
Abstract
Palladium(II) complexes have been obtained from the reactions of the iminophosphine ligands, (L1–L7), respectively, with [PdCl2(COD)] and [PdMeCl(COD)] in CH2Cl2 at room temperature. The palladium(II) complexes were characterised using elemental analysis, electro spray ionisation–mass spectrometry (ESI–MS), NMR (1H and 31P), IR spectroscopy and X-ray diffraction studies. Single-crystal X-ray diffraction analysis for complexes 2, 7 and 8 revealed that the complexes exhibited a slightly distorted square planar geometry. In vitro cytotoxic study results show that the palladium complexes exhibit moderate activity and block the proliferation of WHCO1 cells with an IC50 range of 19.02–45.27 μM, and IC50 range of 10.03–68.54 μM for the KYSE450 cell lines.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
During recent years, there has been a growing interest in the chemistry of ligands with both ‘hard’ nitrogen and ‘soft’ phosphorus donor atoms [1–3]. Metal complexes with N and P donor atoms display a variety of coordination possibilities beyond those of P–P or N–N ligands [4]. The hard ligand components can readily dissociate from a soft metal centre generating a vacant site on the metal ion for substrate binding. These ligands show a particular behaviour in binding to soft metal centres such as palladium(II) and platinum(II) that make their complexes good precursors in catalytic processes [5–8]. Among the most studied ligands with this characteristic are the pyridylphosphines and iminophosphines which have been widely reported in complexes with ruthenium [9], palladium [10], rhodium [11] and iridium [12]. In the last 3 decades, the interest towards platinum(II) and palladium(II) complexes containing N and S donor ligands has increased, resulting in the development of metal-based drugs exhibiting high anticancer activity together with reduced toxicity, compared with cisplatin and analogous compounds [13].
The development of palladium anticancer drugs has not been promising, probably because their design has been based on structure–activity considerations generated from platinum antitumor drugs. Bearing in mind that Pd(II) complexes are about 105 times more reactive than their Pt(II) analogues, the low antitumoral activity of Pd compounds has been attributed to very rapid hydrolysis of the leaving groups that dissociate readily in solution, leading to reactive species far from their pharmacological targets [14]. Palladium is a suitable candidate for metallodrugs because it displays structural properties similar to those of platinum and also exhibits promising cytotoxicity.
As part of our continuing interest in the synthesis of transition metal complexes of biological molecules, we have investigated the coordination behaviour of iminophosphines towards palladium(II), with a view to develop new transition metal pharmaceuticals. This paper describes the synthesis, structural characterisation and preliminary biological activity of palladium iminophosphine complexes.
Experimental
All manipulations were carried out under an atmosphere of nitrogen or argon using standard Schlenk techniques. All other glassware was thoroughly dried at 210 °C for at least 4 h prior to use. [PdCl2(COD)], [PdMeCl(COD)], complexes 1, 2 and ligands L1–L7 were prepared according to known literature procedures [15–17]. Melting points were determined on a Kofler hotstage microscope (Reichart Thermovar) and are uncorrected. Microanalysis data were obtained using a Carlo Erba EA1108 elemental analyser. Infrared spectra were recorded on a Perkin-Elmer 1000 FT–IR spectrometer, as KBr discs for solids. All data are given in wavenumbers (cm−1). 1H, 13C and 31P NMR spectra were recorded on a Varian Unity-400 instrument. Mass spectra (EI) were recorded using a JEOL-MATE(II) GC–MS instrument. X-ray intensity data were collected on a Nonius Kappa-CCD diffractometer with 1.5 kW graphite monochromated Mo-Kα radiation.
MTT assay
Thousand five hundred cells were seeded per well in 90 μl DMEM in 96 well plates. Cells were incubated for 24 h, then, test samples were plated at a range of concentrations in 10 μl media, with a final concentration of 0.2 % DMSO. After 48 h of incubation, the cells were observed under a phase contrast microscope and the general appearance of the cells together with confluency status and presence of precipitate if any was recorded.
Ten microlitres of MTT reagent was added per well at the end of the experiment, and the plates incubated for 4 h at 37 °C. Hundred microlitres of solubilisation solution was then added to each well, and plates were incubated at 37 °C overnight. After 16 h, the plates were read at 595 nm on an Anthos microplate reader 2001.
