Notable examples of such derivatives include carboplatin and cis-[PtCl2(1,4-DACH)] (DACH = diaminocyclohexane). Picoplatin is a promising candidate for the treatment of a variety of solid tumours and is unique in that it demonstrates that platinum complexes do not need to contain primary amine groups to have antineoplastic activities [10,11,12,13]. Indeed, we [14] and others [15, 16] have shown that complexes containing iminophosphine ligands are also potential candidates for metal-based anticancer therapy. Iminophosphines are a remarkable family of pro-ligands, whereby varying the organic group of the imine appendage allows for fine-tuning of the physicochemical properties of the corresponding platinum complexes. As part of our ongoing research in this area, we have prepared a series of platinum complexes containing iminophosphine ligands and examined their initial cytotoxic properties against two glioma cell lines using the MTT method, the results of which are presented herein.
Experimental section
Materials and methods
Reagents and solvents used were obtained from Aldrich Chemicals. [PtCl2(η2-coe)]2 (coe = cis-cyclooctene) [17], N-(2-(diphenylphosphino)benzylidene)aniline (1) [18], (E)-N-(2-(diphenylphosphino)benzylidene)-2-methoxyaniline (2) [19], (E)-N-(2-(diphenylphosphino)benzylidene)-4-methoxyaniline (4) [20], (E)-N-(2-(diphenylphosphino)benzylidene)-4-fluoroaniline (7) [21] and complex 8 [14] have been synthesized previously, and any additional data have been added below. NMR spectra were recorded on a JEOL JNM-GSX400 FT NMR (1H: 400 MHz; 13C: 100 MHz; 19F: 376 MHz; 31P: 162 MHz) spectrometer. Chemical shifts (δ) are reported in ppm [relative to residual solvent peaks (1H and 13C) and external CF3CO2H (19F) or H3PO4 (31P)]. Multiplicities are reported as singlet (s), doublet (d), triplet (t), multiplet (m) and overlapping (ov) with coupling constants (J) reported in Hz. Melting points were measured uncorrected with a Stuart SMP30 apparatus. FTIR spectra were obtained with a Thermo Fisher Scientific Nicolet iS5 FTIR spectrometer in ATR mode and are reported in cm−1. Elemental analyses for carbon, hydrogen and nitrogen were carried out at Laboratoire d’Analyse Élémentaire de l’Université de Montréal (Montréal, QC). All reactions were performed under a nitrogen atmosphere in a MBraun LabMaster glovebox.
General procedure for the synthesis of pro-ligands 1–6
To a mixture of 2-(diphenylphosphino)benzaldehyde (500 mg, 1.72 mmol) and the appropriate amine (1.81 mmol) was added formic acid (1 drop) in MeOH (5 mL). The reaction was allowed to proceed at RT for 18 h, at which point the iminophosphine pro-ligand was collected by suction filtration as a pale yellow precipitate. Spectroscopic NMR data were collected in CDCl3 as the pro-ligands decompose in wet DMSO-d6. The spectroscopically pure pro-ligands were used as prepared to make the corresponding platinum(II) complexes.
(E)-N-(2-(Diphenylphosphino)benzylidene)-2-methoxyaniline (2)
Yield: 449 mg (66%); m.p.: 108–109 °C.
(E)-N-(2-(Diphenylphosphino)benzylidene)-3-methoxyaniline (3)
Yield: 490 mg (72%); m.p.: 89–91 °C. 1H NMR (CDCl3) δ: 9.04 (d, J HP = 5.0 Hz, 1H, C(H)=N), 8.18 (ddd, J HH = 7.8 Hz, J HP = 4.1 Hz, J HH = 1.4 Hz, 1H, Ar), 7.45 (app t, J HH = 7.3 Hz, 1H, Ar), 7.36–7.28 (ov m, 11H, Ar), 7.18 (t, J HH = 7.8 Hz, 1H, Ar), 6.91 (ddd, J HH = 7.8 Hz, J HP = 4.6 Hz, J HH = 0.9 Hz, 1H, Ar), 6.72 (ddd, J HH = 8.2, J HP = 2.7, J HH = 0.9 Hz, 1H, Ar), 6.50 (dd, J HH = 7.8, 0.9 Hz, 1H, Ar), 6.43 (ov dd, J HP = 2.3, J HH = 1.8 Hz, 1H, Ar), 3.73 (s, 3H, OCH 3). 13C{1H} NMR (CDCl3) δ: 160.2, 159.1 (d, J CP = 21.0 Hz), 153.1, 139.1 (d, J CP = 16.2 Hz), 138.7 (d, J CP = 20.0 Hz), 136.5 (d, J CP = 9.5 Hz), 134.3, 134.1, 133.6, 131.0, 129.8, 129.0, 128.8 (d, J CP = 6.7 Hz), 128.3 (d, J CP = 3.8 Hz), 113.0, 112.3, 106.4, 55.4. 31P{1H} NMR (CDCl3) δ: −12.4. IR: 3060 (w), 1619 (s, νCN), 1577 (m), 1462 (m), 1434 (m), 1263 (m), 1144 (m), 1035 (m), 744 (m).
(E)-N-(2-(Diphenylphosphino)benzylidene)-4-methoxyaniline (4)
Yield: 551 mg (81%); m.p.: 116–118 °C.