IC50 data analysis
The resulting dose–response curve was analysed by nonlinear regression analysis [nonlinear regression (sigmoidal dose–response with variable slope)] using GraphPad Prism version 4.00 for Windows (GraphPad Software, San Diego California USA, www.graphpad.com) to yield an IC50 value which is specific for the compound against that particular cell line. The formula used is as follows:
where Y is the absorbance reading, X is the concentration of the compound, top is the maximum absorbance, bottom is the minimum absorbance (also the absorbance of the medium blank) and the hillslope is the gradient of the curve.
Each experiment was repeated at least three times.
General procedure for the preparation of palladium(II) dichloride complexes (1–7)
To a solution of the appropriate ligand (L1–L7) in dry CH2Cl2 (10 ml) was added an equimolar amount of [PdCl2(COD)] dissolved in dry CH2Cl2 (10 ml). The reaction was allowed to stir at room temperature for 4 h before reducing the solvent to ca 5 ml and precipitating the products using hexane. The products were filtered off, washed with Et2O and dried under vacuum.
Complex 1
Yellow crystalline solid. Yield 70 %. m.p.: 158–160 °C. C22H22Cl2NPd: Found: C, 51.69 %; H, 4.12 %; N, 2.72 %. Calcd. C, 51.94 %; H, 4.36 %; N, 2.75 %. IR(υC=N, imine: cm−1): −1632(s). 1H-NMR: (400 MHz, CDCl3) δH 8.85 (s, 1H, Ha) 7.89 (m, 3H, ArH) 7.66 (dd, 4H, J = 6.3 Hz, J = 7.9 Hz, ArH) 7.55 (ddd, 7H, J = 2.0 Hz, J = 5.2 Hz, J = 8.5 Hz, ArH) 2.95 (dddd, 2H, J = 5.4 Hz, J = 8.3 Hz, J = 13.8 Hz, J = 16.7 Hz, Hb) 1.18 (m, 2H, Hc) 0.53 (t, 3H, J = 7.3 Hz, Hd) 31P NMR: δ 30.85 (s). EI–MS: m/z 437.30 [M–2Cl]+.
Complex 2
Yellow powder. Yield 82 %. m.p. 168–170 °C. C22H22Cl2NPPd: Found: C, 51.79 %; H, 4.12 %; N, 2.52 %. Calcd. C, 51.94 %; H, 4.36 %; N, 2.75 %. IR(υC=N, imine: cm−1): −1630(s). 1H NMR: (dmso-d6) δH 8.02 (s, 1H) 7.72 (m, 2H, ArH) 7.51 (m, 11H, ArH) 6.94 (ddd, 1H, J = 0.5 Hz, J = 7.7 Hz, J = 10.6 Hz, Ar) 5.57 (dtd, 1H, J = 0.6 Hz, J = 6.4 Hz, J = 13.0 Hz) 1.16 (d, 6H, J = 6.6 Hz). 31P NMR: δ 31.62 (s). EI–MS: m/z 474.68 [M–Cl]+.
Complex 3
Yellow crystalline powder. Yield 75 %. m.p. 200–202 °C. C31H32Cl2NPPd: Found: C, 59.19 %; H 5.12 %; N 2.42 %. Calcd. C, 59.39 %; H, 5.15 %; N 2.23 %. IR(υC=N, imine: cm−1): −1624(s). 1H NMR: (dmso-d6) δH 8.74 (s, 1H, HC = N) 7.58 (m, 14H ArH) 7.16 (m, 7.16, 4H, ArH) 1.19 (d, 6H, J = 6.7 Hz, –CH3) 0.77 (t, 6H, J = 6.3 Hz, –CH3). 13C NMR: (dmso-d6) δ 169.31, 135.28, 133.94 (d, J CP = 11.0 Hz), 132.89, 129.74 (d, J CP = 11.8 Hz), 128.90, 123.80, 28.73, 27.99, 24.31, 23.42. 31P NMR: δ 33.34 (s). EI–MS: m/z 591.44 [M–Cl]+.
Complex 4
Yellow powder. Yield 76 %. m.p.: 200–202 °C. C26H22Cl2NPPd: Found: C, 56.26 %; H 3.72 %; N, 2.72 %. Calcd. C, 56.09 %; H, 3.98 %; N, 2.52 %. IR(υC=N, imine: cm−1): −1627(s). 1H-NMR: (400 MHz, d6-dmso) δH 7.81 (m, 1H) 7.63 (m, 1H) 7.49 (m, 1H) 7.09 (m, 1H) 7.08 (m, 1H) 6.91 (dd, 1H, J = 4.6 Hz, J = 10.9 Hz) 6.49 (dd, 1H, J 1.1 Hz, J = 8.1 Hz) 4.22 (t, 2H, J = 12.6 Hz). 31P NMR: δ 34.11 (s).
Complex 5
Yellow powder. Yield 70 % yield m.p.: 210–212 °C. C25H21N2PPd: Found: C, 53.69 %; H 3.92 %; N 4.92 %. Calcd. C, 53.84 %; H, 3.80 %; N 5.02 %. IR(υC=N, imine: cm−1): −1626(s). 1H-NMR: (400 MHz, d6-dmso) δH 8.82 (s, 1H, Ha) 8.66 (d, 1H, J = 2.3 Hz, Hg) 8.49 (d, 1H, J = 3.8 Hz, Hf), 8.34 (s, 1H, Ha) 7.91 (dd, 1H, J = 4.1 Hz, J = 7.3 Hz, He) 7.68 (m, 2H, ArH) 7.55 (dd, 4H, J = 6.7 Hz, J = 8.3 Hz, ArH) 7.43 (m, 4H, ArH) 7.14 (m, 1H, Hd) 5.45 (s, 2H, Hb) 31P NMR: δ 37.4 (s). EI–MS: m/z 557.75 [M]+.
Complex 6
Yellow powder. Yield 78 %. m.p.: 200–202 °C. C24H20Cl2NOPPd: Found: C, 52.59 %; H, 3.42 %; N, 2.72 %. Calcd. C, 52.72 %; H, 3.69 %; N, 2.56 %. IR(υC=N, imine: cm−1): −1630(s). 1H-NMR: (400 MHz, d6-dmso) 8.72 (s, 1H, Ha) 7.98–8.08 (m, 1H, ArH) 7.75(t, 1H, J = 1.5 Hz, J = 7.7 Hz, ArH) 7.58 (m, 2H, ArH) 7.45 (td, 4H, J = 2.9 Hz, 4.8 Hz, ArH) 7.05 (dd, 1H, J = 8.1 Hz, J = 1.8 Hz, Hf) 7.24 (m, 5H, ArH) 6.55 (d, 1H, J = 2.9 Hz, He) 6.38 (dd, 1H, J = 2.9 Hz, Hd) 5.78 (t, 1H, J = 1.5 Hz, J = 7.7 Hz, ArH) 5.56 (s, 2H, Hb) 31P NMR: δ 32.3(s). EI–MS: m/z 546.71 [M]+.
Complex 7
Orange crystalline powder. Yield 65 %. m.p. 234–235 °C. C24H20Cl2NPPdS: Found: C, 51.48 %; H, 3.32 %; N, 2.72 %, S, 5.86. Calcd. C, 51.22 %; H, 3.58 %; N, 2.49 %, S, 5.70 %. IR(υC=N, imine: cm−1): −1628(s). 1H NMR: (dmso-d6) δH 8.80 (s, 1H) 8.03–7.92 (m, 2H, ArH) 7.86–7.93 (m, 1H, ArH) 7.74 (m, 1H, ArH) 7.52–7.60 (m, 2H, ArH) 7.40 (td, 4H, J = 4.9 Hz, J = 2.9 Hz, ArH) 7.18 (dd, 4H, J = 7.8 Hz, J = 4.9 Hz, ArH) 6.99–7.07 (m, 2H) 6.90 (dd, 1H, J = 2.9 Hz, J = 2.0 Hz) 5.66 (s, 2H). 31P NMR: δ 31.8(s). EI–MS: m/z 562.73 [M]+.