(E)-N-(2-(Diphenylphosphino)benzylidene)-2-fluoroaniline (5)
Yield: 422 mg (64%); m.p.: 110–111 °C. 1H NMR (CDCl3) δ: 9.17 (d, J HP = 5.5 Hz, 1H, C(H)=N), 8.27 (ddd, J HH = 7.8 Hz, J HP = 4.1 Hz, J HH = 1.4 Hz, 1H, Ar), 7.46 (app t, J HH = 7.4 Hz, 1H, Ar), 7.38–7.28 (ov m, 11H, Ar), 7.13–7.00 (ov m, 3H, Ar), 6.93 (ddd, J HH = 7.8 Hz, J HP = 5.0 Hz, J HH = 1.4 Hz, 1H, Ar), 6.70 (ov ddd, J HH = 7.8 Hz, J HF = 7.8 Hz, J HH = 1.4 Hz, 1H, Ar). 13C{1H} NMR (CDCl3) δ: 161.2 (dd, J CP = 23.8 Hz, J CF = 1.9 Hz), 155.4 (d, J CF = 248.0 Hz), 139.8 (d, J = 10.5 Hz), 139.1 (d, J = 17.2 Hz), 138.9 (d, J = 21.9 Hz), 136.2 (d, J = 9.5 Hz), 134.2 (d, J = 20.0 Hz), 133.6, 131.4, 129.2, 129.1, 128.8 (d, J = 7.6 Hz), 128.2 (d, J = 3.8 Hz), 126.9 (d, J = 7.6 Hz), 124.5 (d, J = 3.8 Hz), 121.8, 116.2 (d, J = 20.0 Hz). 19F{1H} NMR (CDCl3) δ: −126.6. 31P{1H} NMR (CDCl3) δ: −13.5. IR: 3049 (w), 1619 (s, νCN), 1578 (m), 1478 (s), 1453 (m), 1279 (m), 1104 (m), 733 (s), 692 (s).
(E)-N-(2-(Diphenylphosphino)benzylidene)-3-fluoroaniline (6)
Yield: 481 mg (73%); m.p.: 103–104 °C. 1H NMR (CDCl3) δ: 9.01 (d, J HP = 5.5 Hz, 1H, C(H)=N), 8.17 (ddd, J HH = 7.8 Hz, J HP = 4.1 Hz, J HH = 1.4 Hz, 1H, Ar), 7.46 (ov ddd, J HH = 7.4, 7.4, 0.9 Hz, 1H, Ar), 7.38–7.28 (ov m, 11H, Ar), 7.22 (m, 1H, Ar), 6.93 (ddd, J HH = 7.8 Hz, J HP = 4.6 Hz, J HH = 0.9 Hz, 1H, Ar), 6.85 (ov dddd, J HH = 7.8 Hz, 7.8 Hz, J HF = 2.3 Hz, J HH = 1.4 Hz, 1H, Ar), 6.66 (ddd, J HH = 7.8, 1.8, 0.9 Hz, 1H, Ar), 6.55 (ov ddd, J HF = 10.1 Hz, J HH = 1.8 Hz, J HH = 1.8 Hz, 1H, Ar). 13C{1H} NMR (CDCl3) δ: 163.2 (d, J CF = 245.0 Hz), 159.9 (d, J CP = 21.9 Hz), 153.5 (J = 8.6 Hz), 139.0 (d, J = 20.0 Hz), 138.8 (d, J = 16.2 Hz), 136.3 (d, J = 8.6 Hz), 134.2 (d, J = 20.0 Hz), 133.6, 131.3, 130.2 (d, J = 8.6 Hz), 129.1, 129.0, 128.8 (d, J = 7.6 Hz), 128.4 (d, J = 3.8 Hz), 116.8 (d, J = 1.9 Hz), 112.7 (d, J = 21.0 Hz), 108.2 (d, J = 22.9 Hz). 19F{1H} NMR (CDCl3) δ: −113.2. 31P{1H} NMR (CDCl3) δ: −12.5. IR: 3053 (w), 1597 (s, νCN), 1583 (m), 1476 (m), 1433 (m), 1252 (m), 945 (m), 695 (s).
Synthesis of (E)-N-(2-(diphenylphosphino)benzylidene)-4-fluoroaniline (7)
A THF (10 mL) solution of 2-(diphenylphosphino)benzaldehyde (500 mg, 1.72 mmol) and 4-fluoroaniline (191 mg, 1.72 mmol) was allowed to react in the presence of activated 3 Å molecular sieves (5 g) over a period of 7 days. The mixture was filtered, and solvent was removed under vacuum to afford an orange oil. The oil was dissolved in hexane (5 mL) and stored at −30 °C. The resulting precipitate was collected by suction filtration to afford 7 as a pale yellow solid. Yield: 514 mg (78%); m.p.: 83–84 °C. 13C{1H} NMR (CDCl3) δ: 161.3 (d, J CF = 243.1 Hz), 158.7 (d, J CP = 21.0 Hz), 147.7 (d, J = 2.9 Hz), 139.1 (d, J = 16.2 Hz), 138.7 (d, J = 20.0 Hz), 136.5 (d, J = 8.6 Hz), 134.2 (d, J = 20.0 Hz), 133.7, 131.1, 129.1, 128.8, 128.7, 128.3 (d, J = 3.8 Hz), 122.5 (d, J = 8.6 Hz), 115.8 (d, J = 21.9 Hz).