General procedure for the preparation of palladium(II) chloromethyl complexes (8–11)
To the appropriate ligand (L4–L7) in dry CH2Cl2 (10 ml) was added [PdMeCl(COD)] also in dry CH2Cl2 (10 ml) in an equimolar amount. The reaction was allowed to stir at room temperature for ca 2 h before reducing the solvent to ca 5 ml and precipitating out the products with hexane, filtering under gravity and washing the precipitate with dry Et2O and drying under vacuum for 4 h.
Complex 8
Pale orange powder. Yield 85 %. m.p.: 168–170 °C. C27H25ClNPPd: Found: C, 60.72 %; H, 4.51 %; N, 2.81 %. Calcd. C, 60.46 %; H, 4.70 %; N, 2.61 %. IR(υC=N, imine: cm−1): −1628(s). 1H-NMR: (400 MHz, CDCl3) δH 8.81 (s, 1H, Ha) 7.91 (dd, 1H, J = 4.3 Hz, J = 46.3 Hz, ArH) 7.83 (t, J = 7.6 Hz, ArH) 7.69 (t, 1H, J = 7.4 Hz, ArH) 7.56 (m, 2H, ArH) 7.45 (dt, 4H, J = 2.3 Hz, 7.6 Hz, ArH) 7.29 (m, 1H, ArH) 7.18 (m, 8H, ArH) 7.09 (dd, 1H, J = 7.7 Hz, J = 10.5 Hz, ArH) 5.10 (s, 2H, Hb) 0.18 (d, 3H, J = 1.2 Hz, CH 3). 31P NMR: δ 38.5 (s). EI–MS: m/z 521.75 [M–CH3]+.
Complex 9
Pale yellow powder. Yield 85 %. m.p.: 178–180 °C. C26H24ClN2PPd: Found: C, 58.19 %; H, 4.12 %; N, 5.54 %. Calcd. C, 58.12 %; H, 4.50 %; N, 5.21 %. IR(υC=N, imine: cm−1): −1628(s). 1H NMR: (400 MHz, CDCl3) δH 8.80 (d, 1H, J = 1.6 Hz, Hf) 8.63 (s, 1H, Ha) 8.47 (dd, 1H, J = 1.6 Hz, J = 4.8 Hz, Hg) 7.88 (ddd, 1H, J = 1.3 Hz, J = 4.1 Hz, J = 7.6 Hz, ArH) 7.66 (m, 2H, ArH) 7.79 (tt, 1H, J = 1.4 Hz, J = 7.6 Hz, ArH) 7.52 (m, 2H, ArH) 7.39 (ddd, 4H, J = 2.0 Hz, J = 5.1 Hz, J = 7.3 Hz, ArH) 5.41 (s, 2H, Hb) 7.11 (m, 6H) 0.26 (d, 3H, J = 3.3 Hz, CH 3). 31P NMR: δ 37.4 (s) EI–MS: m/z 537.13 [M]+.
Complex 10
Yellow powder. Yield 75 %. m.p.: 195–196 °C. C25H23ClNOPPd: Found: C, 57.19 %; H, 4.12 %; N, 2.72 %. Calc. C, 57.07 %; H, 4.40 %; N, 2.66 %. IR(υC=N, imine: cm−1): −1631(s). 1H-NMR: (400 MHz, CDCl3) δH 8.65 (s, 1H, Ha) 7.86 (ddd, 1H, J = 1.2 Hz, J = 4.1 Hz, J = 7.4 Hz, ArH) 7.77 (t, 1H, J = 7.5 Hz, ArH) 7.66 (t, 1H, J = 7.5 Hz, ArH) 7.51 (m, 6H, ArH) 7.34 (m, 4H, ArH) 7.18 (m, 1H, Hf) 6.43 (m, 1H, Hd) 5.39 (s, 2H, Hb) 0.23 (d, 3H, J = 3.2 Hz, CH 3). 31P NMR: δ 37.6 (s). EI–MS: m/z 526.03 [M]+.
Complex 11
Pale yellow powder. Yield 80 %. m.p.: 186–188 °C. C25H23ClNPPdS: Found: C, 55.19 %; H, 4.08 %; N, 2.38 %; S, 6.05. Calcd. C, 55.36 %; H, 4.27 %; N, 2.58 %; S, 5.91. IR(υC=N, imine: cm−1): −1629(s). 1H-NMR: (400 MHz, CDCl3) δH 8.76 (s, 1H, Ha) 7.87 (m, 1H) 7.80 (t, 1H, J = 7.5 Hz) 7.69 (t, 1H, J = 7.5 Hz, Hf) 7.54 (dd, 2H, J = 6.5 Hz, J = 8.2 Hz, ArH) 7.44 (m, 5H, ArH) 7.17 (td, 5H, J = 6.1 Hz, J = 12.2 Hz, ArH) 7.07 (d, 1H, J = 2.4 Hz, He) 6.96 (dd, 1H, J = 3.5 Hz, J = 5.1 Hz, Hd) 5.57 (s, 2H, Hb) 0.25 (d, 3H, J = 3.3 Hz, CH 3) 31P NMR: δ 37.6 (s). EI–MS: m/z 531.8 [M–CH3]+.
X-ray crystal structure determination
Crystals suitable for single-crystal X-ray diffraction for complexes 2, 7 and 8 were obtained by slow evaporation of a dmso-d6-CH2Cl2 solution of the complex at room temperature. All X-ray intensity data were collected on a Nonius Kappa-CCD diffractometer with 1.5 kW graphite monochromated Mo-Kα radiation.
The structures were solved by direct methods using SHELXS-97 and refined employing full-matrix least squares with the programme SHELXL-97 refining on F2. Packing diagrams were produced using the programme PovRay and graphic interface X-seed [18]. Crystallographic data for the structure determinations are listed in Table 1.
Results and discussion
Treatment of the iminophosphine ligands (L1–L7), with [PdCl2(COD)] in CH2Cl2 at room temperature afforded the palladium iminophosphine complexes 1–7, respectively, and palladium methylchloride complexes 8–11 were obtained from the reaction of ligands (L4–L7) (Scheme 1).

Synthesis of palladium dichloride complexes
The reaction was allowed to stir in dry CH2Cl2 at room temperature for 8 h. The solvent was reduced and product precipitated out with Et2O. Further precipitation was achieved by allowing the products to crystallize slowly at −16 °C giving pale yellow/orange crystals in reasonable yields. These palladium methylchloride complexes are much more soluble than the palladium dichloride complexes.
The ligands show a distinctive stretching frequency, ν(C=N) at between 1,629 and 1,636 cm−1 which agrees with previously reported values for iminophosphine ligands [19]. Upon complexation, the peaks shift to lower frequencies than in the ligands at 1,624–1,630 cm−1. This is due to increased electron density on the metal upon coordination of the imine moiety to the metal centre.
The 1H NMR spectra of complexes 8–11 showed imine protons in the region δ 8.63–8.81 ppm. The observed upfield shifts of 0.21–0.28 ppm with respect to the free ligands further confirmed coordination of the imine nitrogen to the metal centre. A downfield shift of δ 0.18–0.25 ppm was also observed for the methylene signals, due to the coordination of the adjacent imine nitrogen thereby deshielding these protons. No significant chemical shifts were observed for the olefinic signals of the furyl and the thiophenyl with respect to those of the free ligands, suggesting that these groups did not participate in bonding with the metal centre. The 31P NMR spectra of complexes 8–11 showed the expected downfield shift to δ 37.4–38.5 ppm with respect to the free ligands which appeared at δ −13.2 to −13.9 ppm, due to coordination of the phosphine moiety to the palladium metal centre. The appearance of one signal in the 31P NMR spectra also suggests that only one species had been formed.
Structural description of complexes 2, 7 and 8
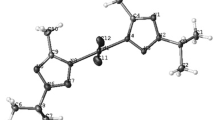
The selected bond lengths and angles for complexes 2 [25], 7 and 8 [26] are summarised in Tables 2, 3 and 4. Their molecular structures are shown in Figs. 1, 2 and 3.