General synthesis of iminophosphineplatinum(II) complexes 9–14
To a stirred THF (5 mL) suspension of [PtCl2(η 2-coe)]2 (200 mg, 0.27 mmol) was added a THF (2 mL) solution of the appropriate iminophosphine pro-ligand (0.55 mmol). The reaction was allowed to proceed at RT for 18 h, at which point a yellow–orange precipitate was collected by suction filtration. The final purification steps for each complex are detailed below. Spectroscopic NMR data were collected in DMSO-d6 due to the poor solubility of the complexes in CDCl3.
Synthesis of 9
Washing with hexane (3 × 5 mL) afforded 9 as a yellow–orange solid. Yield: 296 mg (83%); m.p.: 198–200 °C. 1H NMR (DMSO-d6) δ: 8.82 (s, J HPt = 97.6 Hz, 1H, C(H)=N), 8.04 (m, 1H, Ar), 7.84 (m, 2H, Ar), 7.62–7.54 (ov m, 6H, Ar), 7.49–7.44 (ov m, 4H, Ar), 7.21 (t, J = 7.3 Hz, 1H, Ar), 7.12 (m, 1H, Ar), 7.08 (d, J = 7.8 Hz, 1H, Ar), 7.02 (d, J = 8.2 Hz, 1H, Ar), 6.91 (t, J = 7.8 Hz, 1H, Ar), 3.64 (s, 3H, OCH 3). 13C{1H} NMR (DMSO-d6) δ: 168.0 (d, J CP = 6.7 Hz), 152.2, 142.2, 138.1 (d, J CP = 7.6 Hz), 136.8 (d, J CP = 12.4 Hz), 135.4 (d, J CP = 7.6 Hz), 134.2 (d, J CP = 11.4 Hz), 133.6 (d, J CP = 27.7 Hz), 132.5, 129.3 (d, J CP = 11.4 Hz), 127.2, 126.5, 125.9, 121.5, 120.9, 119.9, 112.6, 56.4. 31P{1H} NMR (DMSO-d6) δ: 3.8 (J PPt = 3720 Hz). IR: 3061 (w), 1613 (m, νCN), 1493 (m), 1436 (s), 1258 (m), 1100 (s), 1018 (m), 690 (s). Anal. calcd. for C26H22NCl2OPPt (661.42 g/mol) (%): C 47.21, H 3.35, N 2.12; found: C 47.48, H 3.66, N 2.32.
Synthesis of 10
Washing with hexane (3 × 5 mL) afforded 10 as a yellow solid. Yield: 265 mg (74%); m.p.: 268–270 °C. 1H NMR (DMSO-d6) δ: 8.83 (s, J HPt = 97.1 Hz, 1H, C(H)=N), 8.11 (m, 1H, Ar), 7.86 (ov dd, J = 7.8, 7.4 Hz, 1H, Ar), 7.80 (ov dd, J = 7.8, 7.4 Hz, 1H, Ar), 7.62–7.55 (ov m, 6H, Ar), 7.47–7.42 (ov m, 4H, Ar), 7.24 (ov dd, J = 8.2, 7.8 Hz, 1H, Ar), 7.02 (app t, J = 7.8, 7.8 Hz, 1H, Ar), 6.94 (d, J = 8.2 Hz, 1H, Ar), 6.88 (s, 1H, Ar), 6.84 (d, J = 8.2 Hz, 1H, Ar), 3.73 (s, 3H, OCH 3). 13C{1H} NMR (DMSO-d6) δ: 167.2 (d, J CP = 6.7 Hz), 159.3, 154.0, 137.9 (d, J CP = 8.6 Hz), 137.1 (d, J CP = 13.4 Hz), 135.2 (d, J CP = 7.6 Hz), 134.2 (d, J CP = 11.4 Hz), 133.4, 133.0, 132.8, 129.5 (d, J CP = 11.4 Hz), 128.7, 125.8, 125.1, 121.9, 121.3, 117.0, 113.7, 110.5, 56.0. 31P{1H} NMR (DMSO-d6) δ: 5.7 (J PPt = 3660 Hz). IR: 3054 (w), 1598 (m, νCN), 1479 (m), 1435 (s), 1289 (s), 1273 (m), 1141 (m), 1102 (m), 693 (s). Anal. calcd. for C26H22NCl2OPPt (661.42 g/mol) (%): C 47.21, H 3.35, N 2.12; found: C 46.88, H 3.47, N 2.44.
Synthesis of 11
Complex 11 was isolated as a yellow solid by recrystallization from a solution of CH2Cl2: hexane (10 mL: 5 mL) stored at 5 °C. Yield: 211 mg (59%); m.p.: 262–263 °C. 1H NMR (DMSO-d6) δ: 8.78 (s, J HPt = 87.9 Hz, 1H, C(H)=N), 8.08 (dd, J HH = 6.4 Hz, J HP = 4.1 Hz, 1H, Ar), 7.84 (ov dd, J HH = 7.8, 7.3 Hz, 1H, Ar), 7.78 (ov dd, J HH = 7.8, 7.4 Hz, 1H, Ar), 7.62–7.54 (ov m, 6H, Ar), 7.47–7.42 (ov m, 4H, Ar), 7.38 (d, J HH = 9.2 Hz, 2H, Ar), 7.03 (dd, J HP = 10.5, J HH = 7.8 Hz, 1H, Ar), 6.88 (d, J = 9.2 Hz, 2H, Ar), 3.73 (s, 3H, OCH 3). 13C{1H} NMR (DMSO-d6) δ: 166.1 (d, J CP = 6.7 Hz), 159.2, 146.4, 137.4, 137.3 (d, J CP = 22.0 Hz), 134.9 (d, J CP = 8.6 Hz), 134.3 (d, J CP = 10.5 Hz), 133.4, 132.8 (2C), 129.5 (d, J CP = 11.5 Hz), 125.7, 125.6, 125.1, 122.0, 121.4, 113.7, 55.9. 31P{1H} NMR (DMSO-d6) δ: 5.8 (J PPt = 3660 Hz). IR: 3057 (w), 1608 (m, νCN), 1504 (m), 1437 (s), 1256 (s), 1020 (m), 765 (m), 692 (s). Anal. calcd. for C26H22NCl2OPPt (661.42 g/mol) (%): C 47.21, H 3.35, N 2.12; found: C 47.38, H 3.55, N 2.54.
Synthesis of 12
Compound 12 was purified by recrystallization from CH2Cl2 (5 mL) stored at 5 °C. Yield: 238 mg (68%); m.p.: 241–243 °C. 1H NMR (DMSO-d6) δ: 9.01 (s, J HPt = 85.2 Hz, 1H, C(H)=N), 8.08 (dd, J HH = 8.7 Hz, J HP = 4.2 Hz, 1H, Ar), 7.84 (dd, J HH = 6.8, J HF = 2.3 Hz, 2H, Ar), 7.63–7.53 (ov m, 6H, Ar), 7.50–7.45 (ov m, 4H, Ar), 7.30–7.13 (ov m, 5H, Ar). 13C{1H} NMR (DMSO-d6) δ: 169.6 (d, J CP = 6.7 Hz), 154.8 (d, J CF = 246.0 Hz), 140.7 (d, J = 11.5 Hz), 138.6 (d, J = 8.6 Hz), 136.4 (d, J = 13.4 Hz), 135.9 (d, J = 7.7 Hz), 134.3 (d, J = 10.5 Hz), 133.8 (d, J = 2.9 Hz), 133.6, 132.7, 129.8 (d, J = 7.7 Hz), 129.4 (d, J = 12.5 Hz), 126.9, 126.6, 125.9, 124.3 (d, J = 3.8 Hz), 121.6, 121.0, 116.2 (d, J = 19.2 Hz). 19F{1H} NMR (DMSO-d6) δ: −123.4. 31P{1H} NMR (DMSO-d6) δ: 4.7 (J PPt = 3690 Hz). IR: 3058 (w), 1597 (m, νCN), 1493 (m), 1435 (m), 1219 (m), 1103 (s), 690 (s). Anal. calcd. for C25H19NCl2FPPt (649.39 g/mol) (%): C 46.24, H 2.95, N 2.16; found: C 46.52, H 3.14, N 2.45.
Synthesis of 13
Compound 13 was purified by recrystallization from CH2Cl2 (5 mL) stored at 5 °C. Yield: 252 mg (72%); m.p.: 253–255 °C. 1H NMR (DMSO-d6) δ: 8.88 (s, J HPt = 85.2 Hz, 1H, C(H)=N), 8.11 (dd, J HH = 6.4 Hz, J HP = 4.1 Hz, 1H, Ar), 7.89–7.80 (ov m, 2H, Ar), 7.62–7.55 (ov m, 5H, Ar), 7.49–7.44 (ov m, 3H, Ar), 7.42 (m, 1H, Ar), 7.24 (ov dd, J HF = 12.4 Hz, J HH = 8.2 Hz, 2H, Ar), 7.15–7.05 (ov m, 2H, Ar), 7.03–6.96 (ov m, 2H, Ar). 13C{1H} NMR (DMSO-d6) δ: 168.2 (d, J CP = 6.7 Hz), 161.7 (d, J CF = 244.4 Hz), 154.3 (d, J = 9.6 Hz), 138.3 (d, J = 8.6 Hz), 136.8 (d, J = 12.5 Hz), 135.5 (d, J = 8.6 Hz), 134.3 (d, J = 11.4 Hz), 133.5, 133.2 (d, J = 2.9 Hz), 132.8, 130.3 (d, J = 8.6 Hz), 129.6 (d, J = 11.5 Hz), 125.9, 125.2, 121.9, 121.3, 121.1, 115.2 (d, J = 21.1 Hz), 112.2 (d, J = 24.9 Hz). 19F{1H} NMR (DMSO-d6) δ: −107.6. 31P{1H} NMR (DMSO-d6) δ: 5.3 (J PPt = 3640 Hz). IR: 3052 (w), 1591 (m, νCN), 1484 (m), 1436 (m), 1255 (m), 1102 (m), 1079 (s), 693 (s). Anal. calcd. for C25H19NCl2FPPt (649.39 g/mol) (%): C 46.24, H 2.95, N 2.16; found: C 46.66, H 2.53, N 2.57.