Molecular structure of complex 2. All non-hydrogen atoms were presented with ellipsoidal model with probability level 40 %. The asymmetric unit contains the organometallic compound and a DMSO solvent molecule

Molecular structure of complex 7. All non-hydrogen atoms were presented with ellipsoidal model with probability level 40 %

Molecular structure of 8 showing the atomic numbering scheme. All non-hydrogen atoms were presented with ellipsoidal model with probability level 40 %
The coordination around the palladium is slightly distorted from the ideal square planar geometry. The main distortion is the NPdP bite angle of 86.13(5)° similar to other palladium complexes with iminophosphines [20]. The Pd–P distance (2.2189(6) Å) is within the expected range and the length of the carbon–nitrogen double bond is also within the expected range.
The molecular structure revealed a slightly distorted square planar geometry around the palladium metal centre. The ligand was shown to bind in the expected K2−P^N fashion with a bite angle P(4)-Pd(1)-N(24) of 86.38(9)°. The angle deviated slightly from the expected 90°, presumably due to the strain imposed by the six-membered chelate ring P(4)-C(17)-C(22)-C(23)-N(24)-Pd(1). This reduction in the bite angle was compensated for by an increase in the Cl(2)-Pd(1)-P(4) angle of 91.49(8)°. This deviation of the bite angle from 90° has been observed for similar complexes with iminophosphines [20–22].
The Pd–P distances of 2.1940(7) Å are within the expected range and close to the values determined for the monohalide complex [PdMeCl(L)] (2.1925(9) Å and in the dihalide complexes of the same ligand studied by Coleman et al. [23, 24]. The Pd–N distances are similar to those found for Pd(II) complexes in the same series. The methyl group is trans to the nitrogen atom of the ligand. The torsion angle Pd(1)-P(4)-C(17)-C(22) = 39.9(2) Å indicates that the =CHC6H4- unit lies above the PdMeCl(P,N) plane.
Preliminary biological evaluation
The palladium complexes synthesised were evaluated for their cytotoxic activity using the MTT assay to determine IC50 values against the oesophageal cancer cell lines WHCO1 and KYSE450 for selected complexes. Dose–response curves for each of the complexes were performed against the cell lines. Each experiment was performed in triplicate.
In vitro anticancer activity
The cytotoxicity of the palladium complexes was examined. In WHCO1 cell lines, complex 2 had the highest activity with an IC50 value of 19.02 μM and the least active was complex 10 with an IC50 value of 45.27 μM. In comparison with cisplatin which has an IC50 of 15–18 μM in WHCO1 cell lines, the palladium complexes described here displayed moderate activity against oesophageal cancer cell lines. In KYSE 450 cells, highest activity was observed with complex 2 with an IC50 of 10.03 μM and lowest activity with complex 10 with an IC50 of 68.54 μM as shown in Table 5.
The palladium complexes 1 and 2 were the dichlorides which were very soluble in DMSO as compared with the other dichloride complexes 3–7 which were insoluble. Complexes 8–11 were the chloromethyl derivatives and these were very soluble in DMSO. Introduction of a methyl group into the dichloride complexes increased the solubility of the complexes.
Conclusion
Palladium dichloride and methyl chloride complexes have been prepared and characterised using standard spectroscopic and analytical techniques. Single-crystal X-ray diffraction revealed that in complexes 2, 7 and 8, there is a slightly distorted square planar geometry around the palladium metal centre and the Pd–P distances are within the expected ranges. Three complexes displayed moderate to good cytotoxicity for the indicated cell lines. Biological activity of some of the palladium(II) complexes could not be determined due to poor solubility. Further investigations are under way to increase the solubility of the palladium complexes and to explore their anticancer activity.
Supplementary materials
Crystallographic data for the structural analysis have been deposited with the Cambridge Crystallographic Data Centre, CCDC Nos. 825329, 841468 and 818609 for complexes 2, 7 and 8, respectively. Copies of this information may be obtained free of charge from The Director, CCDC, 12 Union Road, Cambridge CB2 1EZ, UK, Fax: +44-1223-336033, e-mail: deposit@ccdc.cam.ac.uk of www: http://www.ccdc.cam.ac.uk.