Synthesis of 14
Complex 14 was isolated as a yellow solid by recrystallization from a solution of CH2Cl2: hexane (10 mL: 5 mL) stored at 5 °C. Yield: 295 mg (84%); m.p.: 260 °C. 1H NMR (DMSO-d6) δ: 8.85 (s, J HPt = 98.3 Hz, 1H, C(H)=N), 8.10 (dd, J HH = 6.4 Hz, J HP = 4.1 Hz, 1H, Ar), 7.86 (app t, J HH = 7.3 Hz, 1H, Ar), 7.81 (app t, J HH = 7.3 Hz, 1H, Ar), 7.63–7.54 (ov m, 6H, Ar), 7.48–7.42 (ov m, 6H, Ar), 7.21 (app t, J = 8.7 Hz, 2H, Ar), 7.06 (dd, J HP = 10.1, J HH = 7.8 Hz, 1H, Ar). 13C{1H} NMR (DMSO-d6) δ: 167.5 (d, J CP = 6.7 Hz), 161.6 (d, J CF = 243.1 Hz), 149.5, 138.0 (d, J = 8.6 Hz), 137.0 (d, J = 13.4 Hz), 135.2 (d, J = 8.6 Hz), 134.3 (d, J = 10.5 Hz), 133.4, 133.1 (d, J = 2.9 Hz), 132.8 (d, J = 1.9 Hz), 129.5 (d, J = 11.4 Hz), 126.5 (d, J = 8.6 Hz), 125.9, 125.3, 121.9, 121.3, 115.4 (d, J = 22.9 Hz). 19F{1H} NMR (DMSO-d6) δ: −113.0. 31P{1H} NMR (DMSO-d6) δ: 5.5 (J PPt = 3650 Hz). IR: 3061 (w), 1609 (m, νCN), 1502 (s), 1436 (m), 1238 (m), 1160 (m), 1103 (s), 835 (m), 690 (s). Anal. calcd. for C25H19NCl2FPPt (649.39 g/mol) (%): C 46.24, H 2.95, N 2.16; found: C 46.18, H 2.52, N 2.34.
Stability of compounds in dimethyl formamide
Solutions of the compounds in wet DMF were monitored by 31P{1H} NMR spectroscopy over a period of 2 days at 37 °C. Evidence of 2-diphenylphosphinobenzaldehyde was observed in solutions of the pro-ligands (demonstrating decomposition to starting materials); therefore, they were not included in the biological testing. As no decomposition of the platinum complexes 8–14 was observed, biological studies of these compounds were initiated.
X-ray crystallography
Crystals of 11 and 11a were grown from a CH2Cl2/CH3Cl: hexane solution stored at RT. Crystals were attached to the tip of a 400 μm MicroLoop with Paratone-N oil. Measurements were made on a Bruker APEXII CCD equipped diffractometer (30 mA, 50 mV) using monochromatic Mo-Kα radiation (λ = 0.71073 Å) at 125 K. The initial orientation and unit cell were indexed using a least-squares analysis of a random set of reflections collected from three series of 0.5° wide scans, 10 s per frame and 12 frames per series that were well distributed in reciprocal space. For data collection, four ω-scan frame series were collected with 0.5° wide scans, 5 (11) or 30 (11a) second frames and 416 frames per series at varying φ angles (φ = 0°, 90°, 180°, 270°). The crystal to detector distance was set to 6 cm, and a complete sphere of data was collected. Cell refinement and data reduction were performed with the Bruker SAINT software, which corrects for beam inhomogeneity, possible crystal decay, Lorentz and polarization effects. Data processing and a multi-scan absorption correction were applied using the APEX2 software package [22]. The structure was solved using direct methods [23]. All non-hydrogen atoms were refined anisotropically. In general, hydrogen atoms were included at geometrically idealized positions with coupled isotropic temperature factors and were fixed (C-H, Ar-H), or in the case of methyl groups, the dihedral angle of the idealized tetrahedral CH3 fragment was allowed to refine. There was a molecule of dichloromethane that was co-crystallized with 11. Figures were made using Ortep-3 for Windows [24].
In the initial models in the case of 11a, there was significant excess electron density around the dichloromethane molecules. After running a variety of models, it was deduced that the electron density was part of a water molecule. In the case of the co-crystallized water molecule, the O-H bonds were fixed at 0.90 Å and the H-H 1,3-distance was set to 1.40 Å and the H atoms were positioned to have a hydrogen bond interaction with Cl of dichloromethane. Attempts to provide a disorder model to account for the excess electron density near the Cl atoms of the dichloromethane molecules were unsuccessful and were thus left as is.
Cell cultures
Human glioma cells Hs683 and T98G were cultured in an incubator at 37 °C and 5% CO2 in DMEM supplemented with 10% FBS (foetal bovine serum) and 1% antibiotics (Thermo Fisher Scientific) and have been previously characterized [25]. Hs683 cells were originally provided by Dr. Adrian Merlo (Basel, Switzerland), while T98G cells were purchased from American Type Culture Collection (ATCC, #CRL-1690).
MTT assays
A total of 10,000 cells were seeded in triplicates in 96-well plates in 200 μL of cell culture medium. Cells were incubated at 37 °C and 5% CO2 for 24 h. Medium was replaced with medium containing 1, 10 or 100 μM of the required complex dissolved in DMF. Cells were incubated for an additional 48 h. DMF was used instead of complexes in control wells. Following incubation, 20 μL of 5 mg/mL MTT in PBS was added to each well. Cells and MTT were incubated for 3 h and subsequently removed. Stained cells were re-suspended in 100 μL of a 1:24 1 M HCl/95% EtOH solution and read at 560 nm on a Varioskan (Scanlab). Experiments were performed as described twice.
LC50 cytotoxicity assays
MTT assays were used to calculate the experimental LC50 values for the compounds. Hs683 or T98G cells were seeded at a density of 10,000 cells per well in 96-well plates and cultured as above for 24 h. Cells were then treated with the complexes at final concentrations of 0.1, 0.5, 1, 10 and 100 μM, 150 and 250 μM. DMF was used to dissolve the compounds at the appropriate concentrations. Each compound, at each final concentration, was assessed in triplicate wells. DMF-only treatment was also assessed in triplicate and served as a control. Cells were incubated at 37 °C and 5% CO2 for 48 h following treatment. Cells were then stained, incubated, re-suspended and absorbance values collected as above. Data were converted to the percentage of cell viability compared to solvent control (DMF only) wells from the same experiment. LC50 values were calculated via the GraphPad Prism 6 software. LC50 experiments were repeated twice.
Results and discussion
In order to overcome some of the limitations inherent within cisplatin therapy, a considerable amount of research has recently focused on the synergistic use of radiotherapy and platinum chemotherapy for the treatment of glioblastomas [26,27,28,29,30,31,32,33]. This unique combination allows for reduction in the platinum dosage, and thereby alleviates some of the problematic side effects. Gliomas are a common and deadly form of brain cancer which are typically classified into four clinical grades, of which glioblastoma multiforme (GBM) is the most aggressive. The median survival period for patients diagnosed with GBM is only 12 months. The tumours that infiltrate into regions of the brain complicate surgical extraction so it is understandable that the quest for efficacious chemotherapeutic agents to treat GBM is of utmost importance in an effort to improve this combination of cancer treatment. As mentioned previously, we have found that platinum(II) complexes bearing iminophosphine ligands containing boronate ester groups showed moderate anticancer activity against two glioma cell lines [14]. With this in mind, we have prepared iminophosphineplatinum complexes 9–14 containing electron-donating methoxy derivatives and electron-withdrawing fluoride groups along with the aniline derivative 8 and examined these species for their cytotoxic properties against two glioma cell lines using the MTT method.
The synthesis of pro-ligands 1–7 was performed under an atmosphere of dinitrogen, since the resulting iminophosphines readily decompose back to the starting materials in the presence of water. Formation of the pro-ligands was confirmed by multinuclear NMR spectroscopy where the aldehyde proton at 10.50 ppm disappears and a new peak is observed at ca. 9 ppm for the imine proton in the 1H NMR spectra. Imine formation is further confirmed by 13C{1H} NMR data, since the aldehyde C=O resonance at 191.8 ppm for 2-diphenylphosphinobenzaldehyde disappears as the appearance of a new peak at ca. 160 ppm for the C=N bonds is observed.
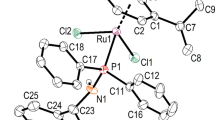
Iminophosphineplatinum(II) complexes (8–14) were prepared in moderate to good yields (Scheme 1) by the addition of pro-ligands 1–7 to THF suspensions of [PtCl2(η2-coe)]2 (coe = cis-cyclooctene). The platinum compounds have been characterized using a number of physical methods, including multinuclear NMR and FTIR spectroscopy, as well as elemental analysis. Coordination of the iminophosphine to the metal centre was confirmed by 1H NMR spectroscopy where an upfield shift is observed for the imine proton from around 9 ppm to ca. 8.4 ppm (in CDCl3) with associated 195Pt satellites (avg. J HPt = 92 Hz). Upon coordination, a significant shift is also observed in the 31P{1H} spectra, from roughly −13 ppm for the free pro-ligands to around 5 ppm for complexes 8–14. Furthermore, the 195Pt satellites observed with these resonances display coupling constants (J PPt = 3640 to 3720 Hz) which are well within the range reported for related species. The decrease in the νC=N stretching vibration band in the IR spectra from ~1619 cm−1 for the free iminophosphines to ~1603 cm−1 is indicative of coordination of the imine moiety to a metal centre [34, 35]. We carried out a single-crystal X-ray diffraction study on the para-methoxy derivative 11 in order to confirm the solid-state structure of these complexes, the molecular structure of which is shown in Fig. 1. Crystallographic data are provided in Table 1 and selected bond distances and angles in Table 2. The molecule assumes a distorted square planar geometry about the platinum atom as is evidenced by the bond angles N(1)-Pt(1)-Cl(1) and P(1)-Pt(1)-Cl(2) which are 176.0(2)° and 171.54(7)°, respectively, and are significantly less than 180°. The ligand coordinates to the metal in a κ2-P,N fashion where the stronger trans-effect of the diphenylphosphino group over the amine is observed, as the Pt(1)-Cl(1) distance of 2.293(2) Å is noticeably shorter than that of the Pt(1)-Cl(2) distance of 2.372(2) Å [36]. The nitrogen–platinum and phosphorus–platinum bond distances of 2.051(7) Å and 2.197(2), respectively, are similar to those reported in other platinum systems [37,38,39]. For instance, the nitrogen–platinum and phosphorus–platinum bond distances in the related complex (κ2-P,N-2-Ph2PC6H4C(H) = N-2,6-iPr2C6H3)PtCl2 are 2.0421(18) and 2.2128(6) Å, respectively [15]. The corresponding N(1)-Pt-P(1) bond angle in this related complex is 89.80(5)°, whereas the same angle in 11 is 86.4(3). The imine N(1)-C(1) distance of 1.287(11) Å in 11 is in the range of accepted carbon–nitrogen double bonds. A single crystal of the minor product 11a was also solved by an X-ray diffraction study. The molecule structure consists of two ligands coordinated to the platinum dication surrounded by two outer-sphere Cl- anions and solvent molecules (CHCl3, CH2Cl2 and H2O). The molecular structure of the dication is presented in Fig. 2, with crystallographic data in Table 1 and selected bond lengths and angles in Table 3. The Pt(1)-N(1) distance of 2.121(2) is similar to that of 11, and the C(19)-N(1) distance of 1.292(4) is indicative of a carbon–nitrogen double bond. The platinum centre exists in a slightly distorted square planar configuration, as demonstrated by the sum of angles around Pt of 360.95(34)°. The ligands are arranged so that the phosphine and imine fragments are trans to one another. The Pt-N and Pt-P bond lengths are 2.120(2) and 2.2509(7) Å, respectively, and are much longer than observed in 11, presumably due to either the steric effects of the two iminophosphine ligands, or due to differing trans-effects when compared to the Cl ligands in 11. Related cis-diaminophosphineplatinum(II) complexes have been reported previously [40,41,42,43,44,45,46]. For instance, the disparate Pt-N and Pt-P bond lengths in [Pt{(NC5H4)[(Ph2PNH)C(=NH)]-4}{(NC5H4)[(Ph2PNH)C(=N)]-4}][BF4] [44] are 2.07(2)/2.03(2) and 2.218(6)/2.246(6) Å, respectively. The coordination chemistry of a wide array of P,N-ligands with late metals is an area of considerable interest [47,48,49].