References
Dilworth JR, Howe SD, Hutson AJ, Miller JR, Silver J, Thomson RM, Harman M, Hursthouse MB (1994) J Chem Soc Dalton Trans (24):3553–3562
Dilworth JR, Hutson AJ, Lewis JS, Miller JR, Zheng Y, Chen Q, Zubieta J, (1996) J Chem Soc Dalton Trans (6):1093–1104
Dilworth JR, Miller JR, Wheatley N, Baker MJ, Sunley G, (1995) J Chem Soc Chem Commun (15):1579–1581
Kapteijn GM, Spee MPR, Grove DM, Kooijman H, Spek AL, van Koten G (1996) Organometallics 15:1405
Barbaro P, Bianchini C, Laschi F, Midollini S, Moneti S, Scapacci G, Zanello P (1994) Inorg Chem 33:1622
Stoccoro S, Chelucci G, Zucca A, Cinellu MAG, Minghetti G Manassero J, (1996) J Chem Soc Dalton Trans (7):1295–1299
Wehman P, van Donge HMA, Hagos A, Kamer PCJ, van Leeuven PWNM (1997) J Organomet Chem 535:183
Bacchi A, Carcelli M, Costa M, Leporati A, Leporati E, Pelagatti P, Pelizzi C, Pelizzi G (1997) J Organomet Chem 535:107
Gao J-X, Ikariya T, Noyoro R (1996) Organometallics 15:1087
Brunner H, Fusrt J (1994) Inorg Chim Acta 220:63
Rauchfuss TB (1978) J Organomet Chem 162:C19
Jeffery JC, Rauchfuss TB, Tucker PA (1980) Inorg Chem 19:3306
Faraglia G, Fregona D, Sitran S, Giovagnini L, Marzano C, Baccichetti F, Casellato U, Graziani R (2001) J Inorg Biochem 83:31
Gill DS (1984) In: Hacker MP, Douple EB, Krakoff IH (eds) Platinum coordination complexes in cancer chemotherapy. Nijhoff, Boston, p 267
Chatt J, Vallarino LM, Venanzi LM (1957) J Chem Soc Part V (678):3413–3416
Bailey CT, Linsesky GCJ (1985) J Chem Edu 62:896
Mogorosi MM, Mahamo T, Moss JR, Mapolie SF, Slootweg JC, Lammertsma K, Smith GS (2011) J Organomet Chem 696:3585–3592
Barbour LJ (2001) X-Seed: a software tool for supramolecular crystallography. J Supromol Chem 1:189
Ghilardi CA, Midollini S, Moneti S, Orlandini A, Scapacci G (1992) J Chem Soc Dalton Trans 23:3371–3376
Sanchez G, Serrano JL, Moral MA, Perez J, Molins E, Lopez G (1999) Polyhedron 18:3057
Ankersmit HA, Bjorn HL, Kooijman H, Spek AL, Vrieze K, van Koten G (1996), Inorga Chim Acta, 252:141–155
Koprowski M, Sebastian RM, Maraval V, Zablocka M, Cadierno V, Donnadieu B, Igau A, Caminade AM, Majoral JP (2002) Organometallics 21:4680
Coleman KS, Green MLH, Pascu SI, Rees NH, Cowley AR, Rees LHJ (2001) Chem Soc Dalton Trans (22):3384–3395
Pascu SI, Coleman KS, Cowley AR, Green MLH, Rees NH (2005) New J Chem 29:385
Chiririwa H, Meijboom R, Omondi B (2011) Acta Cryst E67:m608–m609
Chiririwa H, Meijboom R (2011) Acta Cryst E67:m1498
Acknowledgments
Financial support from the University of Cape Town, the National Research Foundation of South Africa, CANSA, Project AuTEK (Mintek and Harmony Gold, South Africa) and a donation of metal salts from the Anglo Platinum Corporation is gratefully acknowledged.
Author information
Authors and Affiliations
Corresponding author
Additional information
John R. Moss: Deceased.
Rights and permissions
About this article
Cite this article
Chiririwa, H., Moss, J.R., Hendricks, D. et al. Synthesis, characterisation and in vitro evaluation of palladium(II) iminophosphine complexes for anticancer activity. Transition Met Chem 38, 165–172 (2013). https://doi.org/10.1007/s11243-012-9674-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11243-012-9674-4