Synthesis of iminophosphineplatinum(II) complexes 8–14. 1: R1=R2=R3=H; 2: R1=OMe,R2=R3=H; 3: R1=R3=H,R2=OMe; 4: R1=R2=H,R3=OMe; 5: R1=F,R2=R3=H; 6: R1=R3=H,R2=F; 7: R1=R2=H,R3=F; 8: R1=R2=R3=H; 9: R1=OMe,R2=R3=H; 10: R1=R3=H,R2=OMe; 11: R1=R2=H,R3=OMe; 12: R1=F,R2=R3=H; 13: R1=R3=H,R2=F; 14: R1=R2=H,R3=F

The molecular structure of 11 drawn with 50% probability ellipsoids and hydrogen atoms and a molecule of solvent removed for clarity

The molecular structure of the dication 11a drawn with 50% probability ellipsoids and hydrogen atoms and solvent molecules removed for clarity
We then decided to examine the anticancer potential of compounds 8–14 against two glioma cell lines using the MTT method, but not one of these derivatives showed any appreciable bioactivity. These results are in contrast to our previous study, wherein complexes of the type (κ2-P,N-2-Ph2PC6H4C(H)=NC6H4X)PtCl2 (X = Bpin, pin = 1,2-O2C2Me4) showed moderate anticancer activities, suggesting it is the boron group that imparts bioactivity [14].
Conclusion
Seven iminophosphine compounds were prepared from the condensation of 2-diphenylphosphinobenzaldehyde and aniline, and its derivatives containing electron-donating methoxy groups and electron-withdrawing fluorides. The corresponding platinum(II) complexes were formed by addition of the pro-ligands to [PtCl2(η2-coe)]2 (coe = cis-cyclooctene). All new pro-ligands and metal complexes were fully characterized by multinuclear NMR and FTIR spectroscopy, and elemental analysis for the platinum species. A single-crystal X-ray diffraction study was performed for 11, the platinum complex containing the para-methoxy iminophosphine ligand. The structure of the unique cis-diiminophosphineplatinum(II) dication 11a was also determined through a single-crystal X-ray diffraction study. Platinum compounds 8–14 showed minimal cytotoxic effects against two glioma cell lines.
Supplemental material
Full supplemental crystallographic data in CIF format have been deposited with the Director, Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge, CB2 UK (fax: +44 1223 336033 or e-mail: deposit@ccdc.cam.ac.uk or www.ccdc.cam.ac.uk) and are available on request, quoting deposition numbers 1557055 (11) and 1557056 (11a).
References
Hannon MJ (2007) Pure Appl Chem 79:2243
Barry NPE, Sadler PJ (2013) ACS Nano 7:5654
de Biasi AR, Villena-Vargas J, Adusumilli PS (2014) Clin Cancer Res 20:5384
Medici S, Peana M, Nurchi VM, Lachowicz JI, Crisponi G, Zoroddu MA (2015) Coord Chem Rev 284:329
Nogales V, Reinhold WC, Varma S, Martinez-Cardus A, Moutinho C, Moran S, Heyn H, Sebio A, Barnadas A, Pommier Y, Esteller M (2016) Oncotarget 7:3084
Doucette KA, Hassell KN, Crans DC (2016) J Inorg Biochem 165:56
Johnstone TC, Suntharalingam K, Lippard SJ (2016) Chem Rev 116:3436
Dasari S, Tchounwou PB (2014) Eur J Pharmacol 740:364
Siddik ZH (2003) Oncogene 22:7265
Wheate NJ, Walker S, Craig GE, Oun R (2010) Dalton Trans 39:8113
Posadas I, Alonso-Moreno C, Bravo I, Carrillo-Hermosilla F, Garzón A, Villaseca N, López-Solera I, Albaladejo J, Ceña V (2017) J Inorg Biochem 168:46
Altoum AOS, Alhoshani A, Alhosaini K, Altaf M, Ahmad S, Popoola SA, Al-Saadi AA, Sulaiman AA, Isab AA (2017) J Coord Chem 70:1020
Hamilton G, Olszewski U (2013) Expert Opin Drug Metab Toxicol 9:1381
St-Coeur P-D, Kinley S, Vogels CM, Decken A, Morin P Jr, Westcott SA (2017) Can J Chem 95:207
Chiririwa H, Moss JR, Hendricks D, Smith GS, Meijboom R (2013) Polyhedron 49:29
Chiririwa H, Moss JR, Hendricks D, Meijboom R, Muller A (2013) Transit Met Chem 38:165
Shaver MP, Vogels CM, Wallbank AI, Hennigar TL, Biradha K, Zaworotko MJ, Westcott SA (2000) Can J Chem 78:568
Chen X, Femia FJ, Babich JW, Zubieta J (2001) Inorg Chim Acta 315:147
Crochet P, Gimeno J, Borge J, García-Granda S (2003) New J Chem 27:414
Antonaroli S, Crociani B (1998) J Organomet Chem 560:137
Scrivanti A, Matteoli U, Beghetto V, Antonaroli S, Crociani B (2002) Tetrahedron 58:6881
Bruker (2008) APEX2 version 2008.5. Bruker AXS, Inc., Madison
Sheldrick GM (2008) Acta Cryst A 64:112
Farrugia LJ (1997) J Appl Cryst 30:565
Ishii N, Maier D, Merlo A, Tada M, Sawamura Y, Diserens AC, Van Meir EG (1999) Brain Pathol 9:469
Margiotta N, Denora N, Ostuni R, Laquintana V, Anderson A, Johnson SW, Trapani G, Natile G (2010) J Med Chem 53:5144
Charest G, Paquette B, Fortin D, Mathieu D, Sanche L (2010) J Neurooncol 97:187
Gwak H-S, Shingu T, Chumbalkar V, Hwang Y-H, DeJournett R, Latha K, Koul D, Yung WKA, Powis G, Farrell NP, Bögler O (2011) Int J Cancer 128:787
Maksimović-Ivanić D, Mijatović S, Mirkov I, Stošić-Grujičić S, Miljković D, Sabo TJ, Trajković V, Kaluđerović GN (2012) Metallomics 4:1155
Mihajlović LE, Savić A, Poljarević J, Vučković I, Mojić M, Bulatović M, Maksimović-Ivanić D, Mijatović S, Kaluđerović GN, Stošić-Grujičić S, Milijković D, Grgurić-Šipka S, Sabo TJ (2012) J Inorg Biochem 109:40
Soares MA, Mattos JL, Pujatti PB, Leal AS, dos Santos WG, dos Santos RGJ (2012) Radioanal Nucl Chem 292:61
Yildirim H, Köçkar F, Nakīboğlu C (2012) Afr J Biotech 11:12422
Ahn M-J, D’Cruz A, Vermorken JB, Chen J-P, Chitapanarux I, Dang HQT, Guminski A, Kannarunimit D, Lin T-Y, Ng WT, Park K-U, Chan ATC (2016) Oral Oncol 53:10
Mogorosi MM, Mahamo T, Moss JR, Mapolie SF, Slootweg JC, Lammertsma K, Smith GS (2011) J Organomet Chem 696:3585
Antonels NC, Therrien B, Moss JR, Smith GS (2009) Inorg Chem Commun 12:716
Doherty S, Knight JG, Scanlan TH, Elsegood MRJ, Clegg W (2002) J Organomet Chem 650:231
Chiririwa H, Ntuli F, Muzenda E, Muller A (2013) Transit Met Chem 38:393
Motswainyana WM, Onani MO, Madiehe AM, Saibu M, Thovhogi N, Lalancette RA (2013) J Inorg Biochem 129:112
Blacker AJ, Clarke ML, Loft MS, Mahon MF, Williams JMJ (1999) Organometallics 18:2867
Burger S, Therrien B, Süss-Fink G (2003) Eur J Inorg Chem 3099
Hedden D, Roundhill DM, Fultz WC, Rheingold AL (1986) Organometallics 5:336
Swanson RA, Haywood RS, Gibbons JB, Cordova KE, Patrick BO, Moore C, Rheingold AL, Daley CJA (2011) Inorg Chim Acta 368:74
Hii KK, Perera SD, Shaw BL, Thornton-Pett M (1994) J Chem Soc. Dalton Trans 1:103
Wong W-K, Sun C, Wong W-T (1997) J Chem Soc. Dalton Trans 18:3387
Pascu SI, Coleman KS, Cowley AR, Green MLH, Rees NH (2005) New J Chem 29:385
Margraf G, Pattacini R, Messaoudi A, Braunstein P (2006) Chem Commun 29:3098
Durran SE, Elsegood MRJ, Hammond SR, Smith MB (2007) Inorg Chem 46:2755
Crochet P, Gimeno J, García-Granda S, Borge J (2001) Organometallics 20:4369
Ramírez P, Contreras R, Valderrama M, Carmona D, Lahoz FJ, Balana AI (2008) J Organomet Chem 693:349
Acknowledgements
Thanks are gratefully extended to Mount Allison University, Saint Mary’s University, Université de Moncton, the Natural Science and Engineering Research Council of Canada (SAW, JM, PJM) and the Canada Research Chair Programme (SAW) for financial support. We also thank Deman Durant (Mount Allison University) for his expert technical assistance, and anonymous reviewers are thanked for their very helpful comments.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
St-Coeur, PD., Adams, M.E., Kenny, B.J. et al. Synthesis and characterization of iminophosphineplatinum(II) complexes of the type (κ2-P,N-2-Ph2PC6H4C(H)=NC6H4X)PtCl2 (X = OMe, F). Transit Met Chem 42, 693–701 (2017). https://doi.org/10.1007/s11243-017-0176-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11243-017-0176-2